
Abstract
Background
Bacterial kidney disease (BKD), caused by Renibacterium salmoninarum, is a bacterial disease of fish, which is both geographically widespread and difficult to control. Previously, application of various molecular typing methods has failed to reliably discriminate between R. salmoninarum isolates originating from different host species and geographic areas. The current study aimed to utilize multilocus variable number tandem repeats (VNTR) to investigate inter-strain variation of R. salmoninarum to establish whether host-specific populations exist in Atlantic salmon and rainbow trout respectively. Such information would be valuable in risk assessment of transmission of R. salmoninarum in a multispecies aquaculture environment.
Results
The present analysis utilizing sixteen VNTRs distinguished 17 different haplotypes amongst 41 R. salmoninarum isolates originating from Atlantic salmon and rainbow trout in Scotland, Norway and the US. The VNTR typing system revealed two well supported groups of R. salmoninarum haplotypes. The first group included R. salmoninarum isolates originating from both Atlantic salmon and rainbow trout circulating in Scottish and Norwegian aquaculture, in addition to the type strain ATCC33209T originating from Chinook salmon in North America. The second group comprised isolates found exclusively in Atlantic salmon, of mainly wild origin, including isolates NCIB1114 and NCIB1116 associated with the original Dee disease in Scotland.
Conclusions
The present study confirmed that VNTR analysis can be successfully applied to discriminate R. salmoninarum strains. There was no clear distinction between isolates originating from Atlantic salmon and rainbow trout as several haplotypes in group 1 clustered together R. salmoninarum isolates from both species. These findings indicate a potential exchange of pathogens between Atlantic salmon and rainbow trout in Scottish and Norwegian aquaculture during the last 20 years. In a scenario of expansion of rainbow trout farming into the marine environment, appropriate biosecurity measures to minimize disease occurrence are advised. The present results also suggest that R. salmoninarum isolates circulating in European aquaculture over the last 20 years are genetically distant to the wild strains originally causing BKD in the rivers Dee and Spey.
Similar content being viewed by others


Background
Renibacterium salmoninarum[1] is a Gram-positive bacterium, belonging to the Micrococcus-Arthrobacter subgroup of the actinomycetes [2–4] and the causative agent of bacterial kidney disease (BKD), a chronic systemic disease of salmonid fish in both marine and freshwater environments [5]. Bacterial kidney disease was first reported in wild Atlantic salmon (Salmo salar) in the Rivers Dee and Spey (Scotland, United Kingdom) in 1930 [6, 7] and similar disease signs were reported from North America in 1935 in brook trout (Salvelinus fontinalis), brown trout (Salmo trutta) and rainbow trout (Oncorhynchus mykiss) [8, 9]. Renibacterium salmoninarum has an intracellular lifecycle and transmission, both horizontally through contact with infected fish/water or vertically inside fish ova, has been confirmed in many salmonid species [10–14]. Recent epidemiological studies have identified an association between the spread of BKD and anthropogenic activities [15, 16].
Bacterial kidney disease is geographically widespread and has been reported from most countries where salmonid fish are cultured or naturally occurring. The disease is known to have the potential to cause high mortalities [17, 18] and represents one of the most difficult bacterial diseases of fish to control due to its slow progression and lack of effective treatment. In Scotland, farmed Atlantic salmon and rainbow trout may be infected in both seawater and freshwater environments [19], although the contribution of wild fish to infection transmission is considered low [16].
Sensitive R. salmoninarum typing tools are required to improve BKD control through identification of sources of infection and transmission routes. Molecular typing methods, such as direct sequencing of the intergenic spacer (ITS1 and ITS) of ribosomal RNA and randomly amplified polymorphic DNA (RAPD) analysis [20–23], have shown a limited power to distinguish R. salmoninarum strains. Amplification of length polymorphisms in the tRNA intergenic spacer (tDNA-ILPs) has, however, offered improved discriminatory power with some potential for identification of R. salmoninarum isolates known to come from the same hatchery [23].
In Scotland, BKD and infection with R. salmoninarum are regulated under The Aquatic Animal Health (Scotland) Regulation 2009. From the available farm data, it appears that BKD persists longer on rainbow trout farms [24], compared with Atlantic salmon farms [16, 19]. To date, all typing systems have failed to distinguish between R. salmoninarum strains originating from Atlantic salmon and rainbow trout [20, 22, 23], suggesting that individual isolates may represent a risk to both host species. Confirmation of this, applying a more sensitive typing tool, would be beneficial, for example, in a scenario of an expansion of rainbow trout sea water aquaculture. Application of appropriate biosecurity measures could then be applied to minimise risk of pathogen transmission.
In recent years, multilocus variable number tandem repeat analysis, based on amplification of short repetitive DNA sequences, has been found to be a rapid and simple typing technique that enables differentiation of bacterial strains displaying otherwise low genomic variation. The method has been used to discriminate between closely related strains of various human pathogenic microorganisms such as Clostridium difficile[25], Bartonella henselae[26], or Streptococcus agalactiae[27] as well as fish pathogenic species such as Francisella noatunensis[28]. The primary purpose of this study was therefore to investigate the genetic variation in R. salmoninarum isolated from Atlantic salmon and rainbow trout farms in Scotland using multilocus variable number tandem repeat analysis (VNTR). Additional samples from other countries were also included in the present study to put any observed variation into context and identify whether the present VNTR typing scheme can distinguish between R. salmoninarum collected from different geographic areas.
Results
Characterization of tandem repeat loci
In total, 32 tandem repeat loci were identified using either the Microorganisms Tandem Repeat Database or Tandem Repeats Finder (Additional file 1: Table S1). All loci were successfully amplified in 41 R. salmoninarum isolates (Additional file 2: Table S2) and sequences were analyzed for polymorphism (differences in number of tandem repeat units) (Accession numbers KF903677-KF904322). Sixteen of 32 studied loci were polymorphic (Table 1). The 16 monomorphic loci were excluded from the VNTR genotyping scheme.
The allelic diversity ranged from two (BKD 92, 396, 494, 526, 1023, 1850 and 3038) to eight different alleles (BKD 2126) per locus. The largest observed variation in allele size was found in locus BKD2126 which varied between five to eleven repeats (Table 1). The VNTR typing system has a discriminatory power value of 0.81 and seventeen different haplotypes of R. salmoninarum were distinguished using 16 combined polymorphic VNTRs (Table 1, Table 2). A VNTR typing system relying on only six combined loci (BKD23, BKD305, BKD694, BKD1506, BKD1935, BKD2126) also sufficiently recognized 17 R. salmoninarum haplotypes, with the same discriminatory power value of 0.81.
Phylogenetic relationships among R. salmoninarumisolates inferred from VNTRs
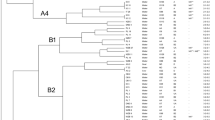
The phylogenetic relationships among the R. salmoninarum strains inferred from 16 polymorphic VNTRs are illustrated in Figure 1. Two distinct groups comprising haplotypes A-L (group 1) and M-Q (group 2) were supported with a high bootstrap value (92%). Group 1 comprised R. salmoninarum from both Atlantic salmon and rainbow trout farmed in Scotland and Norway, recovered over a period of more than 40 years. This group also includes the type strain of R. salmoninarum ATCC33209T, recovered from a Chinook salmon (Oncorhynchus tshawytscha) in Oregon (USA) in the early 1970s. Strain B represented the most common haplotype, comprising 18 R. salmoninarum isolates from Atlantic salmon and rainbow trout farmed in Scotland and Norway over a period of more than 20 years. Strain B was one of five closely related strains (A, B, C, D, E) differing from each other at a single locus.

Relationship among the observed haplotypes described in Table2. Group 1 includes haplotypes A to L and Group 2 includes haplotypes M to Q. Bootstrap values indicate the level of support for clusters if higher than 50%.
Group 2 represents R. salmoninarum isolates obtained uniquely from Atlantic salmon originating from Scotland and Norway. These isolates differed from group 1 at loci BKD396 and BKD1935. A moderately supported cluster within group 2, comprising strains O-Q, represented isolates exclusively from wild Atlantic salmon, including the Dee disease isolates NCIMB 1114 and 1116 associated with first occurrence of BKD in Scotland.
Similar clustering of R. salmoninarum isolates into two main groups was achieved using the eBURST algorithm based on either 16 or 6 polymorphic loci (Figure 2A,B). Using 16 polymorphic loci, a large radial cluster of 7 closely related haplotypes (A-G) was defined. Haplotype B was assigned as the most parsimonious “founder” of this group. Group 2 haplotypes occurred as a single pair O/P representing the Dee disease isolates and three singletons (L,M,N). Using eBURST, a loss in resolving power of the genotyping system was observed when the number of polymorphic loci included was reduced to 6 (Figure 2B).

eBURST diagram of R. salmoninarum population derived from the allelic variation in (A) 16 polymorphic loci or (B) 6 polymorphic loci.
The present VNTR typing scheme was also applied to investigate whether Scottish R. salmoninarum isolates can be distinguished from isolates originating from Norway. Within group 1, some association with country of origin was observed for haplotypes A, C, and G, uniquely obtained from Scottish aquaculture, while haplotype E represented R. salmoninarum from Norway. On the contrary, the most common haplotype B contained isolates obtained from aquaculture establishments in both countries.
Discussion
The present study describes development and application of a VNTR typing system for R. salmoninarum, a bacterium affecting salmonid aquaculture worldwide and discusses the potential implications for disease management. In comparison to other genotyping methods used to study R. salmoninarum such as RAPD, tDNA-ILPs [20–23], multilocus VNTR typing offers a considerable improvement. Using a combination of sixteen VNTRs, 17 different haplotypes can be identified among 41 R. salmoninarum isolates. The discriminatory power of the present combined VNTR scheme was high, characterized by HGDI index of 0.81, indicating that two unrelated isolates will on 81% of occasions fall into different haplotypes. In contrast, no more than nine different tDNA-ILP profiles could be identified from a much larger collection of R. salmoninarum isolates [23]. In addition, VNTR represents a more reproducible typing system in comparison to techniques relying on random amplification under low-stringency parameters and accurate data from individual isolates can readily be shared between different laboratories. Although the discriminatory power of VNTR when applied to R. salmoninarum is lower than has been achieved with some human pathogenic bacteria such as Bartonella or Streptococcus[26, 27], these later studies are based on significantly larger data sets usually gathered from wider geographic areas. If a larger R. salmoninarum data set becomes available in future, the VNTRs described in the present study should be applied to test its ability to trace disease outbreaks and connect individual infected farms with a source of infection.
The developed VNTR typing system separated the studied isolates into two well-supported groups. Group 1 clustered together 12 out of 17 R. salmoninarum haplotypes, including a wide range of isolates from Scotland, Norway and North America, from three different species of salmonid fish, spanning the period between 1974 and 2009. Several haplotypes of group 1 (B, D, E and G) comprised multiple isolates causing disease in both Atlantic salmon and rainbow trout, suggesting a relatively common historical transfer of the pathogen between these fish species. On the other hand, some association was found between rainbow trout and R. salmoninarum haplotype A and between Atlantic salmon and R. salmoninarum haplotypes C, F, H, I and L-Q. However, with the exception of haplotypes A and C, these haplotypes were represented by single isolations.
The present study concludes that using a data set of 41 isolates representing bacterium circulating in Scotland over a period of more than 20 years, there was no consistent division of R. salmoninarum isolates into two host specific populations. This result is consistent with the possibility that individual R. salmoninarum strains can infect both host species in environments where both species co-occur. The transfer of R. salmoninarum free stock to the marine environment could in theory eliminate disease transmission. However, the possibility that a carrier would be not detected, as a consequence of a potentially low infection prevalence and low diagnostic sensitivity of tests for asymptomatic stock, have to be considered [29]. The spatial separation of marine rainbow trout and Atlantic salmon farms into separate disease management areas in marine environment, as described in [16], can further reduce the risk of pathogen transfer between host species.
All previous R. salmoninarum typing systems have failed to reliably discriminate between European and US isolates [20, 22, 23]. This study identified the type strain ATCC33209T originating from Oregon (US) as a unique strain (haplotype J) and its position within group 1 supports previous studies suggesting intercontinental spread of this pathogen related to anthropogenic activities such as movement of fish during expansion of the rainbow trout industry in Europe [30]. The present genotyping system could, however, clearly separate R. salmoninarum strains M-Q (group 2), indicating an origin not associated with ATCC33209T. The majority of these strains were of wild origin and have not been reported in wild or farmed fish since their original description in the 1930s. The locus BKD1935 was previously described by [22] referred to as the Exact Tandem Repeat A (ETR-A). It was demonstrated that the ETR-A can successfully separate the wild-fish isolates such as NCIMB1114 and NCIMB1116 (tandem repeat 1) from the farmed isolates such as MT452 and MT1363 (tandem repeat 2). Further investigation on a larger data set, focusing on loci BKD396 and BKD1935, which are solely responsible for differentiation between groups 1 and 2, might bring more insight into a relationship between farmed and wild R. salmoninarum strains and confirm the origin of R. salmoninarum in Scottish aquaculture.
Conclusions
Cross-species infectivity of R. salmoninarum strains also has wider implications for marine ecosystems; including possible transfer of R. salmoninarum from farmed to wild fish or vice versa. In Scotland, recent studies provided evidence of a relatively low prevalence of R. salmoninarum in wild fish captured in close proximity to farms, suggesting that the transmission of this pathogen between wild and farmed fish is limited [16, 31]. However, this scenario might not apply for other regions or countries such as England or Norway [32] and the described VNTR typing system can be utilized to identify and understand farmed and wild fish interactions in terms of R. salmoninarum transmission if a larger data set should become available.
Methods
Preparation of Renibacterium isolates and DNA extraction
Twenty-five R. salmoninarum isolates from confirmed disease outbreaks on Scottish farms were selected for this study. Number and selection of Scottish R. salmoninarum isolates represents the geographic range, habitat, frequency of disease outbreaks in the salmonid aquaculture sector, supply of fish stock and takes into account difficulties of bacteria culturing from asymptomatic fish and resuscitation of archived material. In addition, 14 Norwegian isolates and two isolates derived from the first successful cultivation of R. salmoninarum from the River Dee [7] were included. Isolate details including country of origin, date of isolation, host species and environment are summarized in Additional file 2: Table S2.
For Scottish strains, lyophilised cultures were resuscitated onto Mueller-Hinton L-cysteine agar (MHCA) containing polymyxin-B-sulphate, D-cycloserine, oxolinic acid and cycloheximide and incubated at 15°C for several weeks to allow growth. Suspensions of culture in 0.85% sterile saline were made to a turbidity of McFarland 1, serially diluted to 10-4 in sterile saline and 0.1 ml volume of the dilutions spread onto MHCA to achieve single colony purification. The Gram appearance and purity of individual colonies was confirmed before sub-culturing onto fresh MHCA and additional checks for purity and identity (API ZYM, ELISA using the Bios Chile kit) were carried out. At 4–6 weeks, each agar culture was scraped from the plate and suspended in sterile saline before pelleting at 2,400 × g. Genomic DNA was extracted from bacterial cultures using the MagAttract DNA mini M48 kit (Qiagen) and quantified using a ND-1000 Nanodrop Spectrophotometer (NanoDrop Technologies). For Norwegian strains, cryo-preserved isolates (−80°C) were resuscitated on kidney disease (KD) medium [33] followed by KD broth culture to an approximate turbidity of McFarland 1 prior to extraction of genomic DNA using the Gentra Puregene cell kit (Qiagen).
Tandem repeat identification and amplification
The complete genome sequence of R. salmoninarum reference strain ATCC33209T[4] (Accession number NC_010168) was utilized to identify the repetitive DNA sequence regions using the Microorganisms Tandem Repeat Database (http://minisatellites.u-psud.fr) [34] and Tandem Repeats Finder (TRF version 4.03) (http://tandem.bu.edu) [35]. Tandem repeats with at least two repeat units per locus and a repeat unit length of between 4 and 80 bp were selected for further analysis.
Primers for amplification of each locus were designed using OligoPerfect™ Designer (http://tool.invitrogen.com) and their specificity tested using BLAST (blastn) searches. Loci were amplified using the primer pairs listed in Additional file 1: Table S1. Each reaction consisted of 1 × PCR buffer (Bioline), 1.5 mM MgCl2, 200 μM dNTPs, 10 μM of each primer, 1 U BioTaq (Bioline) in a final volume of 20 μl. The cycling conditions were 35 cycles of: 95°C for 1 min, 50 or 55°C (see Additional file 1: Table S1) for 1 min, 72°C for 1 min, followed by a final elongation step of 72°C for 5 min. Amplified products were visualized on a 1% ethidium bromide-stained agarose gel (Invitrogen) and purified using ExoSAP IT or ExoStar 1-Step (GE Healthcare). Approximately 15 ng of purified PCR product was sequenced, utilising the same primers as in the amplification reaction using the GenomeLab DTCS Quick Start kit (Beckman Coulter) and the automated CEQ8800 DNA Sequencer (Beckman Coulter).
Tandem repeat analysis
Each type (size) of repeat, identified by sequencing, at each locus was assigned a unique allele identifier. Data were imported from a Microsoft Office Excel 2003 generated comma-separated-value data file and analysed using version 2.14.0 of the R statistical computing environment [36]. The permutations of alleles across 16 polymorphic loci were used to define distinct haplotypes.
Gross differences between haplotypes (identified using 16 polymorphic loci) were measured with the Hamming distance [37] and used to construct an unrooted neighbor-joining tree using version 1.6-0 of the supplementary R package phangorn [38]. To simplify interpretation of results, haplotypes were named A-Q on the basis of their respective position in the phylogenetic tree. Support for clusters was evaluated using the bootstrap test of phylogeny (1000 repeats) and clusters with values of less than 50% collapsed [39]. The clustering of very closely related haplotypes, defined as those differing at only one locus, was examined using eBURST v 3.0 [40]. Homoplasy and extent of recombination events were investigated using Splits Decomposition, as implemented in Splitstree v 4 [41], by depicting conflicting signals in the data caused by recombination events. The resulting network was consistent with the phylogenetic analysis, and no reticulation was evident, indicating that the evolutionary relationships have not been affected by recombination or homoplasy (data not shown).
The discriminatory power of a typing system was estimated using the Hunter-Gaston discriminatory index HGDI [42]. The index provides a probability that two randomly sampled unrelated isolates will be placed into different typing groups/haplotypes. The minimum number of loci required to distinguish all the strains was determined.
References
Sanders JE, Fryer JL: Renibacterium salmoninarum gen. nov., sp. nov., the causative agent of bacterial kidney disease in salmonid fishes. Int Syst Bacteriol. 1980, 30: 496-502. 10.1099/00207713-30-2-496.
Gutenberger SK, Giovannoni SJ, Field KG, Fryer JL, Rohovec JS: A phylogenetic comparison of the 16S rRNA sequence of the fish pathogen, Renibacterium salmoninarum, to gram-positive bacteria. FEMS Microbiol Lett. 1991, 77: 151-156. 10.1111/j.1574-6968.1991.tb04339.x.
Koch CF, Rainey FA, Stackebrandt E: 16S rDNA studies on members of Arthrobacter and Micrococus: and aid for their future taxonomic restructuring. FEMS Microbiol Lett. 1994, 123: 167-172. 10.1111/j.1574-6968.1994.tb07217.x.
Wiens GD, Rockey DD, Wu Z, Chang J, Levy R, Crane S, Chen DS, Capri GR, Burnett JR, Sudheesh PS, Shipma MJ, Burd H, Bhattacharyya A, Rhodes LD, Kaul R, Strom MS: Genome sequence of the fish pathogen Renibacterium salmoninarum suggests reductive evolution away from an environmental Arthrobacter ancestor. J Bacteriol. 2008, 190: 6970-6982. 10.1128/JB.00721-08.
Evelyn TPT: Bacterial kidney disease – BKD. Bacterial Diseases of Fish. Edited by: Inglis V, Roberts RJ, Bromage NR. 1993, Oxford, United Kingdom: Blackwell Scientific Publications, 177-195.
Mackie TJ, Arkwright JA, Pryce-Tannatt TE, Mottram JC, Douglas Johnston WD, Menzies WJM, Martin W: Second Interim Report of the Furunculosis Committee. 1933, London: Ministry of Agriculture and Fisheries
Smith IW: Freshwater and Salmon Fisheries Research. The Occurrence and Pathology of Dee Disease. 1964, Edinburgh: Her Majesty’s Stationery Office, 34, 34
Belding DL, Merrill B: A preliminary report upon a hatchery disease of the Salmonidae. Trans Am Fish Soc. 1935, 65: 76-84. 10.1577/1548-8659(1935)65[76:APRUAH]2.0.CO;2.
Fryer JL: Bacterial kidney disease of salmonid fish. Ann Rev Microbiol. 1981, 35: 273-298. 10.1146/annurev.mi.35.100181.001421.
Mitchum DL, Sherman LE: Transmission of bacterial kidney disease from wild to stocked hatchery trout. Can J Fish Aquat Sci. 1981, 38: 547-551. 10.1139/f81-077.
Bruno D, Munro ALS: Observation on Renibacterium salmoninarum and the salmonid egg. Dis Aquat Org. 1986, 1: 83-87.
Evelyn TPT, Prosperi-Porta L, Ketcheson JE: Experimental intra-ovum infection of salmonid eggs with Renibacterium salmoninarum and vertical transmission of the pathogen with such eggs despite their treatment with erythromycin. Dis Aquat Org. 1986, 1: 197-202.
Balfry SK, Albright LJ, Evelyn TPT: Horizontal transfer of Renibacterium salmoninarum among farmed salmonids via the faecal-oral route. Dis Aquat Org. 1996, 25: 63-69.
McKibben CL, Pascho RJ: Shedding of Renibacterium salmoninarum by infected chinook salmon Oncorhynchus tschawytscha. Dis Aquat Org. 1999, 38: 75-79.
Murray AG, Munro LA, Wallace IS, Peeler EJ, Thrush MA: Bacterial kidney disease: an assessment of risk to Atlantic salmon from infection in trout farms and other sources. Scottish Marine Freshwater Sci. 2011, 2 (3): 1-80.
Murray AG, Munro LA, Wallace IS, Allan CET, Peeler EJ, Thrush MA: Epidemiology of Renibacterium salmoninarum in Scotland and the potential for compartmentalised management of salmon and trout farming areas. Aquaculture. 2012, 324–325: 1-13.
Murray CB, Evelyn TPT, Beacham TD, Barner LW, Ketcheson JE, Prosperi-Porta L: Experimental induction of bacterial kidney disease in Chinook salmon by immersion and cohabitation challenges. Dis Aquat Org. 1992, 12: 91-96.
Starliper CE, Smith DR, Shatzer T: Virulence of Renibacterium salmoninarum to salmonids. J Aquat Anim Health. 1997, 9: 1-7. 10.1577/1548-8667(1997)009<0001:VORSTS>2.3.CO;2.
Bruno D: Prevalence and diagnosis of bacterial kidney disease (BKD) in Scotland between 1990 and 2002. Dis Aquat Org. 2004, 59: 125-130.
Grayson TH, Cooper LF, Atienzar FA, Knowles MR, Gilpin ML: Molecular differentiation of Renibacterium salmoninarum isolates from worldwide locations. Appl Environ Microbiol. 1999, 65: 961-968.
Grayson TH, Alexander SM, Cooper LF, Gilpin ML: Renibacterium salmoninarum isolates from different sources possess two highly conserved copies of the rRNA operon. A van Leeuw. 2000, 78: 51-61. 10.1023/A:1002745129625.
Grayson TH, Atienzar FA, Alexander SM, Cooper LF, Gilpin ML: Molecular diversity of Renibacterium salmoninarum isolates determined by randomly amplified polymorphic DNA analysis. Appl Environ Microbiol. 2000, 66: 435-438. 10.1128/AEM.66.1.435-438.2000.
Alexander SM, Grayson TH, Chambers EM, Cooper LF, Barker GA, Gilpin ML: Variation in the spacer regions separating rTNA genes in Renibacterium salmoninarum distinguishes recent clinical isolates from the same location. J Clin Microbiol. 2001, 39: 119-128. 10.1128/JCM.39.1.119-128.2001.
Murray AG, Hall M, Munro LA, Wallace IS: Modelling management strategies for a disease including undetected sub-clinical infection: Bacterial kidney disease in Scottish salmon and trout farms. Epidemics. 2011, 3: 171-182. 10.1016/j.epidem.2011.10.002.
Wei HL, Kao CW, Wei SH, Tzen JTC, Chiou CS: Comparison of PCR ribotyping and multilocus variable-number tandem-repeat analysis (MLVA) for improved detection of Clostridium difficile. BMC Microbiol. 2011, 11: 217-10.1186/1471-2180-11-217.
Monteil M, Durand B, Bouchouicha R, Petit E, Chomel B, Arvand M, Boulouis H-J, Haddad N: Development of discriminatory multiple-locus variable number tandem repeat analysis for Bartonella henselae. Microbiol. 2007, 153: 1141-1148. 10.1099/mic.0.2006/001164-0.
Haguenoer E, Baty G, Pourcel C, Lartigue M-F, Domelier A-S, Rosenau A, Quentin R, Mereghetti L, Lanotte P: A multi locus variable number of tandem repeat analysis (MLVA) scheme for Streptococcus agalactiae genotyping. BMC Microbiol. 2011, 11: 171-10.1186/1471-2180-11-171.
Brevik ØJ, Ottem KF, Nylund A: Multiple-locus, variable number of tandem repeat analysis (MLVA) of the fish-pathogen Francisella noatunensis. BMC Vet Res. 2011, 7: 5-10.1186/1746-6148-7-5.
Hall LM, Wallace IS, Munro LA, Walker A, Murray AG: Epidemiology informs policy regarding surveillance of a notifiable disease of salmonids. Epidemiol et Santé Anim. 2011, 59–60: 392-394.
Munro ALS, Waddell IF: Growth of salmon and trout farming in Scotland. Development in Fisheries Research in Scotland. Edited by: Bailey RS, Parrish BB. 1987, England: Fishing News Books Ltd, 246-263.
Wallace IS, Munro LA, Kilburn R, Hall M, Black J, Raynard RS, Murray AG: A report on the effectiveness of cage and farm-level fallowing of the control of bacterial kidney disease and sleeping disease on large cage-based trout farms in Scotland. http://www.scotland.gov.uk/Resource/Doc/356407/0120447.pdf,
Chambers E, Gardiner R, Peeler EJ: An investigation into the prevalence of Renibacterium salmoninarum in farmed rainbow trout, Oncorhynchus mykiss (Walbaum), and wild fish populations in selected river catchments in England and Wales between 1998 and 2000. J Fish Dis. 2008, 31: 89-96. 10.1111/j.1365-2761.2007.00868.x.
Ordal EJ, Earp BJ: Cultivation and transmission of etiological agent of kidney disease in salmonid fishes. Proc Soc Eptl Biol Med. 1956, 92: 85-88. 10.3181/00379727-92-22392.
Denoeud F, Vergnaud G: Identification of polymorphic tandem repeats by direct comparison of genome sequence from different bacterial strains: a web-based resource. BMC Bioinforma. 2004, 5: 4-10.1186/1471-2105-5-4.
Benson G: Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acid Res. 1999, 27: 573-580. 10.1093/nar/27.2.573.
Development Core Team R: R: Language and Environment for Statistical Computing. 2011, Vienna: R Foundation for Statistical Computing
Hamming RW: Error detecting and error correcting codes. Bell Syst Technic J. 1950, 29: 147-160. 10.1002/j.1538-7305.1950.tb00463.x.
Schliep KP: Phangorn: phylogenetic anlaysis in R. Bioinformatics. 2011, 27: 592-593. 10.1093/bioinformatics/btq706.
Felsenstein J: Confidence limit on phylogenies: an approach using bootstrap. Evolution. 1985, 39: 783-791. 10.2307/2408678.
Feil EJ, Bao CL, Aanensen DM, Hanage WP, Spratt BG: eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004, 186: 1518-1530. 10.1128/JB.186.5.1518-1530.2004.
Huson DH, Bryant D: Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006, 23: 254-267.
Hunter PR, Gaston MA: Numerical index of the discriminatory ability of typing systems: an application of Simpsons’s index of diversity. J Clin Microbiol. 1988, 26: 2465-2466.
Acknowledgements
The authors would like to thank Drs. Sandy G. Murray and David Bruno (Marine Scotland Science, Aberdeen, United Kingdom) for valuable comments which greatly improved the manuscript draft.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors contributed to the study design. IM, NB, DM, and SJW contributed to molecular studies. UM and DJC prepared bacterial cultures. IM, EJF and MH analysed the molecular data. IM wrote the manuscript and BN, DJC, EJF, UM, DJV and MH revised the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12866_2013_2156_MOESM1_ESM.doc
Addition al file 1: Table S1: List of amplified and analysed tandem repeat loci within the R. salmoninarum genome. (DOC 56 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Matejusova, I., Bain, N., Colquhoun, D.J. et al. Multilocus variable-number tandem-repeat genotyping of Renibacterium salmoninarum, a bacterium causing bacterial kidney disease in salmonid fish. BMC Microbiol 13, 285 (2013). https://doi.org/10.1186/1471-2180-13-285
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-13-285