
Abstract
Background
Mutations in the FKBP10 gene were first described in patients with Osteogenesis imperfecta type III. Two follow up reports found FKBP10 mutations to be associated with Bruck syndrome type 1, a rare disorder characterized by congenital contractures and bone fragility. This raised the question if the patients in the first report indeed had isolated Osteogenesis imperfecta or if Bruck syndrome would have been the better diagnosis.
Methods
The patients described here are affected by severe autosomal recessive Osteogenesis imperfecta without contractures.
Results
Homozygosity mapping identified FKBP10 as a candidate gene, and sequencing revealed a base pair exchange that causes a C-terminal premature stop codon in this gene.
Conclusions
Our study demonstrates that FKBP10 mutations not only cause Bruck syndrome or Osteogenesis imperfecta type III but can result in a severe type of isolated Osteogenesis imperfecta type IV with prenatal onset. Furthermore, it adds dentinogenesis imperfecta to the spectrum of clinical symptoms associated with FKBP10 mutations.
Similar content being viewed by others

Background
Osteogenesis imperfecta (OI) is a rare monogenic disorder that shows both clinical and genetic heterogeneity. The disorder is characterized by bone fragility that varies in severity between the different OI subtypes. In the majority of patients the disorder is caused by dominant mutations in the procollagen genes COL1A1 and COL1A2 [1–4]. Even rarer than the dominant disorders are the autosomal recessive forms of OI for which several genes are known by now. These genes are mainly involved in the processing, assembly and trafficking of procollagen chains, a multi-step process that involves a large number of critical post-translational modifications. Homozygosity for mutations in CRTAP, LEPRE1, PPIB, or OSTERIX has been shown to cause mostly severe types of OI that result from overmodification of procollagen chains and their retention in the rER [2–7]. Autosomal recessive brittle bone disease without procollagen overmodification can be caused by mutations in either SERPINH1 or FKBP10 genes that encode the collagen chaperone-like proteins HSP47 and FKBP65, respectively [1, 3]. Basic pathomechanisms of OI initiated by mutations in these genes might include a failure to mature type I procollagen trimers and increased protease sensitivity [3, 8]. The pathomechanisms of another OI gene, SERPINF1, are still unknown [9]. Mutations in FKBP10 were first described in patients with a moderate to moderate/severe form of OI type IV (OMIM 610968) [1]. Two follow up reports presented patients in which FKBP10 mutations were associated with Bruck syndrome type 1, a rare disorder characterized by congenital contractures and bone fragility [10–12]. The question was raised if the patients in the first report indeed had isolated OI or if Bruck syndrome would have been the better diagnosis [11, 12]. We here describe three patients showing that FKBP10 mutations not only can cause isolated OI but that the spectrum of clinical symptoms is much broader than previously described.
Methods
Patients
The three affected brothers, their sister and their parents provided informed consent for molecular testing, clinical assessment and the publication of their photographs. Anonymous blood samples from 80 healthy German medical students were used as controls. The experiments were performed in accordance to the Helsinki Declaration and approved by the Ethics committee of the University of Munich.
Homozygosity mapping and FKBP10 sequencing
Genomic DNA samples of family members I1-2, II1-2, and II4-5 were genotyped using the Affymetrix Axiom™ Genome-Wide Human Array, featuring 567,096 SNP markers (Affymetrix, Santa Clara, CA, USA). Extended runs of homozygosity (ROHs) shared by the three affected brothers were assessed in comparison to the unaffected family members using the program HomozygosityMapper (http://www.homozygositymapper.org) [13]. Prioritization of candidate genes within the homozygous region was performed by the program GeneDistiller (http://www.genedistiller.org) [14] and by comparison to PubMed entries (http://www.ncbi.nlm.nih.gov/pubmed). The complete coding sequence of the FKBP10 gene was amplified and directly sequenced.
Results
Clinical findings
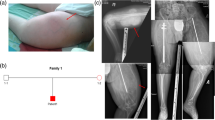
The three affected brothers, aged 46 (II1), 44 (II4), and 43 (II5) years, were born to healthy, unrelated parents. There is no family history of bone diseases or major malformations. The oldest brother was born without fractures but showed delayed motor development during his first year of life. He had his first fracture at age 9 months. The second brother was born with multiple fractures. Multiple fractures of the upper extremities at birth were also suspected in the youngest brother but not confirmed by X-ray. So far each of the brothers already had 70-100 fractures and breaks still occur. Severe progressive kyphoscoliosis has caused reduced lung capacity in all three brothers, and they are wheel-chair bound due to long bone deformities. None of the brothers showed contractures or webbing at birth. The brothers are of short stature (120-140 cm, <3 %tile) with remarkable hypermobile joints (Figure 1). Relevant lab results such as alkaline phosphatase (73 U/l) are within the normal range. The primary teeth were discoloured and rapidly wore down, thus showing typical signs of dentinogenesis imperfecta (Figure 2). The permanent teeth are less affected. Blue sclera were noted in childhood but are no longer present.

Genetic studies. A, Pedigree of the OI family. Black pedigree symbols, affected brothers, homozygous; symbols marked by black dots, unaffected family members that are heterozygous for the mutation; unmarked symbols, unaffected family members that were not available for analysis. B, Genome wide plot of Homozygosity Mapper. Red bars indicate regions with extended ROH at chromosomal regions 11q25 and 17q21.2, shared by the three affected brothers. Homozygosity Score threshold 0.8; HSA, homo sapiens chromosome. C, Sequencing results. Parts of FKBP10 exon 7 are shown. Left side, OI patient II1 homozygous for Arg403X; right side, healthy sister (II2) heterozygous for Arg403X.

Clinical phenotype. A-B, extended left arm II1, II5. Upper arm is severely shortened in II1 and instabilized by pseudarthrosis in II5. Webbing and contractures are absent; C, joint hypermobility, demonstrated by II4; D, Radiographs of left leg and E, left side of thorax showing severe kyphoscoliosis (II4); F, Dentiogenesis imperfecta (primary teeth) in II5.
Genetic studies
Homozygosity mapping detected two candidate ROHs in the chromosomal regions 11q25 and 17q21, respectively (Figure 1B). The ROH on 17q21.2 (17: 39727018-44200191, rs1010594-rs9910284, NCBI Genome Build 37.3) harbors the FKBP10 gene encoding the FKBP65 protein (Additional File 1, Figure S1). Sequencing revealed a homozygous nucleotide exchange in exon 7 that changes an arginine codon into a stop codon (c.1207C>T, FKBP65-p.(Arg403*) (Figure 1C).
Discussion
We identified a homozygous C-terminal premature stop codon (FKBP65-p.Arg403X) in the FKBP10 gene causing OI in three affected siblings of a family with autosomal recessive inheritance. Both parents derived from two neighbouring small villages in Bavaria, suggesting a restricted genetic variability and a potential accumulation of recessive founder mutations in this rather isolated population. Although estimates of the relative relationship by identity-by-state allele sharing did not indicate consanguinity (see Additional File 1), homozygosity mapping was successfully applied to identify a causative homozygous FKBP10 founder mutation. Our study demonstrate that homozygosity mapping represents a suitable diagnostic approach to map recessive OI genes even in outbred populations [15].
The first FKBP10 mutations were found in families that were described as having OI but for which no contractures or webbing were reported [1]. Shaheen et al. reviewed the figures in the original publication and questioned the absence of webbing [11, 12]. Both contractures and webbing were present in their own patients as well as in all but two of the patients reported by Kelley et al.[10] The patients were therefore diagnosed with Bruck syndrome type 1, a distinct form of OI that is characterized by brittle bone disease and arthrogryposis multiplex congenita. Interestingly, case 2 and 4 in the report from Kelley et al. had OI but did not fulfil the criteria for Bruck syndrome [10]. In our family none of the three brothers had any contractures at birth or showed webbing. Thus, taken together, the previously reported patients and the family described here demonstrate that FKBP10 can cause either Bruck syndrome or isolated OI. Additional families need to be collected to answer the question if these two clinical phenotypes are associated with specific mutations, or if isolated OI and Bruck syndrome can occur in the same family.
So far it is not known if the severity of the disorder is correlated to the position of the mutation within the FKBP10 gene. Most patients carry frame shifting mutations that are located in the N-terminal half of the gene and are likely to cause nonsense mediated decay (NMD) (1). Interestingly, the affected brothers described here are homozygous for a C-terminal premature stop codon that, according to its vicinity to intron 7, might not initiate NMD [16, 17]. The resulting truncated protein would still have the protein-binding PPIase domains that are necessary for its chaperone function but would miss the putative rER-retention sequence (HEEL). The chaperone function might have destructive effects if exhibited outside the rER.
With the exception of some of the patients described by Alanay et al. [1] most previously published patients with FKBP10 mutations had a moderate to moderate-severe form of the disorder that is best classified as OI type III. The data summarized in Table 1 show that fractures mostly started a few weeks after birth, sometimes even later, but not pre- or perinatally [1, 10–12]. Progressive skeletal deformities were rarely described. In our family at least one, but more likely two of the affected brothers were already born with multiple fractures. Subsequently, all three brothers developed extreme deformities of their long bone and spines, fulfilling the criteria for severe OI (Figure 2). Furthermore, dentinogenesis imperfecta was present in all three brothers but has so far not been described as a feature of patients with FKBP10 mutations [1, 10–12]. The combination of symptoms in our family does not match the classification of OI type III but rather suggests that this family belongs to the subgroup of patients with OI type IV that show progressive skeletal deformities and dentinogenesis imperfecta.
Conclusions
Taken together, our study shows that a severe, progressive course of OI type IV with pre- or perinatal onset of fractures and dentinogenesis imperfecta has to be added to the spectrum of clinical features that can be associated with FKBP10 mutations. We also demonstrate that homozygosity mapping represents a suitable diagnostic approach to map recessive OI genes even in outbred populations.
Abbreviations
- FKBP10 :
-
FK506-binding protein
- HEEL:
-
rER-retention sequence
- OI:
-
osteogenesis imperfecta
- NMD:
-
nonsense mediated decay
- rER:
-
rough endoplasmatic reticulum
- ROHs:
-
runs of homozygosity.
References
Alanay Y, Avaygan H, Camacho N, Utine GE, Boduroglu K, Aktas D, Alikasifoglu M, Tuncbilek E, Orhan D, Bakar FT, Zabel B, Superti-Furga A, Bruckner-Tuderman L, Curry CJ, Pyott S, Byers PH, Eyre DR, Baldridge D, Lee B, Merrill AE, Davis EC, Cohn DH, Akarsu N, Krakow D: Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2010, 86: 551-559. 10.1016/j.ajhg.2010.02.022.
Cabral WA, Chang W, Barnes AM, Weis M, Scott MA, Leikin S, Makareeva E, Kuznetsova NV, Rosenbaum KN, Tifft CJ, Bulas DI, Kozma C, Smith PA, Eyre DR, Marini JC: Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet. 2007, 39: 359-365. 10.1038/ng1968.
Christiansen HE, Schwarze U, Pyott SM, AlSwaid A, Al Balwi M, Alrasheed S, Pepin MG, Weis MA, Eyre DR, Byers PH: Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am J Hum Genet. 2010, 86: 389-398. 10.1016/j.ajhg.2010.01.034.
Ha-Vinh R, Alanay Y, Bank RA, Campos-Xavier AB, Zankl A, Superti-Furga A, Bonafe L: Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in PLOD2. Am J Med Genet A. 2004, 131: 115-120.
Lapunzina P, Aglan M, Temtamy S, Caparros-Martin JA, Valencia M, Leton R, Martinez-Glez V, Elhossini R, Amr K, Vilaboa N, Ruiz-Perez VL: Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am J Hum Genet. 2010, 87: 110-114. 10.1016/j.ajhg.2010.05.016.
Marini JC, Cabral WA, Barnes AM: Null mutations in LEPRE1 and CRTAP cause severe recessive osteogenesis imperfecta. Cell Tissue Res. 2010, 339: 59-70. 10.1007/s00441-009-0872-0.
van Dijk FS, Nesbitt IM, Zwikstra EH, Nikkels PG, Piersma SR, Fratantoni SA, Jimenez CR, Huizer M, Morsman AC, Cobben JM, van Roij MH, Elting MW, Verbeke JI, Wijnaendts LC, Shaw NJ, Hogler W, McKeown C, Sistermans EA, Dalton A, Meijers-Heijboer H, Pals G: PPIB mutations cause severe osteogenesis imperfecta. Am J Hum Genet. 2009, 85: 521-527. 10.1016/j.ajhg.2009.09.001.
Koide T, Nishikawa Y, Asada S, Yamazaki CM, Takahara Y, Homma DL, Otaka A, Ohtani K, Wakamiya N, Nagata K, Kitagawa K: Specific recognition of the collagen triple helix by chaperone HSP47. II. The HSP47-binding structural motif in collagens and related proteins. J Biol Chem. 2006, 281: 11177-11185. 10.1074/jbc.M601369200.
Becker J, Semler O, Gilissen C, Li Y, Bolz HJ, Giunta C, Bergmann C, Rohrbach M, Koerber F, Zimmermann K, de Vries P, Wirth B, Schoenau E, Wollnik B, Veltman JA, Hoischen A, Netzer C: Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2011, 88: 362-371. 10.1016/j.ajhg.2011.01.015.
Kelley BP, Malfait F, Bonafe L, Baldridge D, Homan E, Symoens S, Willaert A, Elcioglu N, Van Maldergem L, Verellen-Dumoulin C, Gillerot Y, Napierala D, Krakow D, Beighton P, Superti-Furga A, De Paepe A, Lee B: Mutations in FKBP10 cause recessive osteogenesis imperfecta and type 1 bruck syndrome. J Bone Miner Res. 2011, 26: 666-672. 10.1002/jbmr.250.
Shaheen R, Al-Owain M, Sakati N, Alzayed ZS, Alkuraya FS: FKBP10 and Bruck Syndrome: Phenotypic Heterogeneity or Call for Reclassification?. Am J Hum Genet. 2010, 87: 571-10.1016/j.ajhg.2010.09.001.
Shaheen R, Al-Owain M, Sakati N, Alzayed ZS, Alkuraya FS: FKBP10 and Bruck syndrome: phenotypic heterogeneity or call for reclassification?. Am J Hum Genet. 2010, 87: 306-307. 10.1016/j.ajhg.2010.05.020.
Seelow D, Schuelke M, Hildebrandt F, Nurnberg P: HomozygosityMapper-an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009, 37: W593-W599. 10.1093/nar/gkp369.
Seelow D, Schwarz JM, Schuelke M: GeneDistiller-distilling candidate genes from linkage intervals. PLoS One. 2008, 3: e3874-10.1371/journal.pone.0003874.
Hildebrandt F, Heeringa SF, Rüschendorf F, Attanasio M, Nürnberg G, Becker C, Seelow D, Huebner N, Chernin G, Vlangos CN, Zhou W, O'Toole JF, Hoskins BE, Wolf MT, Hinkes BG, Chaib H, Ashraf S, Schoeb DS, Ovunc B, Allen SJ, Vega-Warner V, Wise E, Harville HM, Lyons RH, Washburn J, Macdonald J, Nürnberg P, Otto EA: A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet. 2009, 5: e1000353-10.1371/journal.pgen.1000353.
Hentze MW, Kulozik AE: A perfect message: RNA surveillance and nonsense-mediated decay. Cell. 1999, 96: 307-310. 10.1016/S0092-8674(00)80542-5.
Isken O, Maquat LE: Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 2007, 21: 1833-1856. 10.1101/gad.1566807.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/12/152/prepub
Acknowledgements and Funding
We like to thank Franz Jansen for excellent technical assistance. Written consent for publication was obtained from the patients. This work was supported by the Deutsche Forschungsgemeinschaft (STE16511-2) to OKS.
Author information
Authors and Affiliations
Corresponding author
Additional information
competing interests
The authors declare that they have no competing interests.
Authors' contributions
OST planed the molecular genetic studies, collected the clinical data and drafted the manuscript. EA participated in the clinical and molecular part of the study. HT performed the homozygosity mapping. TS participated in the study' design and carried out the statistical analysis. All authors read and approved the final manuscript.
Electronic supplementary material
12881_2011_903_MOESM1_ESM.DOC
Additional file 1: Supplementary Figure S1: Runs of homozygosity at 17q21.2. Supplementary Figure S2: Graphical relative relationship of individuals with different degrees of relationship. (DOC 227 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Steinlein, O.K., Aichinger, E., Trucks, H. et al. Mutations in FKBP10 can cause a severe form of isolated Osteogenesis imperfecta. BMC Med Genet 12, 152 (2011). https://doi.org/10.1186/1471-2350-12-152
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-12-152