
Abstract
Background
Macaca fascicularis (cynomolgus or longtail macaques) is the most commonly used non-human primate in biomedical research. Little is known about the genomic variation in cynomolgus macaques or how the sequence variants compare to those of the well-studied related species, Macaca mulatta (rhesus macaque). Previously we identified single nucleotide polymorphisms (SNPs) in portions of 94 rhesus macaque genes and reported that Indian and Chinese rhesus had largely different SNPs. Here we identify SNPs from some of the same genomic regions of cynomolgus macaques (from Indochina, Indonesia, Mauritius and the Philippines) and compare them to the SNPs found in rhesus.
Results
We sequenced a portion of 10 genes in 20 cynomolgus macaques. We identified 69 SNPs in these regions, compared with 71 SNPs found in the same genomic regions of 20 Indian and Chinese rhesus macaques. Thirty six (52%) of the M. fascicularis SNPs were overlapping in both species. The majority (70%) of the SNPs found in both Chinese and Indian rhesus macaque populations were also present in M. fascicularis. Of the SNPs previously found in a single rhesus population, 38% (Indian) and 44% (Chinese) were also identified in cynomolgus macaques. In an alternative approach, we genotyped 100 cynomolgus DNAs using a rhesus macaque SNP array representing 53 genes and found that 51% (29/57) of the rhesus SNPs were present in M. fascicularis. Comparisons of SNP profiles from cynomolgus macaques imported from breeding centers in China (where M. fascicularis are not native) showed they were similar to those from Indochina.
Conclusion
This study demonstrates a surprisingly high conservation of SNPs between M. fascicularis and M. mulatta, suggesting that the relationship of these two species is closer than that suggested by morphological and mitochondrial DNA analysis alone. These findings indicate that SNP discovery efforts in either species will generate useful resources for both macaque species. Identification of SNPs that are unique to regional populations of cynomolgus macaques indicates that location-specific SNPs could be used to distinguish monkeys of uncertain origin. As an example, cynomolgus macaques obtained from 2 different breeding centers in China were shown to have Indochinese ancestry.
Similar content being viewed by others


Background
The cynomolgus macaque (M. fascicularis) is used widely in biomedical research, advancing the study of simian immunodeficiency virus (SIV) pathogenesis [1–3], transplantation biology [4, 5], diabetes [6], and alcohol research [7], among others. Currently, more cynomolgus macaques are imported for use in biomedical research in the United States than are any other non-human primate species. While recent efforts have established many genetic tools for the study of the rhesus macaque, including the complete genome sequence [8], a microsatellite mapping set [9], and a collection of SNPs [10, 11], very few genetic resources are available for cynomolgus macaque genetic research [12]. Specifically, the M. fascicularis genome sequence, gene expression arrays and a SNP map are not yet available to advance complex trait analysis in cynomolgus macaques.
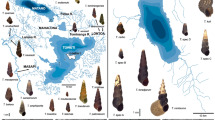
Based upon morphological and zoogeographic studies, the cynomolgus macaque and the rhesus macaque are posited to be descendants from a common ancestor inhabiting Indonesia approximately 1.8 million years ago (MYA) [13]. The cynomolgus macaque range currently extends from Indonesia northward through the Philippines and Mainland Southeast Asia, including Myanmar, Thailand and the Indochinese region of Cambodia, Vietnam, Laos (Figure 1) [14, 15]. The divergence of the continental and Indonesian animals is estimated to have occurred between 0.4–1.2 million years ago, though periodic hypothermals over the last 250,000 years created land bridges that could have permitted migration between regions [16, 17]. An isolated population of cynomolgus macaques inhabits the island of Mauritius. This population is widely considered to have descended from a small number of animals that arrived by trading ships 400–500 years ago [18, 19]. This conclusion is supported by analysis of the major histocompatibility complex (MHC) allele distribution in the Mauritian animals, which carry a very limited number of alleles and haplotypes relative to other macaque populations [3, 20, 21]. Mitochondrial DNA (mtDNA) analysis has shown the Mauritian monkeys to be similar to those of Indonesia and the Philippines [22], and together with Y chromosome sequences, suggest they are specifically derived from Sumatra [23]. Finally, M. fascicularis obtained from captive breeding centers in China are commonly used in research throughout the world. However, cynomolgus macaques are not native to China, and thus are descendants of animals imported from elsewhere.

Geographic ranges of rhesus and cynomolgus macaques. Geographic locations of rhesus (M. mulatta) and cynomolgus (M. fascicularis) macaques included in this study are shown in capital letters. Colored shading indicates the approximate distributions for the two species (adapted from Fooden, 1980; and see Fooden, 2000).
The current range of the rhesus macaque extends from Eastern China through Western India and Pakistan. Within this territory, Indian and Chinese rhesus sub-populations have diverged into genetically distinct subpopulations, as assessed by both mtDNA analysis [24] and MHC allele distributions [25–27]. Recent genomic studies have shown that the vast majority (69%) of SNPs identified in Indian and Chinese rhesus macaque are private to one of the two subpopulations, and that the Chinese population has nearly twice as many SNPs as the Indian rhesus population [10, 11]. The dramatic difference in genetic heterogeneity between the Chinese and Indian rhesus macaques has been attributed to both a large expansion of the Chinese population and a contraction of the Indian rhesus population [11].
M. fascicularis and M. mulatta co-exist in a limited geographic range in Mainland SE Asia. The precise evolutionary history of the macaques in the overlapping range is uncertain. For example, mtDNA sequence comparisons of both species cluster as discrete haplogroups [22, 28], consistent with complete lineage sorting and a lack of interspecies hybridization. However the limited genomic sequence comparisons to date, including portions of 4 autosomal loci and 2 Y chromosomal loci, suggest a closer relationship of the species than that predicted by mtDNA analysis [29–31], and suggests possible contemporary gene-flow between the macaques in Indochina [31]. In addition, analysis of the MHC class II region has revealed a high level of allele sharing between rhesus and cynomolgus macaques, though no evidence of identical MHC class II haplotypes [26].
We recently reported the discovery and analysis of Chinese and Indian rhesus macaques SNPs in portions of 94 genes [10]. Here we identified the composition and distribution of SNPs from cynomolgus macaques in 10 of those same gene regions. In addition, a SNP array of rhesus polymorphisms in 53 genes was used to genotype cynomolgus macaque DNAs from 5 different geographic regions (Indochina, Indonesia, Philippines, Mauritius and breeding centers in China). We found 51% of the polymorphisms were present in both cynomolgus and rhesus macaques. These findings suggests that the SNPs identified through genomic discovery efforts in rhesus macaque will have direct benefit to the development of cynomolgus macaque SNP research tools as well.
Results
We sequenced the DNA of 20 cynomolgus macaques, 5 each from Indochina, Indonesia, Mauritius and the Philippines, and compared the sequences to those from the same chromosomal regions of 10 Indian and 10 Chinese rhesus macaques. In each case, the 3' end of a gene was amplified, producing an average DNA fragment of 545 base pairs. The gene regions selected include a representative range of rhesus SNP densities (3–14 SNPs/region) and population distributions of Chinese and Indian rhesus SNPs (63:28). Comparing the M. fascicularis and M. mulatta sequences, we did not identify any fixed base differences that distinguished the macaque species. The only variants detected were polymorphic. There were a total of 69 SNPs in the cynomolgus macaque DNAs, with individual genes having between 2 and 13 polymorphisms (Figure 2a) (see Additional file 1). This SNP density was almost identical to that of the rhesus macaque, which had 71 SNPs in the same regions.

Comparisons of M. fascicularis and M. mulatta SNP densities and distributions in 10 genomic regions. a) The genes in which 3' regions were sequenced in 20 M. fascicularis and 20 M. mulatta are shown. The number of SNPs found in each species and the number found common to both species are indicated. b) The distribution of SNPs in M. fascicularis compared with those found in the same gene regions of M. mulatta. The SNPs identified in M. mulatta (blue) are grouped into those private to just one rhesus population and those shared by both Chinese and Indian rhesus. The yellow bars indicate the number of SNPs found in M. fascicularis.
A comparison of the SNPs identified in these 10 gene regions revealed a high level of overlap between rhesus and cynomolgus macaques. In total, 52% (36/69) of the SNPs in M. fascicularis were also found in rhesus macaques (Figure 2a). All loci sequenced included at least one shared macaque SNP, with an average of 3.6 (SEM ± 0.57) SNPs per locus overlapping between species. Seventy percent (14/20) of the SNPs present in both Indian and Chinese rhesus populations were also present in cynomolgus macaques (Figure 2b). Comparing the two rhesus subpopulations, 38% of the SNPs identified private to Indian rhesus and 44% of those private to Chinese were also found in cynomolgus DNAs. Indian rhesus has an overall lower level of genetic heterogeneity, and thus has a lower absolute number of SNPs that overlap with M. fascicularis (17), as compared with Chinese rhesus (33) (Figure 3).

Venn diagram of SNPs identified in 10 gene regions. The number and relationship of SNPs identified in M. fascicularis and M. mulatta through DNA sequencing of 10 gene regions. The numbers of SNPs found in both single and combinations of populations are shown.
The DNA sequence analysis revealed the level of genetic variation to be similar in all four M. fascicularis geographic populations: Indonesia (39), Mauritius (31), the Philippines (29), and Indochina (31) (Table 1) (see Additional file 2). Of these, 36 of the alleles were private to a single geographical population; Indochinese animals had the most private SNPs, 15. To determine if animals from the overlapping range of the two macaque species had a larger number of shared SNPs, we compared the variants in the Indochinese M. fascicularis with those from Indonesian animals. We found nearly the same number of SNPs associated with the Chinese rhesus macaques in both Indochinese and Indonesian populations, 19 and 20, respectively (Table 1) (see Additional file 2).
To expand the analysis, DNAs from 100 cynomolgus macaques originating from Indochina (25), Indonesia (20), Mauritius (23), the Philippines (16) and breeding centers in China (16) were genotyped using a SNP array containing previously identified rhesus macaque polymorphisms within the 3'ends of genes [10]. The rhesus SNP array includes 64 SNPs from 53 genes on 18 chromosomes (see Additional file 3). The array incorporates SNPs that have a minimum minor allele frequency (MAF) of 0.15 in at least one rhesus population, and are either unique to Indian rhesus (24), Chinese rhesus (19) or are shared in both Indian and Chinese rhesus populations (21). The genotypes of a representative set of 50 M. fascicularis are shown in Figure 4. This assay included 6 SNPs that had been identified in cynomolgus macaques by direct sequencing, and each genotype was confirmed by the SNP array. Excluding the 6 confirmed SNPs, 50% (29/58) of the rhesus SNPs assayed were also present in cynomolgus macaques. In this sampling, 81% of the SNPs previously identified in both Indian and Chinese rhesus were also found in M. fascicularis, whereas 63% of the Chinese-specific SNPs and 29% of the Indian-specific SNPs were detected in cynomolgus macaques (Table 2).

Genotype analysis of 50 M. fascicularis DNAs using M. mulatta SNPs. The M. fascicularis populations surveyed are shown on the left. The SNPs are identified at the top, and are grouped by the M. mulatta population in which they were originally identified. SNP genotypes identified in M. fascicularis are indicated as homozygous major (green), homozygous minor (red), heterozygous (yellow), and no information is indicated by white.
The genotypes of two SNP loci, MAOA:116 and CCR1:641, are of particular interest. MAOA:116 alleles are largely fixed in the rhesus population, with the C-allele being found in Indian rhesus and the G-allele being found in Chinese rhesus [10]. In the cynomolgus macaques 195/200 chromosomes carried the Indian rhesus C-allele. The rare G-allele was only observed in the Indochinese cynomolgus monkeys. At the CCR1:641 locus, the minor allele frequency was high (0.41) in the Indochinese and Chinese M. fascicularis, however no heterozygous genotypes were detected among the 41 macaques screened from these populations. The genotypes from both the SNP assay and direct sequencing were concordant at this locus.
We also used the SNP array to compare the genotypes of cynomolgus macaques from known geographic ancestry to those of animals imported from two breeding centers in China. Of the 12 SNPs in this assay that were only detected among the 25 Indochinese macaques, 11 were also found in the 16 animals obtained from China. The average minor allele frequency of these 12 specific SNPs was 0.19 in the Indochinese animals and 0.16 in the M. fascicularis from China.
Discussion
We found that approximately half of the SNPs identified in M. fascicularis are also present in M. mulatta. The finding indicates that efforts to identify SNPs in either species will be beneficial in generating resources for both macaque species. The recent publication of the rhesus genome sequence has sparked interest in developing genome-wide SNP arrays for use in biomedical research [8]. Such a rhesus macaque SNP array could be used to genotype M. fascicularis DNAs, which based upon this analysis, would capture about 50% of the genomic SNPs in cynomolgus macaques.
In this sampling, we identified 36 SNPs that appeared unique to a single geographic population of M. fascicularis, suggesting that genetic differences between the populations could underlie unrecognized phenotypic differences between these animals. Since genetic heterogeneity can complicate the reproducibility of results in biomedical studies, it would be prudent to use animals from only a single population of M. fascicularis in a single research study. Towards this goal, population-specific SNPs could be used to verify the ancestry of a research animal when it is uncertain. As example, the population-specific SNPs from this study identified cynomolgus macaques from two breeding centers in China as being of Indochinese descent. An expanded SNP discovery effort would readily identify more population markers, making it possible to identify hybrid M. fascicularis as well.
The finding that approximately half of the SNPs found in M. fascicularis overlap with those found in M. mulatta is remarkable, given that previous analysis showed that only 31% of SNPs in rhesus macaques are shared by both the Indian and Chinese subpopulations [10, 11]. The overlapping SNPs in M. fascicularis include ones that are private to Chinese or Indian rhesus, as well as those that are shared between Indian and Chinese populations. While there are more SNPs from the Chinese rhesus than Indian rhesus macaques present in cynomolgus macaques, there are about twice as many SNPs present in the Chinese rhesus population as whole. An evolutionary bottleneck that reduced the overall genetic diversity of the Indian rhesus macaque has been proposed [11], and such a contraction could also have reduced the representation of ancestral macaque SNPs in Indian rhesus monkeys.
There are several possible explanations for the high percentage of shared variants in these two macaque species. The SNPs identified in both M. fascicularis and M. mulatta could represent that the most ancient SNPs in the fascicularis group, those that predate the divergence of rhesus and cynomolgus macaques. Consistent with this idea, the majority of SNPs found in both Indian and Chinese rhesus macaques were also found in M. fascicularis. Interestingly, although mtDNA analysis supports the divergence of these two species 1.8 MYA [32, 33], the few studies to date of nuclear DNA loci have suggested a closer relationship [29–31]. Our findings also suggest a more complex evolutionary history than that suggested by mtDNA alone.
It is possible that relatively recent gene flow between these two macaque species has contributed to the high rate of overlap between M. mulatta and M. fascicularis SNPs. The Y-chromosome sequence studies of Tosi et al. suggest interspecies hybridization, though only within the current overlapping range of Chinese rhesus macaques and Indochinese cynomolgus macaques [30, 31]. However, in this study we found that Indonesian cynomolgus macaques also share a high percentage of SNPs with the rhesus macaques. Thus if interspecies hybridization contributed to the shared sequences, it likely would have occurred before or during periods of glaciation, when land bridges could have permitted the migration of macaques as far South as Indonesia. Nonetheless, gene flow between Chinese rhesus and M. fascicularis does not explain all of the overlapping SNPs, since there is also evidence of Indian-specific rhesus variants in M. fascicularis. Due to the geographic barriers that separate India and Indonesia, SNPs common to both of these populations are more likely a consequence of either sequence conservation or convergent evolution.
Selective pressure to maintain some of the sequence variants could have contributed to the retention of some SNPs in both M. fascicularis and M. mulatta. Possible evidence of selective pressure can be found within this study. By both direct sequence comparisons and SNP array genotyping, we found that one SNP locus (CCR1:641) had a high minor allele frequency (0.41) in the macaques derived from Indochina and Chinese breeding center, and yet no heterozygous individuals were detected, a striking departure from Hardy-Weinberg equilibrium. This finding could be the consequence of inadequate sample size, or a technical issue that was resolved neither by direct sequencing nor by the SNP array. However it is also possible that a heterozygous genotype at CCR1:641, or at alleles tightly linked to this locus, is associated with decreased survival. This is not implausible, since CCR1 encodes the chemokine receptor 1 protein, which is involved in leukocyte recruitment in response to pathogenic infections [34].
The MAOA:116 locus has fixed alleles in the Indian and Chinese rhesus macaque populations and was included in this study of M. fascicularis. The allele present in > 99% of Indian rhesus (C) was also found in almost all of the M. fascicularis, with the only exceptions being a few individuals from the Indochinese population. This skewed presence of the Indian rhesus allele in the M. fascicularis animals is striking. The MAOA gene encodes monoamine oxidase A, a protein that is involved in the breakdown of neurotransmitters, including norepinephrine and serotonin. Some alleles of MAOA are thought to influence aggressive and impulsive behaviors in primates [35]. Perhaps selective pressure favors different MAOA alleles in varying macaque populations or environments.
There were no fixed alleles detected in this study that distinguish M. fascicularis and M. mulatta, and thus there is no direct evidence of gene replacement between species. Based upon the morphological and anatomical differences between the macaques, one would expect some gene replacement to be present. Additional sequencing of larger regions of genomic DNA will be needed to resolve the rate of allele fixation between these two macaque species.
Conclusion
We found that 52% of the SNPs identified in M. fascicularis are also found in M. mulatta. The high rate of overlap suggests that the evolutionary relationship of rhesus and cynomolgus macaques may be closer than that suggested by previous morphological and mtDNA analysis. It also indicates that future SNP discovery efforts in either macaque species will generate information that will be useful for both species. Future efforts to identify cynomolgus SNPs would not only advance genetic research in this widely used animal model, but would also generate tools for verifying the origins of animals for studies where ancestry is important.
Methods
DNA sources
M. fascicularis DNAs used in this study were obtained from at least 2 sources for each geographic region: 25 from Indochina (21 from Cambodia and 4 from Vietnam; SNBL USA, Everett, WA; Alpha Genesis, Yemassee, SC); 20 from Indonesia (Tinjil Island, founded by animals native to Western Java and Southern Sumatra; Java via SNBL USA, Everett, WA; Worldwide Primates, Miami, Florida); 23 from Mauritius (Charles River Laboratories, Wilmington, MA, via the University of Washington and the University of Wisconsin); 16 from the Philippines (SICONBREC, Tanay, Rizal Province, Luzon Island; Del Mundo Trading, Mandaluyong, (central) Manila, Luzon island; Jan Vacek, Located in ILOILO, on the Island of Panay.); 16 from China (SNBL USA, Everett, WA; Alpha Genesis, Yemassee, SC).
DNA analysis
The 10 gene regions chosen for direct DNA sequence analysis were selected from those previously analyzed in rhesus macaques [10]. In each case, PCR amplification was achieved using Taq Polymerase (Fermentas, Inc., Hanover, MD) in accordance with the manufacturers protocol, along with primers designed from the rhesus macaque genomic sequence (see Additional file 4). Amplification products were separated by agarose gel electrophoresis and isolated using Montage Gel Extraction Kit (Millipore, Inc., Bedford, MA). The DNA fragments were sequenced using the PCR amplification primers and Big Dye Chemistry; the products were separated on a Genetic Analyzer 3130 (Applied Biosystems, Inc., Foster City, CA). Sequence electropherograms were visually inspected and compared using Sequencher 4.7 (GeneCodes, Inc., Ann Arbor, MI).
A custom SNP array was used to genotype 64 previously identified rhesus SNPs, using iPLEX reagents and protocols for multiplex PCR, single base primer extension and generation of mass spectra in accordance with the manufacturer's instructions (Sequenom, Inc., San Diego, CA). The multiplex reactions included 28, 17, 12 and 7 primer sets.
All SNPs identified in this work were deposited in dbSNP and monkeySNP databases [36, 37].
References
Cafaro A, Caputo A, Maggiorella MT, Baroncelli S, Fracasso C, Pace M, Borsetti A, Sernicola L, Negri DR, Ten HP, Betti M, Michelini Z, Macchia I, Fanales-Belasio E, Belli R, Corrias F, Butto S, Verani P, Titti F, Ensoli B: SHIV89.6P pathogenicity in cynomolgus monkeys and control of viral replication and disease onset by human immunodeficiency virus type 1 Tat vaccine. J Med Primatol. 2000, 29: 193-208. 10.1034/j.1600-0684.2000.290313.x.
Reimann KA, Parker RA, Seaman MS, Beaudry K, Beddall M, Peterson L, Williams KC, Veazey RS, Montefiori DC, Mascola JR, Nabel GJ, Letvin NL: Pathogenicity of simian-human immunodeficiency virus SHIV-89.6P and SIVmac is attenuated in cynomolgus macaques and associated with early T-lymphocyte responses. J Virol. 2005, 79: 8878-8885. 10.1128/JVI.79.14.8878-8885.2005.
Wiseman RW, Wojcechowskyj JA, Greene JM, Blasky AJ, Gopon T, Soma T, Friedrich TC, O'Connor SL, O'Connor DH: Simian immunodeficiency virus SIVmac239 infection of major histocompatibility complex-identical cynomolgus macaques from Mauritius. J Virol. 2007, 81: 349-361. 10.1128/JVI.01841-06.
Rood PP, Bottino R, Balamurugan AN, Smetanka C, Ayares D, Groth CG, Murase N, Cooper DK, Trucco M: Reduction of early graft loss after intraportal porcine islet transplantation in monkeys. Transplantation. 2007, 83: 202-210. 10.1097/01.tp.0000250680.36942.c6.
Kean LS, Gangappa S, Pearson TC, Larsen CP: Transplant tolerance in non-human primates: progress, current challenges and unmet needs. Am J Transplant. 2006, 6: 884-893. 10.1111/j.1600-6143.2006.01260.x.
Gee MK, Zhang L, Rankin SE, Collins JN, Kauffman RF, Wagner JD: Rosiglitazone treatment improves insulin regulation and dyslipidemia in type 2 diabetic cynomolgus monkeys. Metabolism. 2004, 53: 1121-1125. 10.1016/j.metabol.2004.03.014.
Grant KA, Bennett AJ: Advances in nonhuman primate alcohol abuse and alcoholism research. Pharmacol Ther. 2003, 100: 235-255. 10.1016/j.pharmthera.2003.08.004.
Gibbs RA, Rogers J, Katze MG, Bumgarner R, Weinstock GM, Mardis ER, Remington KA, Strausberg RL, Venter JC, Wilson RK, Batzer MA, Bustamante CD, Eichler EE, Hahn MW, Hardison RC, Makova KD, Miller W, Milosavljevic A, Palermo RE, Siepel A, Sikela JM, Attaway T, Bell S, Bernard KE, Buhay CJ, Chandrabose MN, Dao M, Davis C, Delehaunty KD, Ding Y, Dinh HH, Dugan-Rocha S, Fulton LA, Gabisi RA, Garner TT, Godfrey J, Hawes AC, Hernandez J, Hines S, Holder M, Hume J, Jhangiani SN, Joshi V, Khan ZM, Kirkness EF, Cree A, Fowler RG, Lee S, Lewis LR, Li Z, Liu YS, Moore SM, Muzny D, Nazareth LV, Ngo DN, Okwuonu GO, Pai G, Parker D, Paul HA, Pfannkoch C, Pohl CS, Rogers YH, Ruiz SJ, Sabo A, Santibanez J, Schneider BW, Smith SM, Sodergren E, Svatek AF, Utterback TR, Vattathil S, Warren W, White CS, Chinwalla AT, Feng Y, Halpern AL, Hillier LW, Huang X, Minx P, Nelson JO, Pepin KH, Qin X, Sutton GG, Venter E, Walenz BP, Wallis JW, Worley KC, Yang SP, Jones SM, Marra MA, Rocchi M, Schein JE, Baertsch R, Clarke L, Csuros M, Glasscock J, Harris RA, Havlak P, Jackson AR, Jiang H, Liu Y, Messina DN, Shen Y, Song HX, Wylie T, Zhang L, Birney E, Han K, Konkel MK, Lee J, Smit AF, Ullmer B, Wang H, Xing J, Burhans R, Cheng Z, Karro JE, Ma J, Raney B, She X, Cox MJ, Demuth JP, Dumas LJ, Han SG, Hopkins J, Karimpour-Fard A, Kim YH, Pollack JR, Vinar T, ddo-Quaye C, Degenhardt J, Denby A, Hubisz MJ, Indap A, Kosiol C, Lahn BT, Lawson HA, Marklein A, Nielsen R, Vallender EJ, Clark AG, Ferguson B, Hernandez RD, Hirani K, Kehrer-Sawatzki H, Kolb J, Patil S, Pu LL, Ren Y, Smith DG, Wheeler DA, Schenck I, Ball EV, Chen R, Cooper DN, Giardine B, Hsu F, Kent WJ, Lesk A, Nelson DL, O'brien WE, Prufer K, Stenson PD, Wallace JC, Ke H, Liu XM, Wang P, Xiang AP, Yang F, Barber GP, Haussler D, Karolchik D, Kern AD, Kuhn RM, Smith KE, Zwieg AS: Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007, 316: 222-234. 10.1126/science.1139247.
Rogers J, Garcia R, Shelledy W, Kaplan J, Arya A, Johnson Z, Bergstrom M, Novakowski L, Nair P, Vinson A, Newman D, Heckman G, Cameron J: An initial genetic linkage map of the rhesus macaque (Macaca mulatta) genome using human microsatellite loci. Genomics. 2006, 87: 30-38. 10.1016/j.ygeno.2005.10.004.
Ferguson B, Street SL, Wright H, Pearson C, Jia Y, Thompson SL, Allibone P, Dubay CJ, Spindel E, Norgren RB: Single nucleotide polymorphisms (SNPs) distinguish Indian-origin and Chinese-origin rhesus macaques (Macaca mulatta). BMC Genomics. 2007, 8: 43-10.1186/1471-2164-8-43.
Hernandez RD, Hubisz MJ, Wheeler DA, Smith DG, Ferguson B, Rogers J, Nazareth L, Indap A, Bourquin T, McPherson J, Muzny D, Gibbs R, Nielsen R, Bustamante CD: Demographic histories and patterns of linkage disequilibrium in Chinese and Indian rhesus macaques. Science. 2007, 316: 240-243. 10.1126/science.1140462.
Kikuchi T, Hara M, Terao K: Development of a microsatellite marker set applicable to genome-wide screening of cynomolgus monkeys (Macaca fascicularis). Primates. 2007, 48: 140-146. 10.1007/s10329-006-0008-z.
Delson E: Fossil macaques, phyletic relationships and a scenario of deployment. The Macaques: Studies In Ecology, Behavior and Evolution. Edited by: Lindburg DG. 1980, New York, Van Nostrand Reinhold, 10-30.
Fooden J: Classification and distribution of living macaques. The Macaques: Studies in Ecology, Behavior and Evolution. Edited by: Lindburg DG. 1980, New York, Van Nostrand Reinhold, 1-9.
Fooden J: Systematic review of the rhesus macaque, Macaca mulatta (Zimmerman, 1780). Field Museum of Natural History, Vol.96. 2000, Chicago, Fieldiana, 180-Publication 1509
Abegg C, Thierry B: Macaque evolution and dispersal in insular south-east Asia. Biol J Linn Soc. 2002, 75: 555-576. 10.1046/j.1095-8312.2002.00045.x.
Voris HK: Maps of Pleistocene sea levels in southeast Asia: shorelines, river systems and time durations. J Biogeog. 2000, 27: 1153-1167. 10.1046/j.1365-2699.2000.00489.x.
Sussman RW, Tattersall I: Behavior and ecology of Macaca fascicularis in Mauritius: a preliminary study. Primates. 1981, 22: 192-205. 10.1007/BF02382610.
Sussman RW, Tattersall I: Distribution, abundance and putative ecological strategy of Macaca fascularis on the island of Mauritius, southwestern Indian Ocean. Folia Primatol. 1986, 46: 28-43.
Krebs KC, Jin Z, Rudersdorf R, Hughes AL, O'Connor DH: Unusually high frequency MHC class I alleles in Mauritian origin cynomolgus macaques. J Immunol. 2005, 175: 5230-5239.
O'Connor SL, Blasky AJ, Pendley CJ, Becker EA, Wiseman RW, Karl JA, Hughes AL, O'Connor DH: Comprehensive characterization of MHC class II haplotypes in Mauritian cynomolgus macaques. Immunogenetics. 2007, 59: 449-462. 10.1007/s00251-007-0209-7.
Smith DG, McDonough JW, George DA: Mitochondrial DNA variation within and among regional populations of longtail macaques (Macaca fascicularis) in relation to other species of the fascicularis group of macaques. Am J Primatol. 2007, 69: 182-198. 10.1002/ajp.20337.
Tosi AJ, Coke CS: Comparative phylogenetics offer new insights into the biogeographic history of Macaca fascicularis and the origin of the Mauritian macaques. Mol Phylogenet Evol. 2007, 42: 498-504. 10.1016/j.ympev.2006.08.002.
Smith DG, McDonough J: Mitochondrial DNA variation in Chinese and Indian rhesus macaques (Macaca mulatta). Am J Primatol. 2005, 65: 1-25. 10.1002/ajp.20094.
Viray J, Rolfs B, Smith DG: Comparison of the frequencies of major histocompatibility (MHC) class-II DQA1 and DQB1 alleles in Indian and Chinese rhesus macaques (Macaca mulatta). Comp Med. 2001, 51: 555-561.
Doxiadis GG, Otting N, de Groot NG, de GN, Rouweler AJ, Noort R, Verschoor EJ, Bontjer I, Bontrop RE: Evolutionary stability of MHC class II haplotypes in diverse rhesus macaque populations. Immunogenetics. 2003, 55: 540-551. 10.1007/s00251-003-0590-9.
Otting N, de Vos-Rouweler AJ, Heijmans CM, de Groot NG, Doxiadis GG, Bontrop RE: MHC class I A region diversity and polymorphism in macaque species. Immunogenetics. 2007, 59: 367-375. 10.1007/s00251-007-0201-2.
Doxiadis GG, Rouweler AJ, de Groot NG, Louwerse A, Otting N, Verschoor EJ, Bontrop RE: Extensive sharing of MHC class II alleles between rhesus and cynomolgus macaques. Immunogenetics. 2006, 58: 259-268. 10.1007/s00251-006-0083-8.
Deinard A, Smith DG: Phylogenetic relationships among the macaques: evidence from the nuclear locus NRAMP1. J Hum Evol. 2001, 41: 45-59. 10.1006/jhev.2001.0480.
Tosi AJ, Morales JC, Melnick DJ: Y-chromosome and mitochondrial markers in Macaca fascicularis indicate introgression with Indochinese M. mulatta and a biogeographic barrier in the Isthmus of Kra. Int J Primatol. 2002, 23: 161-178. 10.1023/A:1013258109954.
Tosi AJ, Morales JC, Melnick DJ: Paternal, maternal, and biparental molecular markers provide unique windows onto the evolutionary history of macaque monkeys. Evolution Int J Org Evolution. 2003, 57 (6): 1419-1435.
Hayasaka K, Fujii K, Horai S: Molecular phylogeny of macaques: implications of nucleotide sequences from an 896-base pair region of mitochondrial DNA. Mol Biol Evol. 1996, 13: 1044-1053.
Morales JC, Melnick DJ: Phylogenetic relationships of the macaques (Cercopithecidae: Macaca), as revealed by high resolution restriction site mapping of mitochondrial ribosomal genes. J Hum Evol. 1998, 34: 1-23. 10.1006/jhev.1997.0171.
Baggiolini M: Chemokines and leukocyte traffic. Nature. 1998, 392: 565-568. 10.1038/33340.
Craig IW: The importance of stress and genetic variation in human aggression. Bioessays. 2007, 29: 227-236. 10.1002/bies.20538.
NCBI Single Nucleotide Polymorphism Database. 2007, [http://www.ncbi.nlm.nih.gov/SNP/]
ONPRC Monkey SNP Database. 2007, [http://monkeysnp.ohsu.edu]
Acknowledgements
This work was supported by grants RR00163 and RR00166, from the National Center for Research Resources, National Institutes of Health. It was also supported in part by SNBL USA, Ltd. (Everett, WA). We thank both the reviewers and other individuals who provided insightful comments on the manuscript, including Carlos Bustamante, Joe Felsenstein, Samone Khouangsathiene, David O'Connor, Eliot Spindel and Roger Wiseman. We also thank Jennifer Dunfield, Christine Howard, Samone Khouangsathiene and Erik McArthur for their assistance in preparing the manuscript. We are grateful for the generous contributions of M. fascicularis DNAs from our colleagues: Roger Wiseman and David O'Connor (Wisconsin National Primate Research Center, Madison, WI) contributed Mauritian cynomolgus DNAs. Nickolas Lerche (California National Primate Research Center, Davis, CA) provided DNAs from Philippine M. fascicularis and Kathy Grant (Oregon National Primate Research Center, Beaverton, OR) provided DNAs from Indonesian animals. Travis Rogers at the Vollum DNA Sequencing Core, Oregon Health Sciences University, preformed the DNA sequencing. Kenneth Beckman, Director of the Functional Genomics Laboratory at Children's Hospital in Oakland Research Institute in Oakland, CA provided assistance with the Sequenom iPLEX SNP assay.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
BF and RCK conceived of this study. SLS preformed all DNA sequence and statistical analyses and contributed to the writing of the manuscript. RCK provided M. fascicularis DNAs and perspective on the project design. RG provided a diverse set of cynomolgus DNAs, as well as insightful review of the results. BF was responsible for the project design, the data analysis and the manuscript preparation. All authors read and approved this manuscript.
Electronic supplementary material
12864_2007_1193_MOESM1_ESM.pdf
Additional file 1: SNP identity and NCBI accession numbers. This file lists the SNPs identified in this study and the associated gene. The NCBI STS (reference sequence) accession numbers for each are shown. (PDF 12 KB)
12864_2007_1193_MOESM2_ESM.pdf
Additional file 2: SNPs identified through sequencing in 10 genomic regions of M. fascicularis and M. mulatto. All SNPs identified in 10 genomic regions by sequencing of 20 M. fascicularis and 20 M. mulatta are shown at the top. Loci highlighted in green showed no variant allele in that population; yellow highlight indicates a SNP was detected, and red indicates that only the minor allele of the SNP variant was detected in the population. (PDF 14 KB)
12864_2007_1193_MOESM3_ESM.pdf
Additional file 3: SNP assay gene summary. The fifty-three genes represented in the SNP genotype assay are listed, along with chromosome location. Those genes that were also analyzed by direct sequencing are noted. (PDF 8 KB)
12864_2007_1193_MOESM4_ESM.pdf
Additional file 4: Sequence information for M. fascicularis. The primer information and reference sequences for the ten gene regions sequenced in M. fascicularis are listed. (PDF 8 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Street, S.L., Kyes, R.C., Grant, R. et al. Single nucleotide polymorphisms (SNPs) are highly conserved in rhesus (Macaca mulatta) and cynomolgus (Macaca fascicularis) macaques. BMC Genomics 8, 480 (2007). https://doi.org/10.1186/1471-2164-8-480
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-8-480