
Abstract
Background
Alexander disease (ALX) is a rare neurological disorder characterized by white matter degeneration and cytoplasmic inclusions in astrocytes called Rosenthal fibers, labeled by antibodies against glial fibrillary acidic protein (GFAP). Three subtypes are distinguished according to age at onset: infantile (under age 2), juvenile (age 2 to 12) and adult (over age 12). Following the identification of heterozygous mutations in GFAP that cause this disease, cases of adult-onset ALX have been increasingly reported.
Case Presentation
We present a 60-year-old Japanese man with an unremarkable past and no family history of ALX. After head trauma in a traffic accident at the age of 46, his character changed, and dementia and dysarthria developed, but he remained independent. Spastic paresis and dysphagia were observed at age 57 and 59, respectively, and worsened progressively. Neurological examination at the age of 60 revealed dementia, pseudobulbar palsy, left-side predominant spastic tetraparesis, axial rigidity, bradykinesia and gaze-evoked nystagmus. Brain MRI showed tadpole-like atrophy of the brainstem, caused by marked atrophy of the medulla oblongata, cervical spinal cord and midbrain tegmentum, with an intact pontine base. Analysis of the GFAP gene revealed a heterozygous missense mutation, c.827G>T, p.R276L, which was already shown to be pathogenic in a case of pathologically proven hereditary adult-onset ALX.
Conclusion
The typical tadpole-like appearance of the brainstem is strongly suggestive of adult-onset ALX, and should lead to a genetic investigation of the GFAP gene. The unusual feature of this patient is the symmetrical involvement of the basal ganglia, which is rarely observed in the adult form of the disease. More patients must be examined to confirm, clinically and neuroradiologically, extrapyramidal involvement of the basal ganglia in adult-onset ALX.
Similar content being viewed by others
Background
Alexander disease (ALX) (OMIM #203450), originally described by Alexander in 1949 [1], is a rare and fatal disease of the central nervous system caused by astrocyte dysfunction [2, 3]. The pathological hallmark of the disease is the accumulation of ubiquitinated intracytoplasmic inclusions in astrocytes, called Rosenthal fibers, which are composed of glial fibrillary acidic protein (GFAP), the main intermediate filament of astrocytes, in association with the small heat shock proteins, HSP27 and αB-crystallin [4].
The clinical features of typical infantile-onset ALX, with onset before the age of two, include megalencephaly, seizures, spastic paresis and psychomotor deterioration with leukoencephalopathy characterized by white matter abnormalities predominating in the frontal lobes. As cases accumulate, however, atypical patients have also been described. Adult-onset ALX, with onset over the age of 12, is characterized by more slowly progressive bulbar or pseudobulbar palsy, spastic paresis, ataxia, palatal myoclonus and essentially normal psychic and intellectual functions. Juvenile-onset ALX, with onset between age 2 and 12, bridges the gap between infantile and adult forms of the disease. However, it is not yet clear whether these three categories are the same disease. The clinical presentations are diverse; the only common feature is the presence of pathologically proven Rosenthal fibers [5, 6].
Owing to the discovery of inclusion bodies indistinguishable from Rosenthal fibers in fatal GFAP transgenic mice overexpressing human GFAP in astrocytes [7], de novo heterozygous mutations in the gene encoding GFAP, the main component of Rosenthal fibers, have been identified in patients with the infantile form of ALX [8]. GFAP gene mutations have also been identified in the juvenile [8, 9] and adult [10, 11] forms. These three clinically diverse forms are now widely accepted to be part of the same spectrum [12].
Each subtype has characteristic MRI findings. Cerebral white matter abnormalities, predominating in the frontal lobes, are typical of the infantile form of ALX [13], whereas nodular brainstem lesions and a kind of "garland" along the ventricular wall are seen, with contrast enhancement, in the juvenile form [14, 15]. The adult form has a unique tadpole-like feature, caused by marked atrophy of the medulla oblongata and cervical spinal cord with an intact pontine base [11].
Here we present a case of sporadic adult-onset ALX with specific MRI findings: a typical tadpole-like brainstem, but also symmetric involvement of the basal ganglia, which is unusual in the adult form of ALX.
Case Presentation
This Japanese patient, with an unremarkable past and no family history had been healthy until he was involved in a traffic accident at the age of 46. He suffered a bilateral brain contusion in the fronto-orbital areas, predominating on the left side. He did not lose his consciousness, but retrograde amnesia was seen. There was no hypoxia. After this accident, his character changed. He became querellous, could no longer manage his shop, and soon retired. He gradually became taciturn and his pronunciation became unclear, but he remained independent. At age 57, he began to drag his left foot as he walked, and this symptom gradually worsened. At the age of 59, progressive dysphagia appeared. He was referred to our hospital at the age of 60. Neurological examination revealed pseudobulbar palsy including aphonia, emotional incontinence and dysphagia, left-side predominant tetraparesis. The tone of the limb muscles was spastic, with bilateral positive Babinski signs. Axial rigidity, bradykinesia and retropulsion were observed, but no tremor or palatal myoclonus. Cerebellar ataxia was ambiguous because of spastic tetraparesis requiring use of a wheelchair, but bilateral gaze-evoked nystagmus was seen. He was clearly demented and angrily refused everything he was asked to do by shaking his head instead of speaking because of aphonia. Further evaluation of his dementia was impossible.
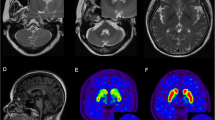
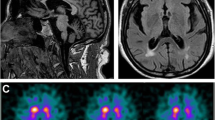
Laboratory tests, including hematology, routine blood chemistry, and analyses of urine and cerebrospinal fluid, were unremarkable. In addition to the bilateral contusion in the fronto-orbital areas, especially on the left side, brain MRI showed a tadpole-like brainstem, caused by marked atrophy from the medulla oblongata to the cervical spinal cord, sparing the pontine base. Marked atrophy of the midbrain tegmentum, mild cerebellar atrophy with a little enlargement of the fourth ventricle, and slight cerebral atrophy were also seen. The typical periventricular lesions (ventricular garlands [15]) and leukoencephalopathy were not seen, however, several lacunae were observed bilaterally in deep white matter. The posterior part of globus pallidus was involved bilaterally, as shown by a signal change without contrast enhancement (Figure 1).

Brain MRI of a patient with adult-onset Alexander disease. a-f: T2-weighted axial images showing marked atrophy of the medulla oblongata (a) with slight cerebellar atrophy (a, b) but little atrophy of the pontine base (b), enlargement of the fourth ventricle (b), atrophy of the midbrain, especially the dorsal part (c), bilateral changes in the posterior part of globus pallidus (d), bilateral lesions of the fronto-orbital areas, predominating on the left, caused by brain contusion (d, e, g, h), moderate cortical atrophy with ventricular enlargement (e, f), and bilateral lacunae in the deep white matter, but no leukoencephalopathy (f). g and k: T1-weighted images of the lesions on axial (g) and sagittal (k) sections. h, i and j: FLAIR images of the lesions on axial (h, i) and coronal (j) sections. The lesions on coronal (j) and sagittal (k) sections are indicated by arrows. Note the absence of ventricular garlands [15]. l: T1-weighted sagittal section showing typical tadpole-like brainstem atrophy, consisting of marked cervico-medullary atrophy with an intact pontine base; note that atrophy of the midbrain tegmentum also contributes to the formation of the tadpole.
With informed consent, the GFAP gene was sequenced, and a heterogeneous missense mutation was detected in exon 5 (c.827G>T), causing a change of arginine to leucine at amino acid position 276 (p.R276L). We have already described this mutation in a patient with pathologically proven hereditary adult-onset ALX [11]. There was no relationship between the present patient and the family previously reported [11]. According to an interview, however, both families originated from the same region of Japan.
Discussion
We have described here a new patient with sporadic adult-onset ALX, and have identified a heterozygous missense mutation in the GFAP gene, c.827G>T, p.R276L, which was already shown to be pathogenic in a case of pathologically proven hereditary adult-onset ALX [11]. This mutation has not been seen for the last 7 years. Thus, this report reconfirms the pathogenetic nature of the mutation and the clinical picture of this form of the disease. So far, the phenotype associated with the R276L mutation is adult-onset spastic ataxia with pseudobulbar symptoms.
Following identification of the GFAP gene as responsible for ALX [8], the adult form has been increasingly reported [10, 11, 15–36]. However, only a few cases have been pathologically proven [11, 16, 17, 21, 28]; the rest were diagnosed as having ALX only by molecular testing. Since missense mutations may only be polymorphisms [37], their pathogenicity must be accepted with caution. Indeed, the E223Q mutation, identified in a patient with neurological deficits and radiological findings atypical for adult-onset ALX [38], is now classified as a polymorphism [21]. Therefore, to avoid this kind of confusion, it is worthwhile defining the typical presentation of adult-onset ALX, including age at onset, cardinal clinical symptoms, neuroradiological findings and clinical course, as described in published reports [see review article, ref [33]].
To date, at least 24 reports of adult onset ALX, including over 40 patients, with 22 different missense mutations in the GFAP gene, have been published (Table 1) [10, 11, 15–36]. There are no sex differences, and half of the cases are familial, consistent with autosomal dominant transmission. The mean age at onset is in the late thirties, although an asymptomatic carrier over age 60 has been described [30]. The cardinal triad of the clinical presentation is pseudobulbar or bulbar palsy, spastic paresis (usually hemiparesis, not paraparesis, at onset), and ataxia, each of which is observed in approximately 70% of patients. Palatal myoclonus is observed in only one third, although it is specific to ALX, and the key finding for a diagnosis, especially in hereditary cases [10]. Mental function is usually preserved, although our patient was obviously demented. Dementia cannot, however, be considered a symptom of adult-onset ALX in this patient, because of his cerebral contusion.
As for the MRI findings [see review article, ref [34]], most of the cases had medullary abnormalities (either signal abnormalities or atrophy), and the marked tadpole-like atrophy of the medulla oblongata and cervical spinal cord with an intact pontine base [11]. We would like to emphasize that not only cervicomedullary atrophy with an intact pontine base, but also severe atrophy of the midbrain tegmentum contributes to the formation of the tadpole. This unusual atrophy is quite specific to adult-onset ALX, and 88% of the patients with adult-onset ALX in the literature showed marked medullary atrophy (Table 1). Thus, awareness of this MRI pattern allows effective selection of the patients who need genetic investigations for mutations in the GFAP gene [34]. Indeed, we could have diagnosed adult-onset ALX in the present patient on the basis of this form of brainstem atrophy.
Approximately half of the patients had deep white matter lesions or periventricular rims, although not always with frontal predominance as in infantile-onset ALX; the absence of these abnormalities is significantly associated with older age at onset (average age at onset; negative 43.7 ± 14.1 (n = 18) vs. positive 30.9 ± 12.8 (n = 18), p = 0.008), consistent with previous study [34]. Similarly, nodular lesions in the brainstem are observed in about half of the patients, and are significantly associated with a younger age at onset (average age at onset; positive 28.2 ± 11.8 (n = 13) vs. negative 43.6 ± 13.9 (n = 18), p = 0.003).
Besides the typical clinical and neuroradiological features, this case of adult-onset ALX is instructive because of the bilateral involvement of the basal ganglia. This is not uncommon in infantile or juvenile-onset ALX, and is one of the radiological criteria for the diagnosis [13], but has rarely been observed in the adult form of the disease. Symmetrical striatal lesions were observed in one patient, however, with hypointensity on T2-weighted MRI [22]. Basal ganglia lesions with hyperintensity on T2-weighted MRI, such as spotty lesions [19] and bilateral lesions in the lateral putamen [30], have occasionally been reported; both of these signs are ambiguous, however, and do not resemble those of our patient. Thus, the symmetrical lesions in our patient are interesting findings in adult-onset ALX, and might be related to a rigid-bradykinesia type parkinsonism, although this is uncertain because spastic paresis masks the parkinsonism. The clinical signs and symptoms of basal ganglia involvement, such as parkinsonism [patient 1 of ref [21]], diffuse bradykinesia [22], and rigidity of the arms [29] are rarely reported. The obvious parkinsonism reported in one patient was induced by valproate [32].
Conclusion
We described here a new sporadic case of genetically confirmed adult-onset ALX. The tadpole-like brainstem atrophy is quite specific for adult-onset ALX. Thus, faced with a patient with progressive spastic ataxia, bulbar or pseudobulbar signs, and the typical tadpole on MRI, a genetic investigation of the GFAP gene is strongly recommended. The unusual feature of our patient is the obvious symmetrical involvement of the basal ganglia, which presumably caused a rigid-bradykinesia type parkinsonism. More cases must be studied to completely elucidate the characteristics of adult-onset ALX.
Consent
Written informed consent was obtained from the patient and his wife for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Abbreviations
- ALX:
-
Alexander disease
- GFAP:
-
glial fibrillary acidic protein
- FLAIR:
-
fluid attenuated inversion recovery
References
Alexander WS: Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain. 1949, 72: 373-381. 10.1093/brain/72.3.373.
Johnson AB: Alexander disease. Neurodystrophies and Neurolipidoses. Edited by: Moser HW. 1996, Amsterdam, Elsevier Science, 22 (66): 701-710. Vinken PJ and Bruyn GW (series Editors): Handbook of Clinical Neurology
Quinlan RA, Brenner M, Goldman JE, Messing A: GFAP and its role in Alexander disease. Exp Cell Res. 2007, 313: 2077-2087. 10.1016/j.yexcr.2007.04.004.
Iwaki T, Iwaki A, Tateishi J, Sakaki Y, Goldman JE: αB-crystallin and 27-kd heat shock protein are regulated by stress conditions in the central nervous system and accumulate in Rosenthal fibers. Am J Pathol. 1993, 143: 487-495.
Riggs JE, Schochet SS, Nelson J: Asymptomatic adult Alexander's disease: Entity or nasological misconception?. Neurology. 1988, 38: 152-154.
Jacob J, Robertson NJ, Hilton DA: The clinicopathological spectrum of Rosenthal fibre encephalopathy and Alexander's disease: a case report and review of the literature. J Neurol Neurosurg Psychiatry. 2003, 74: 807-810. 10.1136/jnnp.74.6.807.
Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M: Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol. 1998, 152: 391-398.
Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A: Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet. 2001, 27: 117-120. 10.1038/87020.
Sawaishi Y, Yano T, Takaku I, Takada G: Juvenile Alexander disease with a novel mutation in glial fibrillary acidic protein gene. Neurology. 2002, 58: 1541-1543.
Okamoto Y, Mitsuyama H, Jonosono M, Hirata K, Arimura K, Osame M, Nakagawa M: Autosomal dominant palatal myoclonus and spinal cord atrophy. J Neurol Sci. 2002, 195: 71-76. 10.1016/S0022-510X(01)00687-6.
Namekawa M, Takiyama Y, Aoki Y, Takayashiki N, Sakoe K, Shimazaki H, Taguchi T, Tanaka Y, Nishizawa M, Saito K, Matsubara Y, Nakano I: Identification of GFAP gene mutation in hereditary adult-onset Alexander's disease. Ann Neurol. 2002, 52: 779-785. 10.1002/ana.10375.
Barkovich AJ, Messing A: Alexander disease. Not just a leukodystrophy anymore. Neurology. 2006, 66: 468-469. 10.1212/01.wnl.0000200905.43191.4d.
van der Knaap MS, Naidu S, Breiter SN, Blaser S, Stroink H, Springer S, Begeer JC, van Coster R, Barth PG, Thomas NH, Valk J, Powers JM: Alexander disease: Diagnosis with MR imaging. AJNR Am J Neuroradiol. 2001, 22: 541-552.
van der Knaap MS, Salomons GS, Li R, Franzoni E, Gutiérrez-Solana LG, Smit LME, Robinson R, Ferrie CD, Cree B, Reddy A, Thomas N, Banwell B, Barkhof F, Jakobs C, Johnson A, Messing A, Brenner M: Unusual variants of Alexander's disease. Ann Neurol. 2005, 57: 327-338. 10.1002/ana.20381.
van der Knaap MS, Ramesh V, Schiffmann R, Blaser S, Kyllerman M, Gholkar A, Ellison DW, van der Voorn JP, van Dooren SJM, Jakobs C, Barkhof F, Salomons GS: Alexander disease. Ventricular garlands and abnormalities of the medulla and spinal cord. Neurology. 2006, 66: 494-498. 10.1212/01.wnl.0000198770.80743.37.
Probst EN, Hagel C, Weisz V, Nagel S, Wittkugel O, Zeumer H, Kohlschütter A: Atypical focal MRI lesions in a case of juvenile Alexander's disease. Ann Neurol. 2003, 53: 118-120. 10.1002/ana.10382.
Stumpf E, Masson H, Duquette A, Berthelet F, McNabb J, Lortie A, Lesage J, Montplaisir J, Brais B, Cossette P: Adult Alexander disease with autosomal dominant transmission. A distinct entity caused by mutation in the glial fibrillary acid protein gene. Arch Neurol. 2003, 60: 1307-1312. 10.1001/archneur.60.9.1307.
Kinoshita T, Imaizumi T, Miura Y, Fujimoto H, Ayabe M, Shoji H, Okamoto Y, Takashima H, Osame M, Nakagawa M: A case of adult-onset Alexander disease with Arg416Trp human glial fibrillary acidic protein gene mutation. Neurosci Lett. 2003, 350: 169-172. 10.1016/S0304-3940(03)00900-5.
Thyagarajan D, Chataway T, Li R, Gai WP, Brenner M: Dominantly-inherited adult-onset leukodystrophy with palatal tremor caused by a mutation in the glial fibrillary acidic protein gene. Mov Disord. 2004, 19: 1244-1248. 10.1002/mds.20161.
Shiihara T, Sawaishi Y, Adachi M, Kato M, Hayasaka K: Asymptomatic hereditary Alexander's disease caused by a novel mutation in GFAP. J Neurol Sci. 2004, 225: 125-127. 10.1016/j.jns.2004.07.008.
Li R, Johnson AB, Salomons G, Goldman JE, Naidu S, Quinlan R, Cree B, Ruyle SZ, Banwell B, D'Hooghe M, Siebert JR, Rolf CM, Cox H, Reddy A, Gutiérrez-Solana LG, Collins A, Weller RO, Messing A, Knaap van der MS, Brenner M: Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander disease. Ann Neurol. 2005, 57: 310-326. 10.1002/ana.20406.
Salvi F, Aoki Y, Nave RD, Vella A, Pastorelli F, Scaglione C, Matsubara Y, Mascalchi M: Adult Alexander's disease without leukoencephalopathy. Ann Neurol. 2005, 58: 813-814. 10.1002/ana.20634.
Salmaggi A, Botturi A, Lamperti E, Grisoli M, Fischetto R, Ceccherini I, Caroli F, Boiardi A: A novel mutation in the GFAP gene in a familial adult onset Alexander disease. J Neurol. 2007, 254: 1278-1280. 10.1007/s00415-006-0361-2.
Ohnari K, Yamano M, Uozumi T, Hashimoto T, Tsuji S, Nakagawa M: An adult form of Alexander disease: a novel mutation in glial fibrillary acidic protein. J Neurol. 2007, 254: 1390-1394. 10.1007/s00415-007-0557-0.
Romano S, Salvetti M, Ceccherini I, De Simone T, Savoiardo M: Brainstem signs with progressing atrophy of medulla oblongata and upper cervical spinal cord. Lancet Neurol. 2007, 6: 562-570. 10.1016/S1474-4422(07)70129-1.
Caroli F, Biancheri R, Seri M, Rossi A, Pessagno A, Bugiani M, Corsolini F, Savasta S, Romano S, Antonelli C, Romano A, Pareyson D, Gambero P, Uziel G, Ravazzolo R, Ceccherini I, Filocamo M: GFAP mutations and polymorphisms in 13 unrelated Italian patients affected by Alexander disease. Clin Genet. 2007, 72: 427-433. 10.1111/j.1399-0004.2007.00869.x.
Sreedharan J, Shaw CE, Jarosz J, Samuel M: Alexander disease with hypothermia, microcoria, and psychiatric and endocrine disturbances. Neurology. 2007, 68: 1322-1323. 10.1212/01.wnl.0000259543.95222.9d.
Huttner HB, Richter G, Hildebrandt M, Blümcke I, Fritscher T, Brück W, Gärtner J, Seifert F, Staykov D, Hilz MJ, Schwab S, Bardutzky J: Acute onset of fatal vegetative symptoms: unusual presentation of adult Alexander disease. Eur J Neurol. 2007, 14: 1251-1255. 10.1111/j.1468-1331.2007.01961.x.
Howard KL, Hall DA, Moon M, Agarwal P, Newman E, Brenner M: Adult-onset Alexander disease with progressive ataxia and palatal tremor. Mov Disord. 2008, 23: 118-122. 10.1002/mds.21774.
Balbi P, Seri M, Ceccherini I, Uggetti C, Casale R, Fundarò C, Caroli F, Santoro L: Adult-onset Alexander disease. Report on a family. J Neurol. 2008, 255: 24-30. 10.1007/s00415-007-0654-0.
Hirayama T, Fukae J, Noda K, Fujishima K, Yamamoto T, Mori K, Maeda M, Hattori N, Shiroma N, Tsurui S, Okuma Y: Adult-onset Alexander disease with palatal myoclonus and intraventricular tumour. Eur J Neurol. 2008, 15: e16-e17. 10.1111/j.1468-1331.2007.02031.x.
Sechi GP, Conti M, Sau GF, Cocco GA: Valproate-induced parkinsonism, glial cells and Alexander's disease. Prog Neuropsychopharmacol Biol Psyciatry. 2008, 32: 1351-1352. 10.1016/j.pnpbp.2008.03.022.
Pareyson D, Fancellu R, Mariotti C, Romano S, Salmaggi A, Carella F, Girotti F, Gattellaro G, Carriero MR, Farina L, Ceccherini I, Savoiardo M: Adult-onset Alexander disease: a series of eleven unrelated cases with review of the literature. Brain. 2008, 131: 2321-2331. 10.1093/brain/awn178.
Farina L, Pareyson D, Minati L, Ceccherini I, Chiapparini L, Romano S, Gambaro P, Fancellu R, Savoiardo M: Can MR imaging diagnose adult-onset Alexander disease?. AJNR Am J Neuroradiol. 2008, 29: 1190-1196. 10.3174/ajnr.A1060.
Kaneko H, Hirose M, Katada S, Takahashi T, Naruse S, Tsuchiya M, Yoshida T, Nakagawa M, Onodera O, Nishizawa M, Ikeuchi T: Novel GFAP mutation in patient with adult-onset Alexander disease presenting with spastic ataxia. Mov Disord. 2009, 24: 1393-1406. 10.1002/mds.22556.
Sueda Y, Takahashi T, Ochi K, Ohtsuki T, Namekawa M, Kohriyama T, Takiyama Y, Matsumoto M: Adult onset Alexander disease with a novel variant (S398F) in the glial fibrillary acidic protein gene. Clin Neurol. 2009, 49: 358-363. in Japanese
Li R, Johnson AB, Salomons GS, van der Knaap MS, Rodriguez D, Boespflug-Tanguy O, Gorospe JR, Goldman JE, Messing A, Brenner M: Propensity for paternal inheritance of de novo mutations in Alexander disease. Hum Genet. 2006, 119: 137-144. 10.1007/s00439-005-0116-7.
Brockmann K, Meins M, Taubert A, Trappe R, Grond M, Hanefeld F: A novel GFAP mutation and disseminated white matter lesions: Adult Alexander disease?. Eur Neurol. 2003, 50: 100-105. 10.1159/000072507.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2377/10/21/prepub
Acknowledgements
We are very grateful to Dr. Merle Ruberg for critical reading of this manuscript. This work was supported by Alexander Disease Research Grants from the Intractable Disease Research Grants, from the Ministry of Health, Labour and Welfare of the government of Japan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
This manuscript was drafted by MN. YT and IN contributed to the references and helped to write the manuscript. JH, HS and KS sequenced the GFAP gene. All authors contributed to the critical review and approval of the final draft.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Namekawa, M., Takiyama, Y., Honda, J. et al. Adult-onset Alexander disease with typical "tadpole" brainstem atrophy and unusual bilateral basal ganglia involvement: a case report and review of the literature. BMC Neurol 10, 21 (2010). https://doi.org/10.1186/1471-2377-10-21
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2377-10-21