
Abstract
Background
Human bocavirus (HBoV) is a newly discovered parvovirus associated with acute respiratory tract illness (ARTI) and gastrointestinal illness. Our study is the first to analyze the characteristics of HBoV-positive samples from ARTI patients with a wide age distribution from Guangzhou, southern China.
Methods
Throat swabs (n=2811) were collected and analyzed from children and adults with ARTI over a 13-month period. The HBoV complete genome from a 60 year-old female patient isolate was also determined.
Results
HBoV DNA was detected in 65/2811 (2.3%) samples, of which 61/1797 were from children (<18 years old) and 4/1014 from adults (≥18 years old). Seasonal peaks of 4.8% and 7.7% were detected in May and June, respectively. 28 of 65 (43.1%) HBoV-positive samples were co-detected with 11/16 other potential pathogens. Mycoplasma pneumoniae had the highest frequency of 16.9% (11/65). Upper and lower respiratory tract illness were common symptoms, with 19/65 (29.2%) patients diagnosed with pneumonia by chest radiography. All four adult patients had systemic influenza-like symptoms. Phylogenetic analysis of the complete genome revealed a close relationship with other HBoVs, and a more distant relationship with HBoV2 and HBoV3.
Conclusions
HBoV was detected from children and adults with ARTI from Guangzhou, southern China. Elderly people were also susceptive to HBoV. A single lineage of HBoV was detected among a wide age distribution of patients with ARTI.
Similar content being viewed by others


Background
Respiratory tract infection etiology is complex and diverse, and new pathogens are continuously being reported. Over the past few years, several novel respiratory viruses including human metapneumovirus (hMPV) [1], severe acute respiratory syndrome (SARS) coronavirus [2], human coronavirus NL63 (HCoV-NL63) [3, 4], and coronavirus HKU1 (HCoV-HKU1) [5–7] have been identified.
In 2005, Allander et al. [8] reported a previously undescribed human parvovirus, human bocavirus (HBoV) that belongs to the genus Bocavirus, in respiratory secretions of children with respiratory tract disease in Sweden. HBoV is a single-stranded deoxyribonucleic acid (DNA) virus with a small genome size of approximately 5.3 kilo-bases (kb), which has three open reading frames (ORF) encoding two non-structural proteins NS1 and NP1, and the two structural proteins VP1 and VP2. VP1 and VP2 are located within the same ORF but have different initiator codon positions [8].
Subsequently, HBoV was reported in respiratory samples from different countries and regions worldwide [9–14], where HBoV was detected in 1.5%-8.3% of respiratory samples from individuals with acute respiratory tract illness (ARTI), especially young children and infants. The virus was also found in stool samples from patients with gastrointestinal illness [15–22]. These reports suggest that HBoV might be associated with upper and lower respiratory disease and gastrointestinal illness throughout the world. In 2009, two viruses closely related to HBoV, named HBoV2 [23] and HBoV3 [24], were found in stool samples, and suggested HBoV diversity.
HBoV infection has recently attracted increasing attention all over the world. However, the incidence and clinical presentation of this infection varies widely, and often involves co-infection with other potential pathogens [9–22]. Such characteristics have led to debate over the role of HBoV as a true pathogen. Therefore, additional evidence and studies are needed throughout the world to gain a better understanding of this virus. In this study, 2811 respiratory samples were collected from patients (with an age range of 9 days to 84 years) with ARTI in Guangzhou, southern China, from November 2009 to November 2010 to analyze the characteristics of HBoV-positive patients.
Methods
Samples in this study were taken as part of standard care. The First Affiliated Hospital of Guangzhou Medical University Ethics Committee approved the experimental design and patient involvement in this study.
Respiratory samples collection
Throat swab samples (n = 2811) were collected from patients with ARTI (presented at least two of the following symptoms: cough, pharyngeal discomfort, rhinobyon, snivel, sneeze, dyspnea) at three hospitals in Guangzhou, southern China between November 2009 and November 2010. Patients' ages ranged from nine days to 84 years, and included 1797 children (<18-years-old) and 1014 adults (≥18-years-old). Clinical characteristics of the patients were recorded for further analysis.
Real-time polymerase chain reaction (PCR) for HBoV detection
DNA from respiratory samples was extracted using a QIAamp DNA Mini Kit (Qiagen), in accordance with the manufacturer's protocol. Taqman real-time PCR primers and probe were designed based on the conserved region of the NP1 gene. Sequences were as follows: forward primer, 5'- GAG AGA GGC TCG GGC TCA TA-3' (2545-2564 nt); reverse primer, 5'- TCG AAG CAG TGC AAG ACG AT-3' (2592-2611 nt); and probe, 5'-FAM- CAT CAG GAA CAC CCA ATC AGC CAC C-BHQ1-3' (2566-2590 nt). Primers and the probe were synthesized by TaKaRa. Premix Ex Taq (Perfect Real Time) real-time PCR reaction buffer was also purchased from TaKaRa. Amplification was conducted using 10 pmol of primers, 3 pmol of probe and 5 μl DNA in a final volume of 25 μl. Cycling conditions included an initial incubation at 94°C for 2 min, followed by 40 cycles of 94°C for 10 sec and 55°C for 35 sec (ABI-7500 real-time PCR instrument,Applied Biosystems). The amplified NP1 gene target sequence (2545-2611 nt) was inserted into the pMD18-T vector (TaKaRa) and used as a positive control for quantification analysis. Sensitivity of the PCR assay was calculated to be 10 copies of plasmid DNA using positive control plasmid diluted gradients.
Detection of common respiratory pathogens in HBoV-positive samples
HBoV DNA positive samples were tested for 16 other potential pathogens, including influenza A virus, influenza B virus, parainfluenza virus (1, 2, 3, 4), respiratory syncytial virus, adenovirus, enterovirus, human metapneumovirus, human coronavirus (229E, OC43, NL63, HKU1), Mycoplasma pneumoniae, and Chlamydia pneumoniae by Taqman real-time PCR, in accordance with the manufacturer's protocol (Guangzhou HuYanSuo Medical Technology Co., Ltd).
Complete genome sequencing
The complete genome of HBoV from a 60-year-old female patient isolate was sequenced and analyzed. Sequencing primer sets were designed according to HBoV genome sequences available in the GenBank database (Table 1). DNA template (2 μl) was added to the Pfu DNA polymerase reaction mixture (Fermentas) in a final volume of 25 μl and PCR amplified. Products were purified after 1% agarose gel electrophoresis using a QIAquick Gel Extraction Kit (Qiagen). The purified DNA was then sequenced (ABI Genetic Analyzer 3130XL) and assembled using DNASTAR-SeqMan software (DNASTAR, http://www.dnastar.com/t-products-lasergene.aspx). PCR amplification and sequencing were conducted at least twice to ensure sequence accuracy.
Phylogenetic analysis
The HBoV complete genome (GU338055) was aligned to other HBoV genomes available in the GenBank database using the Basic Local Alignment Search Tool (BLAST, http://blast.ncbi.nlm.nih.gov/Blast.cgi). Phylogenetic analysis of 18 complete genomes of HBoV, HBoV2 and HBoV3 from different countries and regions, including USA, Sweden, Thailand, Japan, Australia, China, Hong Kong, and Taiwan was conducted using Molecular Evolutionary Genetics Analysis Version 4.0 (MEGA 4.0, http://www.megasoftware.net/). Phylogenetic trees were inferred from VP1/VP2 (3056-5071 nt), NS1 (253-2172 nt), NP1 gene (2410-3069 nt) and complete genome data (1-5299 nt) using the neighbor-joining method, and bootstrap values were calculated from 1000 replicates.
Statistical analysis
For comparison of categorical data, χ2 test and Fisher's exact test where appropriate. All tests were two-tailed and p < 0.05 was considered statistical significant.
Results
Detection of HBoV from patients with ARTI
HBoV DNA positive samples were detected in 65/2811 patients with a total positive rate of 2.3%. In children, positive rates were 4.1% (28/687) for 0-1 year-olds, > 4.8% (16/335) for 1-2 year-olds, 4.4% (10/225) for >2-3 year-olds, 1.7% (2/121) for >3-4 year-olds, 1.3% (1/80) for >4-5 year-olds, and 1.1% (4/349) for >5-17 year-olds (Figure 1), with a total of 3.4% (61/1797). The positive rate of adults (≥18 year-old) was 0.4% (4/1014) (Figure 1).

Distribution of HBoV positive patients among different age groups. *Positive rate (%). The number of HBoV-positive/total patients in each age groups are listed in parenthesis. Co-pathogens: co-detected with other pathogens. Single HBoV: detected with single HBoV, without other 16 potential pathogens.
The 65 positive patients were aged between 54 days and 70 years, comprising 83.1% (54/65) that were ≤ 3 years-old, 10.8% (7/65) that were 4-17 years-old, and 6.2% (4/65) adult patients. Four HBoV-positive adults were 19, 22, 60 and 70 years-old, respectively.
Seasonal distribution of HBoV
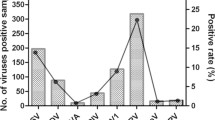
During our 13-month study period, more than 100 samples were collected for detection each month, and the ratio of ≤3 year-old patients ranged from 20.2% to 66.5%. Low positive rates of 0.3% (1/369) (single HBoV: 0, co-pathogen: 0.3%) were detected in March 2010 and 0.7% (1/135) (single HBoV: 0.7%, co-pathogen: 0) in October 2010, while high positive rates of 4.8% (8/166) (single HBoV: 3.0%, co-pathogens: 1.8%) were found in May 2010 and 7.7% (11/143) (single HBoV: 1.4%, co-pathogens: 6.3%) in June 2010 (Figure 2). The positive rates of other seven months fluctuated around the total rate of 2.3% (65/2811) (Figure 2).

Seasonal distribution of HBoV from November 2009 to November 2010. *Positive rate (%). The number of HBoV-positive/total patients in 13 months are listed in parenthesis. Co-pathogens: co-detected with other pathogens. Single HBoV: detected with single HBoV, without other 16 potential pathogens.
Co-detection with common respiratory pathogens
HBoV DNA positive samples were co-detected with 11 of 16 upper referred pathogens in 28/65 (43.1%) of the patients. Mycoplasma pneumoniae had the highest frequency of 16.9% (11/65), followed by RSV with 7.7% (5/65) (Table 2).
The clinical characteristics of HBoV-positive patients
The male:female ratio was 42:23 in the HBoV-positive patients which did not differ significantly from the HBoV-negative patients (p = 0.40). The clinical characteristics of the patients are listed in Table 3. Most patients presented with symptoms of upper respiratory tract illness (URTI), including cough (93.8%) and expectoration (40.0%); 44 (67.7%) patients presented with symptoms of fever (≥38°C); 41 (63.1%) patients had lower respiratory tract illness symptoms (LRTI) and 19 (29.2%) patients were diagnosed as pneumonia by chest radiography; 10 (15.4%) patients had gastrointestinal symptoms; seven (10.8%) patients had systemic influenza-like symptoms (chilly, swirl, headache, myodynia or debilitation); and one (1.5%) patient had herpetic angina.
Abnormal pulmonary breathing sounds and dyspnea were detected in 35 (53.9%) and 30 (46.2%) of 65 patients, respectively (Table 3). Nineteen (29.2%) were diagnosed as pneumonia by chest radiography, and 13/19 (68.4%) pneumonia patients were co-detected with one or two other pathogens, and a statistic difference was found for the symptom of "pneumonia" (p = 0.008) between the two groups "Single HBoV" and "Co-pathogens". Two of seven patients with systemic influenza-like symptom samples were co-detected with influenza A virus-enterovirus (triple pathogens) and influenza B virus (dual pathogens), respectively, while the remaining five patients were detected with single HBoV. All four adult patients (≥18 years old) presented with systemic influenza-like symptoms; three had only HBoV detection and the other 19 year-old female patient was co-detected with influenza B virus.
Complete genome sequencing and phylogenetic analysis
Complete HBoV genome sequences for isolates from a 60 year-old patient were sequenced and submitted to the GeneBank (Accession Number:GU338055). The full length of the genome was 5299 bases and the distribution of A/G/C/T was 32.4%/20.4%/21.8%/25.4%, respectively. Compared to the other HBoV genomes available in the GeneBank database, GU330588 showed a 98% similarity with the HBoV previously described by Allander et al. [8]. Phylogenetic trees were inferred from VP1/VP2, NS1, and NP1 gene data, in addition to the complete genome sequence (Figure 3). GU330588 was similar to other HBoVs, although it displayed obviously sequence variations from HBoV2 and HBoV3. Three HBoV lineages were illustrated in all four phylogenetic trees (Figure 3).

Phylogenetic analysis of HBoV-GU338055 genome (A), VP1/VP2 genes (B), NP1 gene (C) and NS1 gene (D). HBoV/GU338055 is indicated with "●".
Discussion
HBoV is a novel parvovirus first described in 2005 by Allander and colleagues [8]. Since that time, it has been associated with upper and lower-respiratory tract disease and gastrointestinal illness throughout the world. However, most studies were focused on young children and infants [9–14], and only a few papers have described the characteristics of HBoV infection in adult patients [16, 25, 26]. Our study successfully analyzed the characteristics of HBoV-positive samples from ARTI-infected patients with a wide age distribution from Guangzhou, southern China for the first reported time. Similar to previous study [16], the detection rate in pediatric patients (<18 years old) was significantly higher than that in adults (≥18 years old) (p < 0.001), and most HBoV-positive patients were ≤3 years old. HBoV was detected in four adult patients, including 60 and 70 year-old patients. This suggested that older people were also susceptive to HBoV infection, although at much lower positive rates. Four adult patients also presented with systemic influenza-like symptoms, which might suggest that HBoV infection in adults is a more complex and serious problem than in children.
While seasonal peaks of HBoV infection vary among different counties and regions because of climate and other factors, many previous studies suggest a higher detection rate in winter [27, 28]. In our study, a higher frequency of HBoV was observed between May and June during the 13-month testing period (Figure 2). This result was similar to the report of Choi and colleagues, in which a seasonal peak was observed between May and June [29].
Characteristics of HBoV-positive patients in our study were also similar to previous reports [9–14]. The male:female ratio in the HBoV-positive patients did not differ significantly from the HBoV-negative patients. In all clinical characteristics, seven major symptoms (cough, fever (≥38°C), abnormal pulmonary breath sound, dyspnea, expectoration, snivel and pneumonia) had the highest ratio (ratio > 10%) (Table 3) and were common as URTI and LRTI.
As previous studies [13, 29–34], co-detection with other potential pathogens was common in HBoV-positive patients. In this work, 43.1% (28/65) patients were co-detected with other 16 potential pathogens, and there would be higher ratio if human rhinovirus were concerned [16]. Furthermore, not only co-pathogens but also single HBoV groups had a high ratio of major symptoms (Table 3), which might suggest HBoV is an important pathogen in URTI and LRTI. A statistic difference was found for the symptom of "pneumonia" between the two groups "Single HBoV" and "Co-pathogens" (Table 3), which might further suggest HBoV is an important pathogen. Further studies were required to determine whether HBoV played a causative role in these co-infections or acted as an exacerbation factor.
Before 2009, little variation was found in the surface protein of HBoV (VP1-VP2), and some researchers predicted that HBoV infection might only occur after the subsequently development of life-long immunity via the neutralization of the target antibody [16]. However, novel types of HBoV2 and HBoV3 were described successively in 2009 [23, 24], suggested diversity within this group of viruses. Furthermore, we successfully sequenced the complete HBoV genome (GU338055) for an isolate from an adult patient and phylogenetic analysis revealed the existence of three lineages: group I, HBoV; group II, HBoV2; group III, HBoV3, and GU338055 was located in group I.
The frequency and clinical presentations of HBoV infection vary widely [9–14, 16, 25, 26] and are typically associated with other pathogens [13, 29–34]. Such characteristics have subsequently led many scientists to question the role of HBoV as a potential pathogen. This lack of information is largely because of the inability of HBoV to grow in standard cell lines [35]. To confirm the effects of HBoV, more studies are required throughout the world, focusing on various aspects of this infection, including epidemiology, serology, molecular biology, in vitro culture and animal models.
Conclusions
HBoV was detected from children and adults with ARTI from Guangzhou, southern China, and the features were described in this study. HBoV was confirmed in elderly patients (60 and 70 years old), suggesting that older people were also susceptive to HBoV. All four adult patients with HBoV positive in this study presented systemic influenza-like symptoms, which potentially suggest that HBoV infection in adults may develop more serious symptoms than those in children. Phylogenetic analysis suggested that HBoV-GU338055 from an elderly patient is in a single lineage with other HBoVs.
References
van den Hoogen BG, de Jong JC, Groen J, Kuiken T, de Groot R, Fouchier RAM, Osterhaus ADME: A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat Med. 2001, 7: 719-724.
Peiris JSM, Lai ST, Poon LLM, Guan Y, Yam LYC, Lim W, Nicholls J, Yee WKS, Yan WW, Cheung MT, Cheng VCC, Chan KH, Tsang DNC, Yung RWH, Ng TK, Yuen KY: Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003, 361: 1319-1325.
Fouchier RA, Hartwig NG, Bestebroer TM, Niemeyer B, de Jong JC, Simon JH, Osterhaus ADME: A previously undescribed coronavirus associated with respiratory disease in humans. Proc Natl Acad Sci USA. 2004, 101: 6212-6216.
van der Hoek L, Pyrc K, Jebbink MF, Vertheim-Oost W, Berkhout RJM, Wolthers KC, van Dillen PMEW, Kaandorp J, Spaargaren J, Berkhout B: Identification of a new human coronavirus. Nat Med. 2004, 10: 368-373.
Woo PCY, Lau SKP, Tsoi HW, Huang Y, Poon RW, Chu CM, Lee RA, Luk WK, Wong GK, Wong BH, Cheng VC, Tang BS, Wu AK, Yung RW, Chen H, Guan Y, Chan KH, Yuen KY: Clinical and molecular epidemiological features of coronavirus HKU1-associated community-acquired pneumonia. J Infect Dis. 2005, 192: 1898-1907.
Woo PCY, Lau SKP, Chu CM, Chan KH, Tsoi HW, Huang Y, Wong BHL, Poon RWS, Cai JJ, Luk WK, Poon LLM, Wong SSY, Guan Y, Peiris GSM, Yuen KY: Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol. 2005, 79: 884-895.
Lau SKP, Woo PCY, Chu CM, Yip CCY, Tse H, Tsoi HW, Cheng VCC, Lee P, Tang BSF, Cheung CHY, Lee RA, So LY, Lau YL, Chan KH, Yuen KY: Coronavirus HKU1 and other coronavirus infections in Hong Kong. J Clin Microbiol. 2006, 44: 2063-2071.
Allander T, Tammi MT, Eriksson M, Bjerkner A, Lindell AT, Andersson B: Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc Natl Acad Sci USA. 2005, 102 (36): 12891-12896.
Kesebir D, Vazquez M, Weibel C, Shapiro ED, Ferguson D, Landry ML, Kahn JS: Human bocavirus infection in young children in the United States: molecular epidemiological profile and clinical characteristics of a newly emerging respiratory virus. J Infect Dis. 2006, 194: 1276-1282.
Fry AM, Lu X, Chittaganpitch M, Peret T, Fischer J, Dowell S, Anderson LJ, Erdman D, Olsen SJ: Human bocavirus: a novel parvovirus pidemiologically associated with pneumonia requiring hospitalization in Thailand. J Infect Dis. 2007, 195: 1038-1045.
Maggi F, Andreoli E, Pifferi M, Meschi S, Rocchi J, Bendinelli M: Human bocavirus in Italian patients with respiratory diseases. J Clin Virol. 2007, 38: 321-325.
Arden KE, McErlean P, Nissen MD, Sloots TP, Mackay LM: Frequent detection of human rhinoviruses, paramyxoviruses, coronaviruses, and bocavirus during acute respiratory tract infections. J Med Virol. 2006, 78: 1232-1240.
Arnold JC, Singh KK, Spector SA, Sawyer MH: Human bocavirus: prevalence and clinical spectrum at a children's hospital. Clin Infect Dis. 2006, 43: 283-288.
Qu XW, Duan ZJ, Qi ZY, Xie ZP, Gao HC, Liu WP, Huang CP, Peng FW, Zheng LS, Hou YD: Human bocavirus infection, People's Republic of China. Emerg Infect Dis. 2007, 13 (1): 165-168.
Chieochansin T, Thongmee C, Vimolket L, Theamboonlers A, Poovorawan Y: Human bocavirus infection in children with acute gastroenteritis and healthy controls. Jpn J Infect Dis. 2008, 61 (6): 479-481.
Lau SK, Yip CC, Que TL, Lee RA, Au-yeung RKH, Zhou B, So LY, Lau YL, Chan KH, Woo PCY, Yuen KY: Clinical and molecular epidemiology of human bocavirus in respiratory and fecal samples from children in Hong Kong. J Infect Dis. 2007, 196 (7): 986-993.
Lee JI, Chung JY, Han TH, Song MO, Hwang ES: Detection of human bocavirus in children hospitalized because of acute gastroenteritis. J Infect Dis. 2007, 196 (7): 994-997.
Albuquerque MC, Rocha LN, Benati FJ, Soares CC, Maranhão AG, Ramírez ML, Erdman D, Santos N: Human bocavirus infection in children with gastroenteritis, Brazil. Emerg Infect Dis. 2007, 13 (11): 1756-1758.
Cheng WX, Jin Y, Duan ZJ, Xu ZQ, Qi HM, Zhang Q, Yu JM, Zhu L, Jin M, Liu N, Cui SX, Li HY, Fang ZY: Human bocavirus in children hospitalized for acute gastroenteritis: a case-control study. Clin Infect Dis. 2008, 47 (2): 161-167.
Yu JM, Li DD, Xu ZQ, Cheng WX, Zhang Q, Li HY, Cui SX, Jin M, Yang SH, Fang ZY, Duan ZJ: Human bocavirus infection in children hospitalized with acute gastroenteritis in China. J Clin Virol. 2008, 42 (3): 280-285.
Campe H, Hartberger C, Sing A: Role of human bocavirus infections in outbreaks of gastroenteritis. J Clin Virol. 2008, 43 (3): 340-342.
Schildgen O, Muller A, Simon A: Human bocavirus and gastroenteritis. Emerg Infect Dis. 2007, 13 (10): 1620-1621.
Kapoor A, Slikas E, Simmonds P, Chieochansin T, Naeem A, Shaukat S, Alam MM, Sharif S, Angez M, Zaidi S, Delwart E: A newly identified bocavirus species in human stool. J Infect Dis. 2009, 199 (2): 196-200.
Arthur JL, Higgins GD, Davidson GP, Givney RC, Ratcliff RM: A novel bocavirus associated with acute gastroenteritis in Australian children. PLoS Pathog. 2009, 5 (4): e1000391-
Bastien N, Brandt K, Dust K, Ward D, Li Y: Human bocavirus infection, Canada. Emerg Infect Dis. 2006, 12: 848-850.
Kupfer B, Vehreschild J, Cornely O, Kaiser R, Plum G, Viazov S, Franzen C, Tillmann RL, Simon A, Müller A, Schildgen O: Severe pneumonia and human bocavirus in adult. Emerg Infect Dis. 2006, 12: 1614-1616.
Chow BD, Huang YT, Esper FP: Evidence of human bocavirus circulating in children and adults, Cleveland, Ohio. J Clin Virol. 2008, 43 (3): 302-306.
Bastien N, Chui N, Robinson JL, Lee BE, Dust K, Hart L, Li Y: Detection of human bocavirus in Canadian children in a 1-year study. J Clin Microbiol. 2007, 45 (2): 610-613.
Choi EH, Lee HJ, Kim SJ, Eun BW, Kim NH, Lee JA, Lee JH, Song EK, Kim SH, Park JY, Sung JY: The association of newly identified respiratory viruses with lower respiratory tract infections in Korean children, 2000-2005. Clin Infect Dis. 2006, 43 (5): 585-592.
Sloots TP, McErlean P, Speicher DJ, Arden KE, Nissen MD, Mackay IM: Evidence of human coronavirus HKU1 and human bocavirus in Australian children. J Clin Virol. 2006, 35: 99-102.
Regamey N, Frey U, Deffernez C, Latzin P, Kaiser L: Isolation of human bocavirus from Swiss infants with respiratory infections. Swiss Paediatric Respiratory Research Group. Pediatr Infect Dis J. 2007, 26: 177-179.
Kaplan NM, Dove W, Abu-Zeid AF, Shamoon HE, Abd-Eldayem SA, Hart CA: Human bocavirus infection among children, Jordan. Emerg Infect Dis. 2006, 12: 1418-1420.
Foulongne V, Olejnik Y, Perez V, Elaerts S, Rodiere M, Segondy M: Human bocavirus in French children. Emerg Infect Dis. 2006, 12: 1251-1253.
Weissbrich B, Neske F, Schubert J, Tollmann F, Blath K, Blessing K, Kreth HW: Frequent detection of bocavirus DNA in German children with respiratory tract infections. BMC Infect Dis. 2006, 6: 109-
Chow BD, Esper FP: The human bocaviurses: a review and discussion of their role in infection. Clin Lab Med. 2009, 29 (4): 695-713.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2334/11/345/prepub
Acknowledgements
The State Major Infectious Disease Research Program (China Central Government, 2009ZX10004-109) provided financial support for this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RZ and W-KL designed the study. W-KL, Q L, H-XL performed the HBoV DNA testing. D-HC, Z-FY and SQ collected clinical data. All authors participated in the data analysis. W-K L and R Z drafted the manuscript. All authors read and approved the final version of this manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Liu, WK., Chen, DH., Liu, Q. et al. Detection of human bocavirus from children and adults with acute respiratory tract illness in Guangzhou, southern China. BMC Infect Dis 11, 345 (2011). https://doi.org/10.1186/1471-2334-11-345
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2334-11-345