
Abstract
Background
Polymerase chain reaction (PCR)-based techniques are widely used to identify fungal and bacterial infections. There have been numerous reports of different, new, real-time PCR-based pathogen identification methods although the clinical practicability of such techniques is not yet fully clarified.
The present study focuses on a novel, multiplex, real-time PCR-based pathogen identification system developed for rapid differentiation of the commonly occurring bacterial and fungal causative pathogens of bloodstream infections.
Results
A multiplex, real-time PCR approach is introduced for the detection and differentiation of fungi, Gram-positive (G+) and Gram-negative (G-) bacteria. The Gram classification is performed with the specific fluorescence resonance energy transfer (FRET) probes recommended for LightCycler capillary real-time PCR. The novelty of our system is the use of a non-specific SYBR Green dye instead of labelled anchor probes or primers, to excite the acceptor dyes on the FRET probes. In conjunction with this, the use of an intercalating dye allows the detection of fungal amplicons.
With the novel pathogen detection system, fungi, G + and G- bacteria in the same reaction tube can be differentiated within an hour after the DNA preparation via the melting temperatures of the amplicons and probes in the same tube.
Conclusions
This modified FRET technique is specific and more rapid than the gold-standard culture-based methods. The fact that fungi, G + and G- bacteria were successfully identified in the same tube within an hour after the DNA preparation permits rapid and early evidence-based management of bloodstream infections in clinical practice.
Similar content being viewed by others


Background
Bloodstream infections are life-threatening, especially in individuals with serious underlying conditions or an impaired immune system [1]. The number of reported cases of bloodstream infections in the USA, between 1979 and 2002, was 10, 319, 418 and demonstrated an annualized increase of 8.7% [2]. In critically ill patients, the majority of infections are caused by bacteria but fungal infections, although these account for only 4.6% of all infections, have a significant impact on public health. [2]. Mixed fungal/bacterial infections are not uncommon, incidences of combined Candida and bacterial bloodstream infections have been reported in as many as 23% of all episodes of candidaemia [3]. Despite its relatively low frequency, fungal blood stream infections can progress to severe sepsis and septic shock, associated with a drastic rise in mortality; therefore, early and appropriate treatment of such infections is critical [4, 5].
Since molecular diagnosis in sepsis is reliable, and faster than the classical blood-culturing techniques, there has been an increase in interest in methods such as PCR, ligase chain reaction, nucleic acid sequence based amplification, and nested PCR [6, 7]. Nevertheless, these molecular approaches are applied only following the positivity of the blood culture; therefore, they require a substantial amount of elapsed time.
In contrast, the LightCycler PCR assay is fast, reliable and relatively easy to perform - even in small laboratories.
This method is based on a previously-reported fluorescence resonance energy transfer (FRET) technique which involves a distance-dependent interaction between the electronic excited states of two dye molecules [8]. The excitation is transferred from a donor (anchor) molecule to an acceptor (quencher) molecule, without emission of a photon, and has been proved to be an appropriate method for discriminating between the commonly occurring pathogen G + and G- bacteria [9]. The differentiation, via the melting temperature of the overall PCR product and the melting point of the probes, allowed creation subgroups within the G + and G- stains, and this system required less than 4 h, inclusive of the time need for the DNA preparation and the evaluation of the PCR results [10]. Until now, parallel detection of fungal and bacterial infections in a real-time system has been an unresolved problem however there are several tests in the market with the same purpose. Some of them detect bacteria, without fungal identification (Prove-It; Mobidiag, Helsinki, Finland or SeptiTest; Molzym, Bremen, Germany). The Reflex PCR assay (Molzym, Bremen, Germany) includes several steps after the PCR which increases the time required. The SepiFast (Roche; Basel, Switzerland) assay is similar to our system but works with three parallel reaction vessels and a different principle for detection. Furthermore, it requires individual molecular laboratory, equipments and software. Identification of the most common clinically relevant fungi is possible through a simple melting-point analysis relating to the ITS2 (internal transcribed spacer) region. This non-coding region is a highly variable rRNA region that is adaptable for the identification of clinically relevant fungi over a broad range [11]. Measurements are made at 540 nm, and require a non-specific intercalating dye [12].
Real-time PCR detection can be performed by using free dyes or labelled sequence-specific probes. One combination of the two techniques uses unlabelled probes for the amplicon detection and Tm determination [13]. Another parallel application was the combination of TaqMan chemistry and the very new, aspecific dye, BOXTO, as a multiplex PCR [14].
The novelty of our prototype system lies in the use of non-specific SYBR Green dye as a donor molecule, instead of a labelled primer or other specific anchor probe. With this technique, it is possible to examine pathogenic fungi, G + and G- bacteria in a single tube multiplex PCR reaction.
Results and discussion
Discrimination of the fungal, G + and G- bacterial pathogens
DNA samples from all species studied were prepared and amplified successfully with the SYBR Green dye-based method in the LightCycler instrument. Species-specific Tm-s were obtained by melting-point analysis on three detection channels and all pathogens were identified correctly as fungi or G- or G + bacteria (Table 1). On the F1 channel (540 nm), the melting points of all the amplicons (Tm A) were visible, due to the fluorescent signal of the SYBR Green non-specific intercalating dye. On the F2 (640 nm) and F3 (705 nm) channels, the G- and the G + probes (Tm P), respectively, gave fluorescence signals. After the discrimination of the G- and G + strains, the fungal pathogens could be screened, because the fungal strains gave no signal on the F2; F3 channels.
Determination of the bacterial pathogens
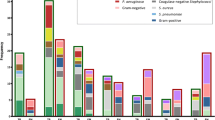
Four G + and nine G- bacterial subgroups could be distinguished through a joint consideration of the melting points of the probes and the melting point of the overall PCR product (Figure 1).

Differentiation of the bacterial pathogens. The group temperatures indicate the entire Tm of the pathogens. The subgroup temperatures are the melting temperatures of the hybridization probes.
S. aureus and S. epidermidis have very close-lying melting temperatures and their species-specific differentiation is not possible via this 16S rRNA sequence (Figure 2). A comparison of the Gene Bank sequences (S. aureus and S. epidermidis NCBI Taxonomy ID: NC_009782.1 and JF_799903.1) of these species revealed a variance of only three base-pairs, none of them in the region where the probe is associated with the DNA. Thus, determination of the clinically relevant Staphylococcus species requires other gene sequences in which the antibiotic resistance can be detected [15]. The situation is the same for the two Enterococcus species [16]. At the same time, S. pyogenes and L. monocytogenes are clearly differentiable.

Melting-peaks of Staphylococcus aureus and Staphylococcus epidermidis. Revealing that it is impossible to differentiate these Staphylococcus species via the Tm data of the amplicons or probes.
Among the G- bacteria, E. coli is one of the most common causative agents of bloodstream infections [17]. Unfortunately, it has almost the same Tm as those of E. cloacae and S. marcescens. Other bacterial strains, such as H. influenza, are clearly differentiable through the melting temperature of the probe (Figure 3) or amplicon. The sensitivity of the reaction was five colony-forming units (CFU) per reaction.

Differentiation of Escherichia coli from Haemophilus influenzae . Although these pathogens have a very similar Tm in the 16S rRNA region, the Tm of the probes are clearly different.
Determination of fungal pathogens
Fourteen frequently-encountered fungal pathogens could be distinguished. The highly variable ITS 2 target sequence allowed correct identification of all of the clinically relevant fungal strains, through the Tm points on the F1 channel [12, 18]. There was no signal on the F2 or F3 channel. The sensitivity of the reaction was 5 CFU per reaction.
The correct differentiation between bacteria and fungi was verified by means of gel electrophoresis, with the help of the amplicon length (fungal amplicons 192–494 bp, bacterial 187 bp).
Determination of the co-infection model
In case of co-infections, there are some limitations in the detection. If the ratios of the different agents are higher than 1:10, the system does not detect the infectious agent which is in lower quantities.
Calibration of the multiplex PCR
All three non-specific dyes (LCGreen, EvaGreen and SybrGreen) excited all of the labelled probes (LCRed640, LCRed705 and Cy5.5). The best results were obtained with the SybrGreen dye.
The determination of Tm is very sensitive to the composition of the PCR reaction mixture, and primarily to the ionic strength. To avoid Tm bias originating from pipetting errors between PCR runs, the application of mastermixes is advisable. One limitation of the method is that the various mastermixes offered by different suppliers differ in reagent composition, which may influence the Tm values.
Naturally, repeated runs with a given mastermix yield reproducible data. In the event of a change of mastermix, however, calibration is necessary to establish the new Tm data on the fungal strains.
The data determined in the present work were obtained with the use of “Fermentas Maxima SybrGreen, no ROX” and five-eight parallel experiments. No false positive samples were found when this method was tested. No significant differences in the melting peak temperatures were observed between different isolates of the same species. The standard deviation of the melting peak temperatures of all 21 references and 93 clinical isolates with bacterial and fungal strains was between 0.08 and 0.88, as listed in Table 1. These data are in concordance with our previous results [19, 20].
Sensitivity and reproducibility
For sensitivity testing of the prototype system, six bacterial and two fungal gDNA preparations were made from artificially infected blood. Eight species, and eight parallel investigations of five dilutions of the bacterial suspensions in blood were, analysed. Of 8 reactions for each species, all were positive with 50 DNA copies, 98.5% were positive with 10 copies, 67.2% were positive with 5 copies and 21.9% were positive with 2 copies (Table 2). All the reactions were carried out within the same parameters described in the section PCR conditions.
Conclusions
Real-time PCR is one of the fastest diagnostic methods currently available. The use of rRNA genes for the detection is based on the conserved 16S rRNA sequences of the bacteria. As regards fungi, the ITS sequence refers to a segment of non-functional RNA, situated between 5.8S and 28S rRNAs. To reproduce the results, it is possible to differentiate between fungi and bacteria, or between fungal species by electrophoresis [21, 22] or melting-point analysis [23]. The Roche LightCycler PCR was specially developed to amplify amplicons under 500 bp. The amplicons amplified by PLK1/PLK2 comprised 187 bp, while the fungal amplicons amplified by ITS86/ITS4 primer pair varied between 192 bp (Geotrichum candidum) and 494 bp (Malassezia furfur), values which are perfectly suited to this instrument profile. In this study, the advantage of the LC system was utilised when FRET technique was used to detect and differentiate the bacterial pathogens. As a novel element, excitation of the fluorescent probes was carried out with the help of a non-specific intercalating dye, this is an uncommon procedure in real-time investigations. It allows parallel detection of fungal pathogens and with bacteria in the same tube. As the result of the use of the multiplex PCR in combination with FRET probes and melting point-analysis, the broad-range identification of many frequent causative agents of bloodstream infections becomes possible within four hours. Sensitivity of pathogen PCR in sepsis is generally between 3 and 100 CFU/mL according to the literature [24]. The sensitivity of our prototype system was five CFU per reaction, which in combination with an efficient preparation is suitable for the detection of bloodstream infections. If commercially available “Midi” preparation kits (i.e.: NucleoSpin Blood L, Macherey-Nagel, Düren, Germany) were used, the sample mateial was 2 mL of blood, the elution volume was 100 μL and finally 5 μL of eluate were used for subsequent PCR. The calculated sensitivity was 50 CFU/mL blood. The sample/eluent ratio was the same in case of midi and maxi preparation kits which means that increased sample volume is not enhancing the sensitivity [25]. The sensitivity of the “gold standard” conventional blood culture technique is one CFU per 10 mL blood sample. Our method is less sensitive. The blood culture technique is not replaceable with molecular techniques so far but the time delay until the adequate therapy can be reduce.
To determine the diagnostic sensitivity and reproducibility of the method, experiments with artificially infected blood were performed. The sensitivity of the PCR was 2 to 10 copies per reaction, which was the same as with cultivated cells. The melting points (TmA and TmP) were the same as we described in Table 1. with “Fermentas Maxima SybrGreen, no ROX”; therefore, human gDNA does not inhibit the reaction and does not modify the melting peaks.
With this method, neither the G + S. aureus and S. epidermidis nor the G- E. coli, E. cloacae and S. marcescens can be distinguished, and additional species-specific probes or primers are necessary for the further differentiation of these species.
Antibiotic resistance cannot be determined directly with this prototype system. The susceptibility testing of resistant E. coli strains can be performed using a PCR-based technique with other 16S rRNA specific primers [26]. Unfortunately, these investigations require a PCR analysis after the identification of the bacteria.
In spite of its limitations, the prompt and reliable information provided by this new diagnostic method on the most common pathogenic bacteria might permit targeted therapy with narrow-spectrum antibiotics, instead of empirically-administered broad-spectrum antibiotics. To confirm these findings in clinical practice, a prospective study is now being designed and engineered.
The incidence of sepsis has been continuously increasing over recent decades, and the early detection of the pathogens can have a great impact on the clinical outcome of infections [27–30] Molecular diagnostic systems allow species identification in less than 24 hours - which is a drastic improvement relative to the gold-standard, culture-based method and Gram staining-based identification methods that yield results in 24 to 72 hours [31, 32].
With the novel method described above (multiplex PCR with the new combination of aspecific dyes and labelled probes), the most common causative agents of bloodstream infections can be detected in two hours, without DNA preparation; therefore, this method offers a huge advantage over traditional FRET-based assays by accurately detecting the Tm of both the probes and the amplicons.
Methods
Reference strains of 17 clinically relevant bacterial species were collected, as typical of the main causative agents of bloodstream infections [33]. Nine reference strains, Staphylococcus aureus ATCC 25923, Staphylococcus epidermidis ATCC 12228, Enterococcus faecalis ATCC29212, Listeria monocytogenes ATCC 4701, Bacteroides fragilis ATCC 25285, Pseudomonas aeruginosa ATCC 27853, Haemophilus influenzae ATCC 49247, Escherichia coli ATCC 25922 and Klebsiella pneumoniae ATCC 700603 were from the American Type Culture Collection. [ATCC]. Streptococcus pyogenes OKI 80002 was from the National Centre for Epidemiology, Hungary [OKI] and Proteus vulgaris HNCMB 60076 was from the Hungarian National Collection of Medical Bacteria [HNCMB]. Furthermore, to confirm the reliability and reproducibility of the technique, clinical strains of S. aureus (n = 4), S. epidermidis (n = 6), S. pyogenes (n = 2), E. faecalis (n = 2), E. faecium (n = 3), L. monocytogenes (n = 1), B. fragilis (n = 2), P. aeruginosa (n = 1), H. influenzae (n = 1), E. coli (n = 5), K. pneumoniae (n = 5), P. vulgaris (n = 3), Stenotrophomonas maltophilia (n = 2), Serratia marcescens (n = 2), Enterobacter aerogenes (n = 2), E. cloacae (n = 2) and Acinetobacter baumannii (n = 3) from the Institute of Clinical Microbiology at the University of Szeged were also included. The species identities of the clinical isolates were confirmed by conventional biochemical methods.
Ten fungal strains were examined. Five reference strains, Candida albicans ATCC 10231 and ATCC 14053, C. tropicalis ATCC 750, C. parapsilosis ATCC 22019 and C. glabrata ATCC 39316, were from the [ATCC], Cryptococcus neoformans IFM 5844 and IFM 5855 were from IFM Quality Services Pty Ltd [IFM], and Aspergillus fumigatus SzMC 2486, A. flavus SzMC 2536 and A. niger SzMC 2761 were from the Szeged Microbiological Collection [SzMC]. Furthermore, clinical strains of C. albicans (n = 14), C. glabrata (n = 5), C. tropicalis (n = 4), C. parapsilosis (n = 5), C. krusei (n = 4), C. quillermondii (n = 4), C. lusitaniae (n = 3), C. norvegensis (n = 1), C. inconspicua (n = 2), C. dubliniensis (n = 2) and Cryptococcus neoformans (n = 2) from the Institute of Clinical Microbiology at the University of Szeged were also tested.
Bacterial DNA purification
The bacterial strains were grown on Columbia agar base under aerobic conditions, except that Bacteroides fragilis was grown under anaerobic conditions. The bacterial DNA was extracted with the QIAamp® DNA Blood Mini Kit (QuiaGene Inc, Chatsworth, Calif., USA), following the manufacturer’s instructions in “Protocols for Bacteria”. One millilitre of log-phase culture suspension, at a concentration of 107 CFU/mL, was used for the preparation. For determination of the sensitivity of the reaction, 100 μL of the serially diluted S. aureus reference strain was used for DNA extraction. The number of bacterial cells was determined by plating aliquots of serially diluted samples onto Columbia agar base.
For lysis of the rigid multilayered G + bacterial cell wall, we used a pre-incubation step with 20 mg/mL lysozyme (in 20 mM Tris · HCl, pH 8.0, 2 mM EDTA, 1.2% TritonX100). The spin protocol for “DNA Purification from Tissues” was followed, after incubation at 30°C for 30 min. The final concentration of DNA was 2.0-13.8 ng/μL, with a ratio A260/A280 = 1.6-1.8 after purification.
Fungal DNA purification
All the fungi were grown on Sabouraud medium. The fungal DNA was extracted from 1 mL of a log-phase culture suspension containing 9.6 × 107 of fungal cells. For determination of the sensitivity of the reaction, 100 μL of the serially diluted C. albicans reference strain was used for DNA extraction. The number of fungal cells was determined by plating aliquots of serially diluted samples onto Sabouraud-glucose medium.
We followed the QIAamp® DNA Mini Kit Protocol for Yeasts. In this case, additional reagents were required for elimination of the complex fungal cell-wall structure: sorbitol buffer (1 M sorbitol, 100 mM EDTA, 14 mM β-mercaptoethanol) [34] was used, and the samples were incubated with lyticase for 30 min at 30°C. Efficient and complete lysis was achieved in 1.5 hour in a shaking water-bath. This purification yielded 2.0–25 μg of DNA in 100 μL of water (2.0–13.8 ng/μL), with A260/A280 = 1.6–1.8.
DNA preparation from infected blood
Samples of 180 μL healthy donor bloods in EDTA vacutainer tubes were infected with 20 μL of log-phase culture suspension at a concentration of 108 CFU/mL bacterial and/or fungal suspensions. Bacterial and fungal cells were quantified, in a Bürker chamber, by viable counts. For the sensitivity testing of the prototype system, the bloods were infected with five dilutions of the log-phase culture suspension at a final volume of 20 μL. The first dilution contained 50 copies in 1 μL template DNA (2.5x104 CFU/mL blood), the second contained 10 copies (5x103 CFU/mL blood), the third 5 copies (2.5x102 CFU/mL blood) and the fourth 2 copies (5x102 CFU/mL blood). The red blood cells were disrupted by lysis buffer [35], the bacterial and fungal cell wall lysed using the freezing-thawing method. After digestion with Proteinase K, the DNA was extraction carried out as reported previously [36].
Bacterial and fungal primer design, FRET probes
Two primer pairs were used for multiplex amplification of bacterial and fungal DNA.
The bacterial primer pair was PLK1 (TAC GGG AGG CAG CAG) forward and PLK2 (TAT TAC CGC GGC TGC T) reverse, which are highly conserved in different groups of bacteria [9] and amplify the 16S rRNA sequence. The PLK2 reverse primer was modified and used without the inner fluorescence labelling. Originally, the labelled primer excited the Gram specific probes. We applied the non-specific SYBR Green dye for excitation; it also serves for visualization of the fungal amplicons. This primer-pair produces a 187 bp fragment in each species.
Previously hybridization probes were used for the Gram classification [10]. ISN2 (5′-CCG CAG AAT AAG CAC CGG CTA ACT CCG T-3′) labelled with LCRed 640 was specific for G-, and ISP3 (5′-CCT AAC CAG AAA GCC ACG GCT AAC TAC GTG-3′) labelled with Cy5.5 was specific for G + bacteria, and the [10] ISP2 probe was labelled with LCRed705 at the 5′ end. The producers offered Cy5.5 dye instead of LCred705. This modified probe was used in our experiments.
The ITS86 forward (GTG AAT CAT CGA ATC TTT GAA C) and the ITS 4 reverse (TCC TCC GCT TAT TGA TAG C) primers were used for detection of the fungi. These primers amplify a 192–494 bp sequence of ITS2 region, which is a highly variable part between the 5.8S and 28S rRNA sequence [37].
Mastermixes/excitation dyes
Different, non-specific intercalating dyes are used for real-time PCR investigations. Most of these are accessible in ready-to-use, mastermix formulae. Our goal was to choose the best dye for excitation of the labelled probes. The tested dyes were LCGreen “LightCycler® 480 High Resolution Melting Master” (Roche Diagnostic GmbH, Mannheim, Germany); SybrGreen “LightCycler® 480 DNA Master SYBR Green I”, (Roche); “IQ™ SYBR® Green Supermix” (Bio-Rad Laboratries, Inc., Hercules, CA, USA); “Maxima™ SYBR Green qPCR Master Mix no ROX” (Fermentas, Vilnius, Lithuania); and EvaGreen (“LC-FastStart DNA Master Hybridization Probes” (Roche) combined with EvaGreen dye (Biotium Inc., Hayward, CA, USA) and “Sso Fast™ EvaGreen® Supermix” (BioRad). All mastermixes were used according to the manufacturer’s instructions.
PCR conditions
PCR was performed using a LightCycler real-time PCR instrument (Roche). The reaction volume of 10 μL contained 1 μL of DNA (with a final concentration of ~10 ng/μL), 1 μM of each of the primers, 0.7 μM of each of the probes, an appropriate amount of mastermix, and 0.2 mM BSA (in the cases of the Fermentas and BioRad mastermixes).
The PCR conditions were as follows: initial denaturation at 95°C for 600 s, followed by 40 cycles of denaturation (95°C for 0 s, 20°C/s), annealing (55°C for 15 s, 20°C/s), and extension (72°C for 20 s, 2°C/s). The emitted fluorescence was measured after the annealing steps. The melting-curve analysis procedure consisted of 1 cycle at 95°C for 10 s, 40°C for 120 s, followed by an increase in the temperature to 95°C at 0.2°C/s. The fluorescence signal (F) was monitored continuously during the temperature ramp, and plotted against temperature (T).
Data analysis
The melting peaks were evaluated using the LightCycler Software V 3.5. The melting-peaks were determined through the manual Tm option on the three detection channels (F1, F2 and F3).
The standard deviation (SD) of the melting-points was calculated from five parallel experiments. The fungal or bacterial samples were verified by gel electrophoresis on 1.5% agarose gel, with the help of a low-range DNA ladder.
The sensitivity of the multiplex PCR calculated from five dilutions of the bacterial suspension.
Authors’ information
ÁH: Doctoral fellow in the Department of Medical Microbiology and Immunobiology, Faculty of Medicine, University of Szeged, Dóm tér 10, Szeged, Hungary, ZP: Chief of Medicine on the Department of Anaesthesiology and Intensive Therapy, Faculty of Medicine, University of Szeged, Semmelweis u. 6, Szeged, Hungary, EU: Chair of the Institute of Clinical Microbiology, Faculty of Medicine, University of Szeged, Semmelweis u. 6, Szeged, Hungary CsV: Chair of the Department of Microbiology, Faculty of Science and Informatics, University of Szeged, Közép fasor 52, Szeged, Hungary FS:Senior research fellow on the Department of Medical Microbiology and Immunobiology, Faculty of Medicine, University of Szeged, Dóm tér 10, Szeged, Hungary,
Abbreviations
- RT PCR:
-
Real-time PCR
- FRET:
-
Fluorescence resonance energy transfer
- LC:
-
LightCycler
- ICU:
-
Intensive Care Unit
- ITS:
-
Internal transcribed spacer
- ATCC:
-
American Type Cell Culture
- IFM:
-
IFM Quality Services Pty Ltd
- SzMC:
-
Szeged Microbiological Collection
- Tm:
-
Melting temperature
- SD:
-
Standard deviation
- Tm A:
-
Melting peak of the amplicon
- Tm P:
-
Melting peak of the probe.
References
Danai PA, Moss M, Mannino DM, Martin GS: The epidemiology of sepsis in patients with malignancy. Chest. 2006, 129: 1432-1440. 10.1378/chest.129.6.1432.
Martin GS, Mannino DM, Eaton S, Moss M: The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003, 348: 1546-1554. 10.1056/NEJMoa022139.
Klotz SA, Chasin BS, Powell B, Gaur NK, Lipke PN: Polymicrobial bloodstream infections involving Candida species: analysis of patients and review of the literature. Diagn Microbiol Infect Dis. 2007, 59: 401-406. 10.1016/j.diagmicrobio.2007.07.001.
Vallés J, Rello J, Ochagavía A, Garnacho J, Alcalá MA: Community-acquired bloodstream infection in critically ill adult patients: impact of shock and inappropriate antibiotic therapy on survival. Chest. 2003, 123: 1615-1624. 10.1378/chest.123.5.1615.
Kumar A, Ellis P, Arabi Y, Roberts D, Light B, Parrillo JE, Dodek P, Wood G, Kumar A, Simon D, Peters C, Ahsan M, Chateau D, Cooperative Antimicrobial Therapy of Septic Shock Database Researc Group: Initiation of inappropriate antimicrobial therapy results in a fivefold reduction of survival in human septic shock. Chest. 2009, 136: 1237-1248.
Leggieri N, Rida A, François P, Schrenzel J: Molecular diagnosis of bloodstream infections: planning to (physically) reach the bedside. Curr Opin Infect Dis. 2010, 23: 311-319.
Carroll NM, Jaeger EE, Choudhury S, Dunlop AA, Matheson MM, Adamson P, Okhravi N, Lightman S: Detection of and discrimination between Gram-positive and Gram-negative bacteria in intraocular samples by using nested PCR. J Clin Microbiol. 2000, 38: 1753-1757.
Didenko VV: DNA Probes Using Fluorescence Resonance Energy Transfer (FRET): Designs and Applications. Biotechniques. 2001, 31: 1106-1121.
Klaschik S, Lehmann LE, Raadts A, Book M, Hoeft A, Stuber F: Real-time PCR for detection and differentiation of Gram-positive and Gram-negative bacteria. J Clin Microbiol. 2002, 40: 4304-4307. 10.1128/JCM.40.11.4304-4307.2002.
Klaschik S, Lehmann LE, Raadts A, Book M, Gebel J, Hoeft A, Stuber F: Detection and differentiation of in vitro-spiked bacteria by real-time PCR and melting-curve analysis. J Clin Microbiol. 2004, 42: 512-517. 10.1128/JCM.42.2.512-517.2004.
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W, Fungal Barcoding Consortium; Fungal Barcoding Consortium Author List: Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci U S A. 2012, 109: 6241-6246. 10.1073/pnas.1117018109.
Somogyvari F, Serly J, Doczi I, Nagy E: Molecular differentiation of most frequent Candida species causing blood-stream infection. Mycoses. 2005, 2: S198-
Zhou L, Myers AN, Vandersteen JG, Wang L, Wittwer CT: Closed-tube genotyping with unlabeled oligonucleotide probes and a saturating DNA dye. Clin Chem. 2004, 50: 1328-1335. 10.1373/clinchem.2004.034322.
Lind K, Ståhlberg A, Zoric N, Kubista M: Combining sequence-specific probes and DNA binding dyes in real-time PCR for specific nucleic acid quantification and melting curve analysis. Biotechniques. 2006, 40: 315-319. 10.2144/000112101.
Kilic A, Basustaoglu AC: Double triplex real-time PCR assay for simultaneous detection of Staphylococcus aureus, Staphylococcus epidermidis, Staphylococcus hominis, and Staphylococcus haemolyticus and determination of their methicillin resistance directly from positive blood culture bottles. Res Microbiol. 2011, 162: 1060-1066. 10.1016/j.resmic.2011.07.009.
Maheux AF, Bissonnette L, Boissinot M, Bernier JL, Huppé V, Bérubé E, Boudreau DK, Picard FJ, Huletsky A, Bergeron MG: Method for rapid and sensitive detection of Enterococcus sp. and Enterococcus faecalis/faeciumcells in potable water samples. Water Res. 2011, 45: 2342-2354. 10.1016/j.watres.2011.01.019.
Bo SN, Bo J, Ning YZ, Zhao Y, Lu XL, Yang JY, Zhu X, Yao GQ: Relationship between time to positivity of blood culture with clinical characteristics and hospital mortality in patients with Escherichia coli bacteremia. Chin Med J (Engl). 2011, 3: 330-334.
Gutzmer R, Mommert S, Küttler U, Werfel T, Kapp A: Rapid identification and differentiation of fungal DNA in dermatological specimens by LightCylerPCR. J Med Microbiol. 2004, 53: 1207-1214. 10.1099/jmm.0.45779-0.
Somogyvari F, Doczi I, Serly J, Suhail A, Nagy E: Rapid discrimination between Candida albicans and Candida dubliniensis by using real-time PCR. Diagn Microbiol Infect Dis. 2007, 58: 367-369. 10.1016/j.diagmicrobio.2007.01.015.
Somogyvari F, Horvath A, Serly J, Majoros H, Vagvolgyi C, Peto Z: Detection of Invasive Fungal Pathogens by Real-time PCR and High-resolution Melting Analysis. In Vivo. 2012, 26: 979-983.
Ferrer C, Colom F, Frasés S, Mulet E, Abad JL, Alió JL: Detection and identification of fungal pathogens by PCR and by ITS2 and 5.8S ribosomal DNA typing in ocular infections. J Clin Microbiol. 2001, 39: 2873-2879. 10.1128/JCM.39.8.2873-2879.2001.
Turenne C, Sanche SE, Hoban DJ, Karlowsky JA, Kabani AM: Rapid identification of fungi by using the ITS2 genetic region and an automated fluorescent capillary electrophoresis system. J Clin Microbiol. 1999, 37: 1846-1851.
Khan Z, Mustafa AS, Alam FF: Real-time LightCycler polymerase chain reaction and melting temperature analysis for identification of clinically important Candida spp. J Microbiol Immunol Infect. 2009, 42: 190-195.
Lehmann LE, Alvarez J, Hunfeld KP, Goglio A, Kost GJ, Louie RF, Raglio A, Regueiro BJ, Wissing H, Stüber F: Potential clinical utility of polymerase chain reaction in microbiological testing for sepsis. Crit Care Med. 2009, 37: 3085-3090. 10.1097/CCM.0b013e3181b033d7.
Bouza E, Sousa D, Rodríguez-Créixems M, Lechuz JG, Muñoz P: Is the volume of blood cultured still a significant factor in the diagnosis of bloodstream infections?. J Clin Microbiol. 2007, 45: 2765-2769. 10.1128/JCM.00140-07.
Waldeisen JR, Wang T, Mitra D, Lee LP: A real-time PCR antibiogram for drug-resistant sepsis. PLoS One. 2011, 6: e28528-10.1371/journal.pone.0028528.
von Lilienfeld-Toal M, Lehmann LE, Raadts AD, Hahn-Ast C, Orlopp KS: Utility of a commercially available multiplex real-time PCR assay to detect bacterial and fungal pathogens in febrile neutropenia. J Clin Microbiol. 2009, 47: 2405-2410. 10.1128/JCM.00491-09.
Vince A, Lepej SZ, Barsic B, Dusek D, Mitrovic Z, Serventi-Seiwerth R, Labar B: LightCycler SeptiFast assay as a tool for the rapid diagnosis of sepsis in patients during antimicrobial therapy. J Med Microbiol. 2008, 57: 1306-1307. 10.1099/jmm.0.47797-0.
Wallet F, Nseir S, Baumann L, Herwegh S, Sendid B: Preliminary clinical study using a multiplex real-time PCR test for the detection of bacterial and fungal DNA directly in blood. Clin Microbiol Infect. 2010, 16: 774-779. 10.1111/j.1469-0691.2009.02940.x.
Bauer M, Reinhart K: Molecular diagnostics of sepsis - Where are we today?. Int J Med Microbiol. 2010, 300: 411-413. 10.1016/j.ijmm.2010.04.006.
Tissari P, Zumla A, Tarkka E, Mero S, Savolainen L, Vaara M, Aittakorpi A, Laakso S, Lindfors M, Piiparinen H, Maki M, Carder C, Huggett J, Gant V: Accurate and rapid identification of bacterial species from positive blood cultures with a DNA based microarray platform: an observational study. Lancet. 2010, 375: 224-230. 10.1016/S0140-6736(09)61569-5.
Cleven BEE, Palka-Santini M, Gielen J, Meembor S, Krönke M, Krut O: Identification and characterization of bacterial pathogens causing bloodstream infections by DNA microarray. J Clin Microbiol. 2006, 44: 2389-2397. 10.1128/JCM.02291-05.
Lucignano B, Ranno S, Liesenfeld O, Pizzorno B, Putignani L, Bernaschi P, Menichella D: Multiplex PCR allows rapid and accurate diagnosis of bloodstream infections in newborns and children with suspected sepsis. J Clin Microbiol. 2011, 49: 2252-2258. 10.1128/JCM.02460-10.
Lim CS, Tung CH, Rosli R, Chong PP: An alternative Candida spp. cell wall disruption method using a basic sorbitol lysis buffer and glass beads. J Microbiol Methods. 2008, 75: 576-578. 10.1016/j.mimet.2008.07.026.
Miller SA, Dykes DD, Polesky HF: A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16: 1215-1218. 10.1093/nar/16.3.1215.
Liu D, Coloe S, Baird R, Pederson J: Rapid mini-preparation of fungal DNA for PCR. J Clin Microbiol. 2000, 38: 471-
Lott TJ, Kuykendall RJ, Reiss E: Nucleotide sequence analysis of the 5.8S rDNA and adjacent ITS2 region of Candida albicans and related species. Yeast. 1993, 9: 1199-1206. 10.1002/yea.320091106.
Acknowledgements
This research was supported by the European Union and the State of Hungary, co-financed by the European Social Fund in the framework of TÁMOP 4.2.4. A/2-11-1-2012-0001 ‘National Excellence Program’.
This publication is supported financially by the project named „TÁMOP-4.2.2. A-11/1/ KONV-2012-0035 – Creating the Center of Excellence at the University of Szeged” is supported by the European Union and co-financed by the European Regional Fund. The authors would like to acknowledge Dr. Gabriella Spengler and Prof. Yvette Mándi whose suggestions helped to improve the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ÁH: helped in the design, performed the experiments, analysed the data and wrote the manuscript. ZP: provided the clinical samples, helped in the analysis and interpretation of the data and revised the manuscript. EU: provided all the clinical bacterial samples and critiqued the manuscript. CsV: have made substantial contributions to concept and design, provided the fungal samples and revised the manuscript. FS: designed all the experiments, participated in the writing of the manuscript, revised the manuscript and gave final approval of the version to be published. All the authors have read and approved the final manuscript.
Zoltán Pető, Edit Urbán, Csaba Vágvölgyi and Ferenc Somogyvári contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Horváth, Á., Pető, Z., Urbán, E. et al. A novel, multiplex, real-time PCR–based approach for the detection of the commonly occurring pathogenic fungi and bacteria. BMC Microbiol 13, 300 (2013). https://doi.org/10.1186/1471-2180-13-300
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-13-300