
Abstract
Background
Peutz-Jeghers syndrome (PJS) is an unusual autosomal dominant disorder characterized by mucocutaneous pigmentation and multiple gastrointestinal hamartomatous polyps. Patients with PJS are at an increased risk of developing multi-organ cancer, most frequently those involving the gastrointestinal tract. Germline mutation of the STK11 gene, which encodes a serine-threonine kinase, is responsible for PJS.
Methods
Using DNA samples obtained from the patient and his family members, we sequenced nine exons and flanking intron regions of the STK11 gene using polymerase chain reaction (PCR) and direct sequencing.
Results
Sequencing of the STK11 gene in the proband of the family revealed a novel 1-base pair deletion of guanine (G) in exon 6 (c.826delG; Gly276AlafsX11). This mutation resulted in a premature termination at codon 286, predicting a partial loss of the kinase domain and complete loss of the C-terminal domain. We did not observe this mutation in both parents of the PJS patient. Therefore, it is considered a novel de novo mutation.
Conclusion
The results presented herein enlarge the spectrum of mutations of the STK11 gene by identifying a novel de novo mutation in a PJS patient and further support the hypothesis that STK11 mutations are disease-causing mutations for PJS with or without a positive family history.
Similar content being viewed by others


Background
Peutz-Jeghers syndrome (PJS; OMIM 175200) is a rare, autosomal dominant disorder characterized by melanocytic macules of the lips, buccal mucosa, and digits, along with multiple gastrointestinal hamartomatous polyps, frequently in the small intestine [1, 2]. Patients with PJS are at an increased risk of developing gastrointestinal cancer and extraintestinal neoplasms involving organs such as the ovaries, testes, breasts, pancreas, lungs, or uterine cervix [3].
Currently, only mutations in the gene STK11 (also known as LKB1; OMIM 602216) at chromosome 19p13.3 have been identified as a cause of PJS [4, 5]. The human STK11 gene encodes a 433 amino acid serine-threonine kinase. STK11 is known to be located both in the nucleus and the cytoplasm of all human tissues [6], and orthologs include mouse LKb1 [7], XEEK1 (Xenopus egg and embryo kinase 1) [8], Caenorhabditis elegans partitioning defective gene 4 (par-4) [9], and drosophila Lkb1 [10].
Loss of the normal allele has been observed in polyps from patients with PJS, and loss of heterozygosity (LOH) has been noted to occur in some tumor tissues, suggesting that STK11 is a tumor suppressor gene [11]. STK11 has been shown to cause apoptosis in intestinal epithelial cells, and is physically associated with p53, regulating specific p53-dependent apoptosis pathways [12]. STK11 is also known to have effects on G1 cell cycle arrest [13], TGF-β signaling [14], polarity [15], and phosphorylating and activating the AMP-activated protein kinase (AMPK) [16].
Screening for point mutations and large deletions by direct sequencing or by multiplex ligation-dependent probe amplification (MLPA) increased the mutation detection rate in the STK11 gene up to 94% [17]. To date, more than 200 different mutations in the STK11 gene have been reported at the Human Gene Mutation Database (HGMD) website http://www.hgmd.cf.ac.uk/ac/all.php and most are small insertions/deletions or missense/nonsense mutations.
We report on a Korean PJS patient with a novel STK11 mutation. During molecular genetic testing for STK11 mutation, we detected a novel small deletion in exon 6, causing a premature stop codon. This mutation was absent in both parents of the patient and was thus a de novo mutation.
Methods
Subjects
The proband was a 13-year-old Korean male. He was diagnosed clinically with PJS at six years of age based on the presence of characteristic mucocutaneous pigmentation of the lips and buccal mucosa, and gastrointestinal hamartomatous polyps after polypectomy (Figure 1). After obtaining informed consent, blood samples were collected from four family members including the patient. Brother and parents of the patient did not consent to endoscopic examinations for evaluation of polyps. Relatives of the patient had no known medical conditions, including mucocutaneous pigmentation or malignancies, and no further study was assessed (Figure 2).

Pathologic findings of the Peutz-Jeghers polyp. The colonic polyp shows hyperplastic mucosal epithelium and arborizing pattern of smooth muscle, consistent with a hamartomatous polyp (Hematoxylin-eosin stain, 200×).

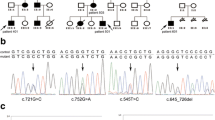
Pedigree of the family with PJS. Circle, female; square, male; black symbol, affected. Asterisk (*) indicates the family member who was available for genetic analysis.
Mutation studies
Four family members including the patient were included in this study after obtaining informed consent. The genomic DNA was isolated from peripheral blood leukocytes using the Wizard Genomic DNA Purification Kit according to the manufacturer's instructions (Promega, Madison, WI, USA). The polyp tissue was not collected for DNA analysis. The STK11 gene was amplified via PCR [by using the appropriate primers as designed by the authors (available upon request)] and a thermal cycler (Model 9700; Applied Biosystems, Foster City, CA, USA). Direct sequencing of all nine coding exons along with the flanking intron regions of the STK11 gene was performed with the Big Dye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems) in conjunction with an ABI Prism 3100 automated genetic analyzer (Applied Biosystems).
Results
Direct sequencing analysis of the proband demonstrated a heterozygous 1-bp deletion of guanine (G) (c.826delG; Gly276AlafsX11) in exon 6 of the STK11 gene, which resulted in a frameshift leading to premature termination of the 433 amino acid protein at the 286th codon, disruption of the kinase domain, and complete loss of carboxyterminal non-catalytic region. This mutation was absent in his family members and 100 control chromosomes (Figure 3).

Identification of the STK11 gene mutation. Direct sequencing of the proband demonstrated a 1-bp deletion (c.826delG) in exon 6 of the STK11 gene, resulting in a frameshift deletion mutation (p.Gly276AlafsX11). The proband's family members did not have the mutation. The localization of the deletion is indicated by the arrow.
Discussion
Germline mutations in the STK11 gene on chromosome 19p13.3 have been shown to be the cause of PJS [4, 5]. Recent study suggests that the STK11 mutation detection rate was above 90% [17]. However, some families with PJS have shown linkage to chromosomal region 19q13.4 [18] and 6 [19]. Human STK11 consists of nine coding exons with a 433 amino acid coding sequence, and one non-coding exon 10 that spans 23 kb [6, 7]. The STK11 protein is mainly comprised of three major domains: the N-terminal non-catalytic domain containing the nuclear localization signal, the catalytic kinase domain important for ATP binding, and the carboxyterminal non-catalytic regulatory domain containing prenylation motif (CAAX-box). Codons 49–309 encode the catalytic kinase domain. The C-terminal non-catalytic region of the STK11 protein is encoded by exon 8 and 9 and encompasses amino acids 309–433.
The patient recruited in this study fulfilled the well-established clinical diagnostic criteria for PJS [20]. The criteria include histopathologically proven hamartomas together with classical mucocutaneous hyperpigmentation and small-bowel polyposis. Therefore, the possibility that this patient is affected with hamartomatous polyposis syndromes other than PJS is highly unlikely. Such syndromes include juvenile polyposis syndrome, PTEN hamartoma tumor syndrome, and Carney complex. In our study, we sequenced the STK11 gene in this patient with PJS. We identified a novel heterozygous 1-bp deletion (c.826delG; Gly276AlafsX11) in exon 6 of the STK11 gene, which resulted in a frameshift leading to premature termination of the 433 amino acid protein at the 286th codon. This mutation was not detected in the STK11 sequencing analysis of his family members, indicating that Gly276AlafsX11 is a novel de novo mutation.
The Gly276AlafsX11 mutation is located in the catalytic kinase domain of STK11 protein, so we hypothesize that this mutation may lead to partial loss of the kinase domain and complete loss of the C-terminal domain. This mutation is novel, but a similar effect of the mutation in the STK11 protein was reported recently [21]. Loss of STK11 protein kinase activity associated with loss of growth suppression function was reported in some mutations in STK11 associated with PJS [4, 22]. Thus, development of the PJS phenotypes is believed to be due to the elimination of the kinase activity of STK11 [23]. The C-terminal domain of STK11 is important for the control of both the AMPK pathway and cell polarity [15]. Mutations leading to loss of the C-terminal domain of STK11, as observed in this case, lead to loss of cell polarity, resulting in the development of malignancies. Taken together, these data suggest that the STK11 mutation in exon 6 may contribute to polyp formation and tumorgenesis through various mechanisms such as loss of growth arrest, apoptosis, and loss of cell polarity. Further studies will be needed to address these questions.
Although an increased cancer risk in patients with PJS is well established [3, 24], data on genotype-phenotype correlation is lacking. Schumacher et al. studied 146 PJS patients and determined that inframe deletions, splice site mutations, and missense mutations in the catalytic kinase domain were rarely associated with cancer. However, it was felt that missense mutations in the C-terminal domain were more frequently associated with malignancies [22]. Recently, however, Hearle et al. studied 419 PJS patients and determined that the type or site of the STK11 mutation did not significantly influence cancer risk [25]. Restricted by the few published papers on this topic, the genotype-phenotype correlation remains to be further investigated.
Conclusion
We enlarged the spectrum of mutations of the STK11 gene by identifying a novel mutation in a Korean patient with PJS. Because of the increased risk of PJS in patients with multi-organ cancers, molecular diagnosis will be an important factor in genetic counseling, clinical management of patients, and tumor screening.
Abbreviations
- PJS:
-
Peutz-Jeghers syndrome
- STK11/LKB1:
-
serine-threonine kinase 11
- XEEK1:
-
Xenopus egg and embryo kinase 1
- LOH:
-
loss of heterozygosity
- AMPK:
-
AMP activated protein kinase
- MLPA:
-
multiplex ligation-dependent probe amplification.
References
Jeghers H, McKusick VA, Katz KH: Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med. 1949, 241 (26): 1031-1036.
Peutz JL: On a very remarkable case of familial polyposis of the mucous membrane of the intestinal tract and nasopharynx accompanied by peculiar pigmentation of the skin and mucous memebrane. Ned Tijdschr Geneeskd. 1921, 10: 134-146.
Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV, Cruz-Correa M, Offerhaus JA: Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000, 119 (6): 1447-1453. 10.1053/gast.2000.20228.
Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Muller O, Back W, Zimmer M: Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998, 18 (1): 38-43. 10.1038/ng0198-38.
Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, et al: A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998, 391 (6663): 184-187. 10.1038/34432.
Yoo LI, Chung DC, Yuan J: LKB1–a master tumour suppressor of the small intestine and beyond. Nat rev Cancer. 2002, 2 (7): 529-535. 10.1038/nrc843.
Smith DP, Spicer J, Smith A, Swift S, Ashworth A: The mouse Peutz-Jeghers syndrome gene Lkb1 encodes a nuclear protein kinase. Hum Mol Genet. 1999, 8 (8): 1479-1485. 10.1093/hmg/8.8.1479.
Su JY, Erikson E, Maller JL: Cloning and characterization of a novel serine/threonine protein kinase expressed in early Xenopus embryos. J Biol Chem. 1996, 271 (24): 14430-14437. 10.1074/jbc.271.24.14430.
Watts JL, Morton DG, Bestman J, Kemphues KJ: The C. elegans par-4 gene encodes a putative serine-threonine kinase required for establishing embryonic asymmetry. Development. 2000, 127 (7): 1467-1475.
Martin SG, St Johnston D: A role for Drosophila LKB1 in anterior-posterior axis formation and epithelial polarity. Nature. 2003, 421 (6921): 379-384. 10.1038/nature01296.
Bardeesy N, Sinha M, Hezel AF, Signoretti S, Hathaway NA, Sharpless NE, Loda M, Carrasco DR, DePinho RA: Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature. 2002, 419 (6903): 162-167. 10.1038/nature01045.
Karuman P, Gozani O, Odze RD, Zhou XC, Zhu H, Shaw R, Brien TP, Bozzuto CD, Ooi D, Cantley LC, et al: The Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell death. Mol Cell. 2001, 7 (6): 1307-1319. 10.1016/S1097-2765(01)00258-1.
Tiainen M, Ylikorkala A, Makela TP: Growth suppression by Lkb1 is mediated by a G(1) cell cycle arrest. Proc Natl Acad Sci USA. 1999, 96 (16): 9248-9251. 10.1073/pnas.96.16.9248.
Smith DP, Rayter SI, Niederlander C, Spicer J, Jones CM, Ashworth A: LIP1, a cytoplasmic protein functionally linked to the Peutz-Jeghers syndrome kinase LKB1. Hum Mol Genet. 2001, 10 (25): 2869-2877. 10.1093/hmg/10.25.2869.
Forcet C, Etienne-Manneville S, Gaude H, Fournier L, Debilly S, Salmi M, Baas A, Olschwang S, Clevers H, Billaud M: Functional analysis of Peutz-Jeghers mutations reveals that the LKB1 C-terminal region exerts a crucial role in regulating both the AMPK pathway and the cell polarity. Hum Mol Genet. 2005, 14 (10): 1283-1292. 10.1093/hmg/ddi139.
Boudeau J, Scott JW, Resta N, Deak M, Kieloch A, Komander D, Hardie DG, Prescott AR, van Aalten DM, Alessi DR: Analysis of the LKB1-STRAD-MO25 complex. J Cell Sci. 2004, 117 (Pt 26): 6365-6375. 10.1242/jcs.01571.
Aretz S, Stienen D, Uhlhaas S, Loff S, Back W, Pagenstecher C, McLeod DR, Graham GE, Mangold E, Santer R, et al: High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum Mutat. 2005, 26 (6): 513-519. 10.1002/humu.20253.
Mehenni H, Blouin JL, Radhakrishna U, Bhardwaj SS, Bhardwaj K, Dixit VB, Richards KF, Bermejo-Fenoll A, Leal AS, Raval RC, et al: Peutz-Jeghers syndrome: confirmation of linkage to chromosome 19p13.3 and identification of a potential second locus, on 19q13.4. Am J Hum Genet. 1997, 61 (6): 1327-1334. 10.1086/301644.
Markie D, Huson S, Maher E, Davies A, Tomlinson I, Bodmer WF: A pericentric inversion of chromosome six in a patient with Peutz-Jeghers' syndrome and the use of FISH to localise the breakpoints on a genetic map. Hum Genet. 1996, 98 (2): 125-128. 10.1007/s004390050173.
Giardiello FM, Welsh SB, Hamilton SR, Offerhaus GJ, Gittelsohn AM, Booker SV, Krush AJ, Yardley JH, Luk GD: Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med. 1987, 316 (24): 1511-1514.
Thakur N, Reddy DN, Rao GV, Mohankrishna P, Singh L, Chandak GR: A novel mutation in STK11 gene is associated with Peutz-Jeghers Syndrome in Indian patients. BMC Med Genet. 2006, 7: 73-10.1186/1471-2350-7-73.
Schumacher V, Vogel T, Leube B, Driemel C, Goecke T, Moslein G, Royer-Pokora B: STK11 genotyping and cancer risk in Peutz-Jeghers syndrome. J Med Genet. 2005, 42 (5): 428-435. 10.1136/jmg.2004.026294.
Hemminki A: The molecular basis and clinical aspects of Peutz-Jeghers syndrome. Cell Mol Life Sci. 1999, 55 (5): 735-750. 10.1007/s000180050329.
Spigelman AD, Murday V, Phillips RK: Cancer and the Peutz-Jeghers syndrome. Gut. 1989, 30 (11): 1588-1590. 10.1136/gut.30.11.1588.
Hearle N, Schumacher V, Menko FH, Olschwang S, Boardman LA, Gille JJ, Keller JJ, Westerman AM, Scott RJ, Lim W, et al: Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006, 12 (10): 3209-3215. 10.1158/1078-0432.CCR-06-0083.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/9/44/prepub
Acknowledgements
We gratefully thank the patient and his family members for their cooperation in the research. Written consent for publication was obtained from the patient and his family members. This work was supported by the National Health Insurance Corporation Ilsan Hospital grant (2007-59).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JY recruited all the subjects investigated, carried out the molecular genetic studies, and drafted the manuscript. Y–KS, K–AL, and C–SK helped with the experiments. JY, Y–JC, and J–GK diagnosed the patient and participated in the editing of the manuscript. J–RC designed and supervised the study. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Yoo, JH., Yoo, JH., Choi, YJ. et al. A novel de novo mutation in the serine-threonine kinase STK11 gene in a Korean patient with Peutz-Jeghers syndrome. BMC Med Genet 9, 44 (2008). https://doi.org/10.1186/1471-2350-9-44
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-9-44