
Abstract
Background
Thermus thermophilus and Deinococcus radiodurans belong to a distinct bacterial clade but have remarkably different phenotypes. T. thermophilus is a thermophile, which is relatively sensitive to ionizing radiation and desiccation, whereas D. radiodurans is a mesophile, which is highly radiation- and desiccation-resistant. Here we present an in-depth comparison of the genomes of these two related but differently adapted bacteria.
Results
By reconstructing the evolution of Thermus and Deinococcus after the divergence from their common ancestor, we demonstrate a high level of post-divergence gene flux in both lineages. Various aspects of the adaptation to high temperature in Thermus can be attributed to horizontal gene transfer from archaea and thermophilic bacteria; many of the horizontally transferred genes are located on the single megaplasmid of Thermus. In addition, the Thermus lineage has lost a set of genes that are still present in Deinococcus and many other mesophilic bacteria but are not common among thermophiles. By contrast, Deinococcus seems to have acquired numerous genes related to stress response systems from various bacteria. A comparison of the distribution of orthologous genes among the four partitions of the Deinococcus genome and the two partitions of the Thermus genome reveals homology between the Thermus megaplasmid (pTT27) and Deinococcus megaplasmid (DR177).
Conclusion
After the radiation from their common ancestor, the Thermus and Deinococcus lineages have taken divergent paths toward their distinct lifestyles. In addition to extensive gene loss, Thermus seems to have acquired numerous genes from thermophiles, which likely was the decisive contribution to its thermophilic adaptation. By contrast, Deinococcus lost few genes but seems to have acquired many bacterial genes that apparently enhanced its ability to survive different kinds of environmental stresses. Notwithstanding the accumulation of horizontally transferred genes, we also show that the single megaplasmid of Thermus and the DR177 megaplasmid of Deinococcus are homologous and probably were inherited from the common ancestor of these bacteria.
Similar content being viewed by others


Background
Deinococcus spp. and Thermus spp. are believed to belong to a distinct branch of bacteria called the Deinococcus-Thermus group [1]. The common origin of these bacteria is supported by the fact that they consistently form a strongly supported clade in phylogenetic trees of ribosomal RNAs and several conserved proteins including ribosomal proteins, RNA polymerase subunits, RecA, and others [1–7]. The Thermus genus currently consists of 14 thermophilic species, whereas the Deinococcus genus includes at least eleven, mostly mesophilic species known for their extreme resistance to γ-irradiation and other agents causing DNA damage, particularly, desiccation [7, 8]. Interestingly, two new species, D. frigens sp. nov., D. saxicola, have been isolated recently from Antarctic rock and soil samples [9]. D. geothermalis and D. murrayi, are considered to be thermophilic (topt~45–50°C) [10].
T. thermophilus (TT) (strain HB27, ATCC BAA-163)and D. radiodurans (DR) (strain R1, ATCC BAA-816) have similar general physiology, both being catalase-positive, red-pigmented, non-sporulating, aerobic chemoorganoheterotrophs [11]. However, the two organisms are dramatically different in terms of stress resistance: DR is one of the most resistant to radiation and desiccation among the characterized organisms and, generally, can survive diverse types of oxidative stress, whereas TT is a thermophile that thrives under thermal stress conditions but is relatively sensitive to radiation and other forms of oxidative stress. Recently, the genomes of T. thermophilus HB27 and HB8 [12, 13] became available for comparison with the previously sequenced genome of D. raduodurans R1 [14].
Despite extensive research, genetic systems underlying both thermophilic adaptations and radiation resistance remain poorly understood. Attempts have been made to detect "thermophilic determinants" in the proteomes of thermophiles using a comparative-genomic approach. This resulted in the delineation of a set of proteins that might be associated with the thermophilic phenotype, although most of these are significantly enriched in thermophiles but not unique to these organisms [15–17]. Other distinctions between thermophilic and mesophilic proteins might be due to differences in their structural properties, such as different amino acid compositions, loop lengths, number of salt bridges, strength of hydrophobic interaction, number of disulfide bonds, and other features [18–25].
Various hypotheses also have been proposed to explain radiation resistance, some postulating the existence of specialized genetic systems, particularly those for DNA repair and stress response [7, 26]. Recently, however, alternative possibilities have been advanced. For example, the post-irradiation adjustment of metabolism of D. radiodurans might prevent production of reactive oxygen species (ROS) by decreasing the number of reactions involving oxygen [27, 28], and high intracellular manganese concentrations of Deinococcus spp. might help scavenge ROS generated during irradiation and post-irradiation recovery [28, 29]. However, these explanations of radiation resistance have received little direct support from comparative-genomic analyses [7, 27, 28].
Several evolutionary processes could potentially contribute to the genome differentiation of TT and DR subsequent to the divergence from the common ancestor: (i) differential gene loss and gain, (ii) acquisition of genes via horizontal gene transfer (HGT) which may be followed by loss of the ancestral orthologous gene (xenologous gene displacement (XGD)), (iii) lineage-specific expansion of paralogous gene families by duplication and/or acquisition of paralogs via HGT; (IV) modification of amino acid composition that could affect protein stability. Here, we experimentally characterize radiation and desiccation resistance of TT in comparison to DR, and, assess the contribution of different evolutionary processes to distinct adaptations of TT and DR, using a variety of comparative-genomic approaches and phylogenetic analysis. We identify the unique feature of the gene repertoires of TT and DR that might contribute to these phenotypic differences, which could be the subject of further experimental work. In addition, we describe the results of a detailed analysis of the proteins predicted to be involved in DNA repair and stress response functions, which are particularly relevant for adaptive evolution of resistance phenotypes.
Results and discussion
Experimental characterization of resistance to gamma-radiation and desiccation, and determination of intracellular Mn/Fe ratio for T. thermophilus
While the radiation- and desiccation-resistant phenotypes of DR and the thermal requirements of both TT and DR have been studied extensively ([7, 12, 30] and references therein), we are unaware of any detailed characterization of the response of TT to irradiation or desiccation. Therefore, we sought to investigate these properties in order to obtain a more complete picture of the differences in the stress response phenotypes of TT and DR. Not unexpectedly, we found that TT was much more sensitive to acute irradiation than DR. The survival curve of TT is similar to that of Esherichia coli K12. For DR, the radiation dose yielding 10% colony forming unit (CFU) survival (D10) is ~16 kGy, whereas for TT and E. coli, the D10 dose is ~0.8 kGy and ~0.7 kGy, respectively (Figure 1). TT is also highly sensitive to desiccation. The 10% CFU desiccation survival frequency of DR is sustained after 30 days, while TT reaches the 10% CFU desiccation survival at ~10 hours (0.4% survival after 24 hours) and, by the 5th day, suffers essentially 100% lethality. The low resistance of TT to desiccation is observed regardless of the temperature and drying rate (see Additional file 1, "Desiccation of TT at 65°C"). The desiccation resistance of E. coli was found to be intermediate between those of TT and DR (Figure 2).

Radiation resistance of T. thermophilus (ATCC BAA-163), D. radiodurans (ATCC BAA-816) and E. coli (K-12 MG1655, provided by M. Cashel, NIH) (60Co irradiation). Standard deviations for the data points are shown.

Desiccation resistance of T. thermophilus (ATCC BAA-163), D. radiodurans (ATCC BAA-816) and E. coli (K-12 MG1655) (room temperature). Note that no surviving cells were obtained for cells desiccated at 65°C. Standard deviations for five data points are shown.
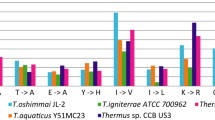
We recently reported a trend of intracellular Mn/Fe concentration ratios in bacterial IR resistance, where very high and very low Mn/Fe ratios correlated with very high and very low resistances, respectively [29]. We have also shown that growing D. radiodurans in conditions which limited Mn(II) accumulation, significantly lowered the cells' IR resistance [29]. These observations led to the hypothesis that the ratio of Mn to Fe in a cell might determine the relative abundance of different ROS induced during exposure to and recovery from IR [28, 29]. At high concentrations, Mn(II) can act as true catalyst of the dismutation of superoxide (O2•-), with Mn cycling between the divalent and trivalent states; Mn redox-cycling scavenges both O2•- and hydrogen peroxide [31]. In this context, we determined the intracellular Mn/Fe ratio of TT using an inductively coupled plasma-mass spectrometry method (ICP-MS), as previously described [29]. TT cells contained 0.211 (± 0.0254) nmol Mn/mg protein and 4.54 (± 0.778) nmol Fe/mg protein. The intracellular Mn/Fe ratio of TT (D10, ~0.8 kGy) is 0.047 compared to 0.0072 for E. coli (D10, ~0.7 kGy), 0.24 for D. radiodurans (D10, ~16 kGy), and <0.0001 for Pseudomonas putida (D10, ~0.25 kGy) [29]. Thus, TT appears to be somewhat more sensitive to acute IR and desiccation than predicted by its Mn/Fe ratio, suggesting the possibility of more complex relationships between these two variables. Notably, scavenging of O2•- by Mn(II) is highly dependent on the availability of H2O2, which in TT would be expected to become limiting during recovery at 65°C because of thermal decomposition [32]; inefficient Mn redox-cycling can lead to Mn(III) accumulation, which is cytotoxic. Both DR and TT encode an ABC- type Mn transporter and a transcriptional regulator that probably regulates Mn homeostasis. Additionally, DR has a NRAMP family Mn transporter, for which there is no ortholog in TT.
Reconstruction of the gene-content tree and the gene repertoire of the common ancestor of Deinococcus and Thermus
Information on the presence-absence of orthologous genes in a set of genomes can be used to produce a gene-content tree [33, 34]. The topology of a gene-content tree may reflect not only the phylogenetic relationships between the compared species but lifestyle similarities and differences as well [33, 35, 36]. Given the dramatic differences in the lifestyles and resistance phenotypes of TT and DR, we were interested to determine whether or not the gene content of TT was most similar to that of DR or those of other thermophilic bacteria or, perhaps, even archaea. To this end, we assigned the proteins encoded in the TT genome to the Clusters of Orthologous Groups of proteins (COGs) [37] and, using the patterns of representation of species in COGs to calculate distances between species, reconstructed a gene-content tree as described previously [35]. In the resulting gene-content tree, which included 62 sequenced genomes of prokaryotes and unicellular eukaryotes, TT and DR were confidently recovered as sister species, and the DR-TT lineage was positioned within a subtree that also included Actinobacteria and Cyanobacteria, several of which are known for their extreme radiation and desiccation resistances [38–40] (Figure 3). For this branch, the topology of the gene-content tree mimics the topologies of trees constructed with other approaches based on genome-wide data [34, 35], indicating that the gene repertoires of these bacteria, and TT and DR in particular, have been diverging, roughly, in a clock-like fashion. To determine which genes were likely to have been lost and gained in each lineage, we reconstructed a parsimonious scenario of evolution from the last bacterial common ancestor (LBCA) to TT and DR, through their last common ancestor. The reconstruction was performed on the basis of the assignment of TT and DR proteins to COGs, together with COG-based phyletic patterns of 62 other sequenced bacterial and archaeal genomes [37], using a previously developed weighted parsimony method [41] (see Methods and Additional file 2). This approach assigns 1,310 genes (COGs) to the DR-TT common ancestor (Figure 4). Of these, 1,081 (~80%) were retained in both TT and DR and belong to their shared gene core. Since TT (2210 genes) has far fewer predicted protein-coding genes than DR (3191 genes), it seems likely that the divergence of the two involved substantial genome reduction in TT and/or genome expansion in DR. However, the reconstruction results suggest that TT has not experienced massive genome reduction although the total gene flux (i.e., the sum total of genes inferred to have been lost and gained) during the evolution of this lineage was considerable, involving ~25% of the gene complement. In contrast, DR gained 272 COGs, with only 59 lost, which indicates substantial genome growth after the DR-TT divergence (Figure 4).

Gene content tree constructed for 66 species included in the COG database on the basis of the patterns of presence-absence in the COGs. The Thermus-Deinococcus clade is marked by bold type. Black, bacteria; yellow, archaea; blue, eukaryotes.

The reconstructed evolutionary scenario for the Thermus-Deinococcus clade. LBCA – Last Bacterial Common Ancestor; DR-TT – the common ancestor of Thermus-Deinococcus clade; TT – T. thermophilus; DR- D. radiodurans. Total number of COGs is shown in boxes for each node. '+' indicates inferred gene (COG) gain, and '-' indicates inferred loss.
We were further interested in determining whether similarities existed among the gene repertoires of TT and two deeply-branching bacterial hyperthermophiles, Aquifex aeolicus (AA) and Thermotoga maritima (TM). We found that genes that are present in TT but not DR are significantly more likely to be present in AA and TM than genes present in DR but not TT (Table 1). In contrast, among the 170 TT genes inferred to have been lost, only 20 were present in both AA and TM. Thus, the gene repertoire of TT has significantly greater similarity with hyperthermophilic bacteria than the gene repertoire of DR, perhaps resulting from direct or parallel HGT (see also below).
The majority of the genes shared by TT and DR encode house-keeping proteins and are widespread in bacteria. Among those, 14 COGs are unusual in that they are not found in any other bacteria but instead are shared by TT and DR with archaea or eukaryotes. This set includes 8 subunits of the Archaeal/vacuolar-type H+-ATPase and six other COGs that consist of characteristic archaeal genes (phosphoglycerate mutase, COG3635; 2-phosphoglycerate kinase, COG2074; SAR1-like GTPase, COG1100; GTP:adenosylcobinamide-phosphate guanylyltransferase, COG2266; Predicted membrane protein, COG3374 ; Predicted DNA modification methylase, COG1041).
As noticed previously, some DR genes that belong to families well-represented in both bacteria and archaea showed clear archaeal affinity [42]. To assess how many genes of apparent "thermophilic" descent (either archaeal or bacterial) might already have been present already in the common DR-TT ancestor, we performed phylogenetic analysis of genes that were assigned to the DR-TT ancestor and had 3 of the 5 best hits to the genes from thermophiles (both archaeal and bacterial; see Additional file 1: "Phylogenetic analysis of the genes of the reconstructed DR-TT common ancestor"). We found that at least 122 genes (~10% of the predicted gene repertoire of the DR-TT common ancestor) of the originally selected 205 genes showed an affinity to thermophilic species, i.e., either a branch of two orthologous genes from DR and TT, or a DR or TT gene (in cases when the respective ortholog apparently was lost in the other lineage) clustered with thermophiles (see Additional file 1, table 1S; and Additional file 6). Due to the fact that many tree topologies are highly perturbed by multiple HGT events and may be inaccurate due to differences in evolutionary rates between lineages, this is only a rough estimate of the number of "thermophilic" genes in the DR-TT ancestor. In particular, it cannot be ruled out that some of the ancestral "thermophilic" genes, which are currently present in TT but not DR (10 such genes out of 122 tested genes), have been acquired by TT via XGD (see Additional file 1, table 1S). Taken together, these observations suggest the possibility of ecological contacts between the DR-TT ancestor and hyperthermophilic archaea and/or bacteria, leading to substantial acquisition of "thermophilic" genes via HGT.
Gene gain and loss in Thermus
Our reconstruction of the evolutionary events that occurred after the divergence of the TT and DR lineages from the common ancestor delineated the sets of genes that likely have been gained and lost by each lineage (Figure 4). We first consider in greater detail the pattern of gene loss and gain in TT. The absence of certain metabolic genes in TT creates gaps in its metabolic pathways, some of which are essential. However, TT is capable of synthesizing all amino acids, nucleotides and a majority of cofactors, suggesting that the gaps are filled by analogous or at least non-orthologous enzymes. Several such cases have been described. For example, TT and DR encode unrelated thymidylate synthases, DR2630 (COG0207) and TTC0731 (COG1351). The classical, folate-dependent thymidylate synthase (COG0207) present in DR is probably ancestral in bacteria and apparently was displaced via HGT in the Thermus lineage by the flavin-dependent thymidylate synthase, typical of archaea and bacterial thermophiles. In other cases, the substituted analogous enzymes or pathways remain uncharacterized. For instance, the displacement of the folate-dependent thymidylate synthase with the flavin-dependent type in the TT lineage and in other bacteria and archaea correlates with the apparent loss of dihydrofolate reductase (folA, COG0262), which catalyzes the last step of the tetrahydrofolate biosynthesis pathway. Since tetrahydrofolate is an essential cofactor, a displacement appears most likely. Recently, it has been shown that the halobacterial FolP-FolC fusion protein complemented a Haloferax volcanii folA mutant [43]. Thus, it appears likely that FolC and FolP in TT and other organisms complement the activity of FolA although the existence of an unrelated, as yet uncharacterized dihydrofolate reductase cannot be ruled out.
In addition, TT does not encode two enzymes for pyridoxal phosphate biosynthesis (pdxK, COG0259 and pdxH, COG2240), while DR has a complete set of enzymes of this pathway. Our reconstructions suggest that pdxK was likely lost in the TT lineage, whereas DR probably independently acquired pdxH. However, the pathway is likely to be functional in both organisms. Since similar gaps in the pyridoxal phosphate biosynthesis are seen in a variety of prokaryotes [44], it appears that, for at least some steps of this pathway, there exists a set of distinct enzymes which remain unidentified.
Some systems apparently were completely lost in the TT lineage. These include the urease complex, the ramnose metabolism pathway, acyl CoA:acetate/3-ketoacid CoA transferase, fructose transport and utilization, and glycerol metabolism. Notably, most of these systems are also absent in thermophilic bacteria and many thermophilic archaea.
In contrast to DR, the genes that appear to have been acquired by TT show a clear connection to the thermophilic lifestyle. In particular, TT seems to have acquired 23 gene families from the set of putative thermophilic determinants [17], whereas the common DR-TT ancestor had 5 genes from the list, and DR seems to have acquired only one (see Additional file 3). The majority of these proteins (17 of 31) are encoded in the TT megaplasmid and 11 belong to the predicted mobile DNA repair system characteristic of thermophiles [16, 45] (Figure 7A; see details below). In addition, TT has acquired 4 "archaeal" genes that are not encoded in any of the genomes of mesophilic bacteria assigned to COGs (peptide chain release factor 1, COG1503; DNA modification methylase, COG1041; and two membrane proteins, COG3462 and COG4645).

A. Organization of genes encoding the putative thermophyle-specific DNA repair system in two Thermus strains and the draft genome of D. geothermalis. The boxes on top of the figure show COG numbers. Genes are shown by block arrows indicating the direction of transcription and identified by their systematic names. For each column of the alignment, the corresponding COG number and predicted function is indicated. D. geothermalis' genes are connected with respective orthologs in both TT strains by a straight line. Generally, orthologous genes are shown by the same color and pattern. The exceptions are the RAMP proteins of COGs 1336, 1367, 1604, 1337 and 1332, which are all shown in pink. Other, more distant RAMPs are also shown in pink, with different patterns [16]. Proteins that do not belong to COGs but have homologs in other species are marked by diamonds. Abbreviations: HEL, predicted helicase, HD nuclease – HD conserved motif containing predicted nuclease conserved region; POL – novel predicted polymerase, RECB – predicted nuclease of RecB family; B. Comparison of gene organization in the region of the megaplasmid coding for reverse gyrase in two TT strains. The shorter gyrase gene in HB27 indicates truncation. Abbreviations: REVGYR, reverse gyrase, MET_PR – predicted metal-dependent protease; REC_DNA – three-domain fusion protein (DnaQ endonuclease, DinG helicase, and RecQ helicase).
The Sox-like sulfur oxidation system is among the group of genes that were apparently acquired in the TT lineage. The TT Sox operon is partly similar to the one identified in AA (see Additional file 1, Figure 2S), and might have been horizontally transferred between the AA and TT lineages with subsequent local rearrangements. The presence of the Sox operon in TT suggests that this bacterium can use reduced sulfur compounds as a source of energy and sulfur. Another system likely acquired by TT is lactose utilization (at least three proteins: LacZ, COG3250; GalK, COG0153; GalT COG1085; GalA, COG3345). This system is also present in TM, which indicates that sugars can be utilized as carbon sources by thermophilic bacteria.
Gene gain and loss in Deinococcus
The DR lineage has apparently acquired many more genes than it has lost (Figure 4). The majority of the genes lost by DR encode enzymes of energy metabolism and biosynthesis of cofactors. One example is the loss of the three subunits of pyruvate:ferredoxin oxidoreductase, which is one of the several known systems for pyruvate oxidation, a key reaction of central metabolism. Another example of gene loss in DR involves the three subunits of NAD/NADP transhydrogenase, which is responsible for energy-dependent reduction of NADP+ [46]. In addition, the DR lineage lost four enzymes of NAD biosynthesis and six enzymes of cobalamine biosynthesis, and consistently, DR is dependent on an exogenous source of NAD for growth [28, 47].
A conspicuous number of genes apparently acquired by the DR lineage encode systems of protein degradation and amino acid catabolism (e.g., urease, DRA0311-DRA0319, and a predicted urea transporter, DRA0320-DRA0324; histidine degradation system, DRA0147-DRA0150; monoamine oxidase, DRA0274; lysine 2,3-aminomutase, DRA0027; kynureninase and tryptophan-2,3-dioxygenase, DRA0338-DRA0339; peptidase E, DR1070, and carboxypeptidase C, DR0964; and D-aminopeptidase, DR1843 (see Additional file 4). A similar trend is observed for the expansion of several protein families in DR, such as secreted subtilisin-like proteases (see below). Additionally, DR acquired two three-subunit complexes of aerobic-type carbon monoxide dehydrogenase (DRA0231-DRA0233 and DRA0235-DRA0237); oxidation of CO by this enzyme might be used as an energy source as shown for some bacteria [48]. Acquisition and expansion of these metabolic systems, together with the loss of certain biosynthetic capabilities, supports the possibility that metabolic restructuring could impact oxidative stress resistance in DR by decreasing the need for high-energy-dependent cellular activities. Energy production through the respiratory chain is one major source of free radicals in the cell [49].
DR has many more genes for proteins involved in inorganic ion transport and metabolism than TT. In particular, DR has acquired the multisubunit Na+/H+ antiporter (7 genes), K+-transporting ATPase (3 genes), and the FeoA/FeoB Fe transport system. This abundance of ion transport systems might be indirectly linked to oxidative stress resistance through regulation of membrane ion gradients and Mn/Fe homeostasis (see above).
DR is more dependent than TT on peptide-derived growth substrates [47] and has a more complex stress response circuitry. Consistent with this, the signal transduction systems of DR, as predicted by genome analysis, are considerably more elaborate than those of TT. In particular, DR has at least 33 COGs (15 probably acquired after the divergence from the common ancestor with TT) related to signal-transduction functions that are not represented in TT as compared to 12 (5 acquired) such COGs in TT. Furthermore, although most of the signal-transduction domains are shared by DR and TT, the domain architectures of the respective multidomain proteins are completely different ([7] and KSM, unpublished observations).
Among the genes apparently acquired by DR, two encode multidomain proteins containing distinct periplasmic ligand-binding sensor domains (DRA0202, COG5278 and DR1174, COG3614). Another protein, DRA0204, contains the CHASE3 domain [50] and is located in a predicted operon with superoxide dismutase (DRA0202), indicating a function in oxidative stress response. The protein DR0724 contains the SARP domain which is involved in apoptosis-related signaling pathways in eukaryotes [51] but its function in bacteria is unknown. The roles of the other signal transduction proteins of DR are even less clear, with the notable exception of a phytochrome-like protein (DRA0050) that apparently was acquired by DR from a bacterial source and has been implicated in UV resistance [52].
Additionally, DR has many genes (25 COGs) encoding systems for microbial defense; TT has only 14 COGs in this category, 13 of which are shared with DR. At least 7 genes for restriction-modification system subunits were specifically acquired by the DR lineage, along with several antibiotic-resistance enzymes. This difference might be linked to the reduced metabolic capabilities of DR, which is dependent on nutrient-rich conditions for growth and, perhaps, encounters more microbial species than TT.
The previous analysis of the DR genome revealed 15 genes that appear to have been horizontally transferred from unexpected sources, such as eukaryotes and viruses [7]; only two of these 15 genes are present in TT, the desiccation-related protein of the ferritin family and the Uma2-like family proteins (see discussion of these proteins below). Two desiccation-related proteins have been shown to be involved in desiccation but not radiation resistance [53]; Ro ribonucleoprotein is apparently involved in UV resistance [54], and topoisomerase IB, while active, has no known role in DR [55]. So far, none of these genes has been linked experimentally with radioresistance in DR.
Identification of xenologous gene displacement by phylogenetic analysis
It is well-established that HGT has made major contributions to the gene repertoires of most thermophilic bacteria as supported by the presence of numerous genes with unexpectedly high similarity to and/or phylogenetic affinity with genes typical of hyperthermophilic archaea [56–60]. These cases include even those proteins that have orthologs in mesophilic bacteria but, as shown by phylogenetic analysis, have clear affinity to archaea or thermophilic bacteria from distant bacterial lineages, which is indicative of XGD [57]. To investigate the impact of HGT from thermophiles leading to XGD on the evolution of the gene repertoire of TT, we determined the taxonomic affiliations of the proteins from the common gene core of TT and DR. We used the taxonomic distribution of best hits in BLAST searches for preliminary identification of HGT candidates, followed by a detailed phylogenetic analysis of selected genes.
As expected, compared to DR, TT has a notable excess in the fraction of best hits to thermophilic bacteria and archaea for both core and non-core proteins (Figure 5A; and see Additional file 5). However, it has been reported that the best BLAST hit does not always accurately reflect phylogeny [61]. In particular, artifacts of best hit analysis may be caused by similar biases in the amino acid composition of proteins in TT and other thermophiles, as demonstrated previously [62, 63]. To assess these effects systematically, we performed phylogenetic analysis of 112 TT proteins and 21 DR proteins from the common core that had their respective best hits in thermophiles (see Additional file 7). Despite the fact that all these trees were built for families in which TT and DR proteins were not mutual best hits in the non-redundant protein sequence database (National Center for Biotechnology Information, NIH, Bethesda), more than half of the trees (69 trees, 52%) recovered a DR-TT clade, 39 of these grouping this clade with mesophiles and 30 with thermophiles (Figure 5B). Nevertheless, the difference of evolutionary patterns of DR and TT came across clearly in this analysis: a reliable affinity with thermophiles was detected for 18 TT proteins and only one DR protein. The former cases are likely to represent HGT into the TT lineage from other thermophiles, whereas the only "thermophilic" gene of DR may involve the reverse direction of HGT, from the DR lineage to Thermoanaerobacter tencongiensis (data not shown).

Taxonomic affinities of TT and DR proteins. A. Distribution of the numbers of best hits to proteins from thermophiles for core and non-core proteins of DR and TT. B. Distribution of phylogenetic affinities of proteins from the DR-TT core that have best hit to proteins from thermophiles.
Amino acid composition bias is known to affect not only sequence similarity searches but phylogeny reconstruction as well [64]. We tested this effect on our data set by comparing the sequence-based maximum likelihood trees to the neighbor-joining trees reconstructed from the amino acid frequencies of corresponding proteins (see Additional file 1, "Influence of amino acid composition on phylogenetic reconstructions"). We found that, in the majority of cases (>80%), the topology of the sequence-based tree was not congruent with that of the amino acid composition tree; thus, the effect of the amino acid composition on the breakdown shown in Figure 5B is unlikely to be substantial. Taken together, these results suggest that XGD involving genes from thermophiles made a measurable contribution to the evolution of the core gene set of TT after the divergence from the common DR-TT ancestor; no such contribution was detected in the case of DR. While this interpretation seems most plausible given the ecological proximity of TT and other thermophiles, it cannot be ruled out that the observed patterns (Figure 5B) are partially explained by XGD with genes from mesophilic bacteria in the DR lineage. More generally, these results emphasize that the taxonomic distribution of best database hits can be taken only as a rough and preliminary indicator of HGT.
Among the cases of potential XGD supported by phylogenetic analysis, there are two ribosomal proteins, L30 and L15, which are encoded by adjacent genes within a conserved ribosomal operon. The proteins encoded by surrounding genes in these operons showed clear affinity to the corresponding DR orthologs (data not shown). In phylogenetic trees of L30 and L15, the TT proteins reliably clustered with orthologs from bacterial hyperthermophiles and not with the corresponding DR orthologs (Figure 6A and 6B). The congruent evolutionary patterns seen with these two ribosomal proteins encoded by adjacent genes suggest that this gene pair has been replaced via XGD in situ, without disruption of the operon organization [65]. Additional cases of apparent XGD, where DR confidently partitions into the mesophilic clade, whereas TT belongs to the thermophilic clade, are shown in Figure 6C, D (in each of these cases, the thermophilic clade has an admixture of mesophilic species whereas the mesophilic clade includes no thermophiles).

Phylogenetic trees for selected TT genes with apparent XGD involving an ortholog from a thermophilic species. Maximum-likelihood unrooted trees were constructed and bootstrap probabilities were computed using the MOLPHY program. Branches with bootstrap probability >70% are marked by black circles. Each leaf is denoted by the standard gene identifier (for the the complete list of correspondence between genes and species, see Additional file 1: "Gene identifiers and species names for figure 6"). The DR and TT genes are boxed. Genes from thermophilic species are shown in red. A. Ribosomal protein L30. B. Ribosomal protein L15. C. Thiamin biosynthesis protein ThiC. D. tRNA thiolation enzyme TtcA.
The apparent lineage-specific HGT in DR and TT was not limited to XGD or to acquisition of genes from thermophiles. Additional examples of various types of HGT supported by phylogenetic analysis are given in Table 2.
Expanded families of paralogs
Most bacterial lineages contain unique sets of expanded paralogous gene families [66]. This notion was borne out by the present comparative-genomic analysis of DR and TT. None of the expanded families that have been detected during the previous detailed analysis of the DR genome [7] was expanded in TT, and many were missing altogether. This strongly suggests that extensive gene duplication and acquisition of new pseudoparalogs via HGT, which led to the expansion of these families in DR, occurred after the divergence from the common ancestor with TT, and could contribute to the specific adaptations of DR (Table 3). Expansion of several other families in DR was revealed in the course of the present comparison with TT. One notable example is the family of predicted membrane-associated proteins (DR2080, DR1043, DR1952, DR1953, DR1738), which are encoded adjacent to transcriptional regulators of the PadR-like family (COG1695; 9 paralogs in DR, none in TT) (Table 3). PadR-like regulators are involved in the regulation of the cellular response to chemical stress agents, derivatives of phenolic acid [67]. Another previously unnoticed paralogous family that is expanded in DR includes proteins containing the MOSC (MOCO sulfurase C-terminal) domain (COG2258, four paralogs in DR and one in TT). These proteins have been predicted to function as sulfur-carriers that deliver sulfur for the formation of sulfur-metal clusters associated with various enzymes [68]. One of these genes (DR0273) forms a predicted operon with genes for a Nudix hydrolase and a monooxygenase, suggesting that these proteins might comprise a distinct stress response/house-cleaning complex.
Several paralogous families are specifically expanded in TT (Table 4). The largest of these (15 paralogs compared to three in DR) is the Uma2 family that is highly expanded in cyanobacteria but otherwise seen in only a few bacteria. The function(s) of these proteins is unknown; the presence of conserved acidic residues suggests that they might be uncharacterized DNA-binding proteins [69]. The expansion of predicted sugar transporters in TT and the paucity of extracellular proteases (including subtilases) is unexpected because it has been shown that TT is predominantly a proteolytic rather than a saccharolytic organism [70]. However, it should be noted that TT, unlike DR, has not been observed to secrete proteases (data not shown).
Notably, several protein families that are expanded in TT but are absent in DR belong to the set of potential thermophilic determinants (HEPN nucleotide-binding domain; predicted phosphoesterases related to the Icc protein) [17] or are expanded in thermophylic archaea (PIN-like nuclease domain, minimal nucleotidyl transferases, UspA-like nucleotide-binding domain) [71], Table 4). In particular, TT has three paralogs of the archaea-specific tungsten-containing aldehyde ferredoxin oxidoreductase (TTC0012, TTC1834, TTP0122, TTP0212), which is the first occurrenceof this enzyme in thermophilic bacteria. However, these enzymes are present in several mesophilic bacteria, and have various substrate specificities and might be involved in sugar, amino acids or sulfur metabolism [72, 73].
Comparison of DNA repair and stress response systems
Comparative analysis of the well-characterized genetic systems for replication, repair and recombination, and related functions in TT and DR shows that fractions of these genes in the respective genomes are very similar (Table 5; see Additional file 1, Table 2S, 3S). The greatest differences were observed among the proteins associated with direct damage reversal (11 in TT versus 26 in DR), which is due to the extraordinary expansion of the NUDIX (MutT-like) family of hydrolases in DR [7]. It should be noted that the majority of these proteins have other substrates than 7,8-dihydro-8-oxoguanine-triphosphate (or diphosphate), which is cleaved by MutT. Consistently, the majority of the NUDIX proteins appear to be "house cleaning" enzymes rather than bona fide components of repair systems [74]. Other notable differences include the apparent loss of the SOS-response transcriptional repressor LexA [12] and another SOS-response protein, endonuclease VII (XseAB) in the TT lineage; these proteins seem to have been lost also by another thermophilic bacterium, AA. In contrast, DR has two LexA paralogs (DRA0344 and DRA0074), but their functions remain unclear. A genetic disruption of DRA0344, the paralog that shows greater similarity to the canonical bacterial LexA protein, does not result in sensitivity to DNA damage or impairment of RecA expression [75]. Photolyase (PhrB) and endonuclease IV (nfo) are among the few DNA repair proteins that probably were acquired by TT after the divergence from the common ancestor with DR. In addition, the catalytic subunit of DNA polymerase III of TT (DnaE, TTC1806) has two inserted inteins, whereas the orthologous DR0507 has none. In general, it seems that the conventional DNA-repair systems of TT and DR are closely related to each other and to the respective systems of other free-living bacteria. Thus, the unique, shared features of these systems do not explain the very large difference observed in resistance between TT and DR species.
However, a conclusion that there are no important differences between the repair systems of TT and DR might be premature. Recently, several additional proteins of DR have been implicated in DNA or RNA repair, either in direct experiments or on the basis of up-regulation following irradiation, complemented with protein sequence analysis. These putative repair enzymes include DRB0094, an RNA ligase [76] that is strongly up-regulated in response to irradiation [27] and might be involved in an uncharacterized RNA repair process; a predicted double-strand break repair complex specific for recovery after irradiation, which consists of DRB0100, a DNA ligase, DRB0098, a protein containing an HD family phosphatase and polynucleotide kinase domains [7]; DRB0099, a predicted phosphatase of the H2Macro superfamily ([27] and KSM, unpublished observations); a double-stranded DNA-binding protein PprA (DRA0346), which stimulates the DNA end-joining reaction in vitro [77]; a predicted DNA single-strand annealing protein DdrA (DR0423) [78]; a regulator of radiation response IrrE (DR0167); a metal-dependent protease fused to a helix-turn-helix domain [79, 80]; and the uncharacterized protein DR0070 that has been shown to be essential for full resistance to acute irradiation [27]. Among these poorly characterized (predicted) repair proteins of DR, only DdrA has an ortholog in the TT genome. In general, these putative repair genes are sparsely represented in bacteria, and it appears most unlikely that they were present in the common ancestor of TT and DR; most likely, these genes were acquired by the DR lineage via HGT after the divergence from the common ancestor with TT, and might have contributed to the evolution of the resistance phenotype. However, functional relevance of these genes to radiation resistance remains to be confirmed because most of the corresponding knockout mutants showed only relatively small to moderate decreases in radiation resistance [27, 78].
Among the unique (predicted) repair enzymes of TT, the most conspicuous ones are the components of the putative thermophile-specific repair system, which are predominantly encoded on the TT megaplasmid (Figure 7A). The functional features of the proteins encoded in this system (COG1203, a DNA helicase, often fused to a predicted HD-superfamily hydrolase; COG1468, a RecB-family exonuclease; COG1353, predicted polymerase) suggest that they are involved in an as yet uncharacterized DNA repair pathway. It has been hypothesized that this novel gene complex might be functionally analogous to the bacterial-eukaryotic system of translesion, mutagenic repair whose central components are DNA polymerases of the UmuC-DinB-Rad30-Rev1 superfamily, which typically are missing in thermophiles [16].
Comparison of proteins comprising various (predicted) systems involved in stress response reveals a greater number and diversity of such proteins in DR, which has 26 COGs with relevant functions that are not represented in TT compared to 3 such COGs in TT (see Additional file 1, Table 4S). Altogether, there are 147 proteins in DR in this category and 86 in TT, suggesting that some of them are additionally expanded in DR (see the section on "Expanded families").
Enzymatic systems of defense against oxidative stress predicted in TT and DR also show important differences. TT has one Mn-dependent superoxide dismutase (SodA) [81] and one Mn-dependent catalase (with no ortholog in DR), whereas DR encodes three superoxide dismutases (one of which is the ortholog of the SodA gene of TT) and three predicted catalases with no TT orthologs [7]. Additionally, DR has a cytochrome C peroxidase (DRA0301) and a predicted iron-dependent peroxidase (DRA0145), enzymes that are likely to provide protection against toxic peroxides [49]. Orthologs of these enzymes are rare among bacteria, suggesting that the Deinococcus lineage acquired them via HGT after the divergence of TT and DR from the common ancestor. Reduction of oxidized methionine residues in proteins is crucial for survival of cells under oxidative stress [82, 83]. Consistent with this idea, two peptide methionine sulfoxide reductases (PMSRs), MsrA (DR1849) and MsrB (DR1378), are encoded in the DR genome [84, 85], whereas none are present in TT. Interestingly, both PMSRs are also missing in Aquifex, Thermotoga and most thermophilic archaea, suggesting at least two possibilities: either this type of oxidative damage is rare at high temperatures or the known PMSRs are replaced by uncharacterized analogous enzymes due to the inefficiency of the former at high temperatures.
Oxidative stress defense mechanisms also might include control of Mn and Fe partitioning in the cell [29]. Proteins of the Dps/ferritin family are required for the storage of iron in a non-reactive state, which prevents iron-catalyzed formation of hydroxyl radicals, thus protecting the cell from iron toxicity (Fenton-type chemistry) [86]. Two Dps-related proteins are encoded in the DR genome (DR2263, DRB0092, COG1528), and it has been shown that one of them (DR2263) protects DNA from both hydroxyl radical cleavage and from DNase I-mediated cleavage [87]. Some proteins homologous to DPS can non-specifically bind DNA and therefore are viewed as DNA-specific protectors [88]. Like most thermophiles, TT has no proteins of this family but encodes a ferritin from another family (TTC1122).
Since desiccation also causes oxidative stress, proteins involved in desiccation resistance belong to the general cellular defense category [89]. Desiccation-related proteins from at least two distinct families have been detected in DR [7]. These desiccation resistance protein families (Lea 76 family, DR0105 and DR1172; and Lea14 family, DR1372) are not represented in TT. However, three TT proteins (TTP0170, TTP0166, TTP0169) are homologs of another desiccation resistance protein that was originally characterized in a plant, Craterostigma plantagineum [90]; DR also has two proteins of this family, DRB0118 and DRA0258. These proteins are distantly related to COG1633 and belong to the ferritin family of iron storage proteins (KSM, unpublished). Highly conserved homologs of these proteins are also present in thermophilic bacteria and archaea. Two desiccation-related proteins (DR1172 and DRB0118) appear to be essential for desiccation resistance but not for radiation resistance in DR [53].
Comparison of the genome partitions of TT and DR
Both TT and DR have multipartite genomes. To examine possible evolutionary relationships between the genome partitions of TT and DR, we analyzed the distribution of symmetrical best hits (putative orthologs) in the single extra-chromosomal element of TT, the pTT27 megaplasmid, and the three smaller genome partitions of DR (small chromosome, DR412; megaplasmid, DR177 and plasmid, CP1; Table 6). The results of this analysis show that pTT27 has a highly significant excess of orthologs on DR177, suggesting that these two megaplasmids are homologous, i.e., probably evolved from a distinct genome partition of the common DR-TT ancestor. Apparently, however, the genomes of the megaplasmids have undergone extensive rearrangements since the divergence from the common ancestor because no conservation of gene order could be identified (data not shown).
Notably, among the putative thermophilic determinants of TT, ~50% are encoded on the megaplasmid (18 out of 36). Of these, 11 belong to the putative mobile thermophile-specific DNA repair system [16, 45] (Figure 7A). Additionally, the megaplasmid carries at least four other genes associated with this system, which have not yet been assigned to COGs (Figure 7A). Moreover, the TT megaplasmid also carries a pseudogene for reverse gyrase, the most conspicuous signature protein of hyperthermophiles [15, 17].
Recently, the genome of another strain of TT (HB8) has been completely sequenced and became available in public databases [13]. A preliminary comparison of the two TT strains (HB27 and HB8) revealed considerable differences in the gene orders and contents of the megaplasmids (see Additional file 1, Figure 3S). Interestingly, these differences in gene content are derived mostly from genes that appear to be associated with the thermophylic lifestyle. In particular, strain HB8 encodes an intact reverse gyrase. Thus, it appears most likely that the gene for reverse gyrase was acquired from a hyperthermophilic source by the TT lineage and was present in the common ancestor of HB27 and HB8 but decayed in the former. Conversely, HB27 encodes a unique, three-domain fusion protein (DnaQ endonuclease, DinG helicase and RecQ helicase), whereas HB8 lacks the DinG and RecQ orthologs (Figure 7B). Furthermore, there are unexpected differences in the organization of predicted thermophile-specific repair systems between the two strains of TT. Specifically, HB27 contains a "gram-positive version" (TTP0132-TTP0136), whereas HB8 has a "proteobacterial version" of these genes (TTHB186-TTHB194) (Figure 7A).
Furthermore, a nearly complete draft genome sequence of Deinococcus geothermalis (DG) has recently become publicly available [91]. Since DG is closely related to DR but is moderately thermophilic, we searched for "thermophilic" genes in DG genome. Using the "thermophilic" protein sequences ([17] and see above) of TT and DR as queries, we identified orthologs of 5 of the 6 DR proteins from this set (four of these are also present in both TT strains) and orthologs of 7 of the remaining 23 "thermophilic" proteins of TT (all components of the predicted thermophilic DNA repair system). These 7 proteins had the respective TT proteins as the best hits, and their monophyly was supported by phylogenetic analysis (data not shown), suggesting that these genes were already present in the genome of the DR-TT common ancestor.
These observations give rise to two hypotheses: (i) the TT megaplasmid is essential for the survival of the organism at high temperatures. Consistent with this idea, we detected an expansion of a two-component toxin-antitoxin system, which consists of a PIN-like nuclease (toxin) and a MazE family transcriptional regulator (antitoxin) [69]. Such a system is known to be responsible for the segregational stability of antibiotic resistance plasmids and other plasmids via selective elimination of cells that have failed to acquire a plasmid copy [92], and/or exclusion of competing plasmids [93]; and (ii) the TT megaplasmid is a dynamic genome compartment and a veritable sink for horizontally transferred genes, some of which might affect the thermophilic phenotype of this bacterium. This is compatible with the considerable differences in gene content observed between the two TT strains.
Specific roles of plasmid-borne genes in recovery from DNA damage have been proposed previously, including class Ib ribonucleotide reductase, periplasmic alkaline phosphatase, and extracellular nuclease, and subsequent analyses revealed at least five other genes implicated in this process (two operons, DRB0098-DRB0100, DRB0094-DRB0084, see above in "Comparison of DNA repair and stress response systems") [27, 76]. There is also a toxin-antitoxin system operon in the DR177 megaplasmid (DRB0012a and another gene located immediately upstream of DRB0012a and currently absent from the genome annotation). Additionally, there are five other toxin-antitoxin systems encoded on DR412, the smaller chromosome of DR, which might be related to maintenance of DR177 megaplasmid in DR. The DR412 chromosome appears to have some special features as well. Numerous genes that apparently have been acquired by DR via HGT after the divergence from the common ancestor with TT and are implicated in various processes of amino acid and nucleotide degradation, map to this genome partition. Thus, the megaplasmids of TT and DR (and the smaller chromosome of DR, DR412) appear to have participated in extensive HGT, which might have been important for the evolution of thermophily and radioresistance, although the repertoires of the respective acquired genes are completely different.
Conclusion
TT and DR share a large core of genes and form a clade in the gene-content tree, which supports the idea that these bacteria form a distinct clade, as indicated previously by phylogenetic analysis of rRNA and various proteins, and that the evolution of their gene complements was, roughly, clock-like. However, major differences between the gene repertoires of TT and DR were observed, indicating that both genomes lost numerous ancestral genes and acquired distinct sets of new genes primarily via HGT. In addition, numerous lineage-specific expansions of paralogous gene families were identified, particularly, in DR.
Some of the differences in the gene repertoires of TT and DR can be linked to the distinctive adaptive strategies of these bacteria. For example, TT appears to have acquired many genes from (hyper)thermophilic bacteria and archaea, whereas DR apparently acquired various genes involved in oxidative stress response and other "house-cleaning" functions from diverse bacterial sources.
The gene content of the TT megaplasmid (pTT27) and the DR megaplasmid (DR177) are sufficiently similar to conclude that they evolved from a common ancestor. To our knowledge, this is the first evidence of persistence of a megaplasmid beyond the genus level. However, the TT megaplasmid also carries many genes whose functions are implicated in the thermophylic phenotype; in particular, components of the predicted thermophile-specific repair system. These megaplasmids are likely to be essential for the survival of both TT and DR, with their maintenance controlled by toxin-antitoxin systems. Furthermore, the substantial differences between the gene repertoires of the megaplasmids of TT strains HB27 and HB8 indicate that this genome partition has been highly dynamic, with high rates of gene loss and HGT events occurring during evolution.
The evolutionary reconstruction based on the parsimony principle is generally compatible with the idea that the common ancestor of TT and DR was a mesophilic bacterium, whereas the thermophylic phenotype of TT evolved gradually via HGT of genes from thermophiles. Conversely, the radiation-desiccation resistance phenotype of DR might have gradually evolved via HGT of genes from other mesophiles, particularly, with highly developed oxidative stress response systems. However, it should be noted that the TT-DR common gene core includes dozens of genes of apparent archaeal origin or, at least, genes with thermophilic affiliation. Moreover, the DG genome encodes a few additional "thermophilic determinants" that are missing in DR but are unlikely to have been transferred from a thermophilic source independently of TT, as shown by comparative-genomic and phylogenetic analyses described here. Thus, acquisition of a considerable number of archaeal genes might have occurred along the evolutionary branch leading to the common ancestor of TT and DR. Accordingly, at this stage, we cannot rule out the possibility that this ancestor was a moderate thermophile rather than a mesophile. Further sequencing of bacterial genomes of the Thermus-Deinococcus clade should allow more definitive comparative-genomic analysis to elucidate the nature of the common ancestor of these bacteria.
Methods
Irradiation and desiccation
Irradiations. Three TT colony-isolates were inoculated individually in liquid TGY (10 g/L Bactotryptone, 1 g/L glucose, 5 g/L yeast extract) and incubated at 70°C. Cells were harvested at OD600 ~0.9, which corresponds to 107 – 108 colony forming units (CFU)/ml; 1 TT cell/CFU. TT cells grown in TGY were examined for their total Mn and Fe content by ICP-MS (see main text). For radiation resistance assays, cells were irradiated without change of broth on ice with 60Co at 6.8 kGy/hour (60Co Gammacell irradiation unit [J. L. Shepard and Associates, Model 109]). At the indicated doses, cultures (3 biological replicates) were appropriately diluted and plated on solid medium (8 g/L Bactotryptone, 4 g/L yeast extract, 3 g/L NaCl, pH 7.3, 2.8% Bactoagar), and CFU counts were determined after 2 days' incubation at 65°C.
Desiccation. Five separate colony-isolates were pre-grown in TGY as for irradiation trials. Cell samples 106–107 cells (25 μl) were transferred to microtiter plates, which were transferred to desiccation chambers containing anhydrous calcium sulfate (drierite) and incubated at room temperature or 65°C. At the indicated times, cells were re-suspended in TGY, and CFU-survival frequencies were determined by dilution-plating on solid medium (65°C).
Genome analysis
The sets of predicted proteins of TT and DR were searched against each other for symmetrical and non-symmetrical hits using PSI-BLAST with expectation (E) value threshold of 10-5. Taxonomic affiliation was determined by best hits in non-redundant database of protein sequences at the National Center of Biotechnology Information (NIH, Bethesda) using BLASTP program [94] with default expectation value (0.01). Assignments to COGs were performed using the COGNITOR program [95] and CDD-search against COG-based profiles [96]. Contradictory assignments were resolved manually. Lineage-specific expansions (LSE) were identified as described previously [66, 97]. The common genomic core was determined as follows: among genes that were not assigned to any COG, orthology relationships between TT and DR were determined via symmetrical best hits. Genes belonging to COGs, shared between TT and DR and having only one ortholog from each of the two genomes were directly assigned to the core. For multiple-paralog COGs, symmetrical best hits between GOG members were used to refine the relationships between TT and DR proteins. Members of the corresponding lineage-specific expansions were added to SymBeT pairs to form many-to-many core clusters. LBCA gene set was determined using an empirical parsimony procedure based on COG phyletic patterns (See Additional file 1: "Reconstruction of the gene set of the Last Bacterial Common Ancestor") which assigned a COG to LBCA if it was present in several diverse bacterial clades. All COGs that were present in LBCA and in TT and/or DR were assigned to the DR-TT ancestor.
Phylogenetic analysis
Multiple alignments for phylogenetic analysis were constructed using the MUSCLE program [98]; columns containing gaps in >30% of the sequences were discarded. Maximum likelihood trees were constructed using the ProtML program of the MOLPHY package by optimizing the least-square trees with local rearrangements [99]. Trees based on amino acid content were constructed from the matrix of Euclidean distances between frequency vectors using the NEIGHBOR program of the PHYLIP package [100]. Support for particular arrangements of species (relationships between DR, TT, thermophiles and mesophiles) was calculated using the bipartition analysis of bootstrap samples from original sequences (see Additional file 1, "Influence of amino acid composition on phylogenetic reconstructions"). The gene-content tree based on COG patterns was constructed using the NEIGHBOR program of the PHYLIP package [100]; the number of COGs shared between two genomes was normalized by the smaller genome size [33].
References
Weisburg WG, Giovannoni SJ, Woese CR: The Deinococcus-Thermus phylum and the effect of rRNA composition on phylogenetic tree construction. Syst Appl Microbiol. 1989, 11: 128-134.
Eisen JA: The RecA protein as a model molecule for molecular systematic studies of bacteria: comparison of trees of RecAs and 16S rRNAs from the same species. J Mol Evol. 1995, 41 (6): 1105-1123. 10.1007/BF00173192.
Rainey FA, Nobre MF, Schumann P, Stackebrandt E, da Costa MS: Phylogenetic diversity of the deinococci as determined by 16S ribosomal DNA sequence comparison. Int J Syst Bacteriol. 1997, 47 (2): 510-514.
Gupta RS, Bustard K, Falah M, Singh D: Sequencing of heat shock protein 70 (DnaK) homologs from Deinococcus proteolyticus and Thermomicrobium roseum and their integration in a protein-based phylogeny of prokaryotes. J Bacteriol. 1997, 179 (2): 345-357.
Gupta RS: Protein phylogenies and signature sequences: A reappraisal of evolutionary relationships among archaebacteria, eubacteria, and eukaryotes. Microbiol Mol Biol Rev. 1998, 62 (4): 1435-1491.
Huang YP, Ito J: DNA polymerase C of the thermophilic bacterium Thermus aquaticus: classification and phylogenetic analysis of the family C DNA polymerases. J Mol Evol. 1999, 48 (6): 756-769.
Makarova KS, Aravind L, Wolf YI, Tatusov RL, Minton KW, Koonin EV, Daly MJ: Genome of the extremely radiation-resistant bacterium Deinococcus radiodurans viewed from the perspective of comparative genomics. Microbiol Mol Biol Rev. 2001, 65 (1): 44-79. 10.1128/MMBR.65.1.44-79.2001.
Battista JR, Earl AM, Park MJ: Why is Deinococcus radiodurans so resistant to ionizing radiation?. Trends Microbiol. 1999, 7 (9): 362-365. 10.1016/S0966-842X(99)01566-8.
Hirsch P, Gallikowski CA, Siebert J, Peissl K, Kroppenstedt R, Schumann P, Stackebrandt E, Anderson R: Deinococcus frigens sp. nov., Deinococcus saxicola sp. nov., and Deinococcus marmoris sp. nov., low temperature and draught-tolerating, UV-resistant bacteria from continental Antarctica. Syst Appl Microbiol. 2004, 27 (6): 636-645. 10.1078/0723202042370008.
Ferreira AC, Nobre MF, Rainey FA, Silva MT, Wait R, Burghardt J, Chung AP, da Costa MS: Deinococcus geothermalis sp. nov. and Deinococcus murrayi sp. nov., two extremely radiation-resistant and slightly thermophilic species from hot springs. Int J Syst Bacteriol. 1997, 47 (4): 939-947.
Holt JG, Krieg NR, Sneath PH, Staley JT, Williams ST: Bergy's Manual of Determinative Bacteriology. 1997, London , Williams@Wilkins, 9
Henne A, Bruggemann H, Raasch C, Wiezer A, Hartsch T, Liesegang H, Johann A, Lienard T, Gohl O, Martinez-Arias R, Jacobi C, Starkuviene V, Schlenczeck S, Dencker S, Huber R, Klenk HP, Kramer W, Merkl R, Gottschalk G, Fritz HJ: The genome sequence of the extreme thermophile Thermus thermophilus. Nat Biotechnol. 2004, 22 (5): 547-553. 10.1038/nbt956.
NCBI, Thermus thermophilus HB8, complete genome. [http://www.ncbi.nlm.nih.gov/genomes/framik.cgi?db=Genome&gi=530]
White O, Eisen JA, Heidelberg JF, Hickey EK, Peterson JD, Dodson RJ, Haft DH, Gwinn ML, Nelson WC, Richardson DL, Moffat KS, Qin H, Jiang L, Pamphile W, Crosby M, Shen M, Vamathevan JJ, Lam P, McDonald L, Utterback T, Zalewski C, Makarova KS, Aravind L, Daly MJ, Fraser CM: Genome sequence of the radioresistant bacterium Deinococcus radiodurans R1. Science. 1999, 286 (5444): 1571-1577. 10.1126/science.286.5444.1571.
Forterre P: A hot story from comparative genomics: reverse gyrase is the only hyperthermophile-specific protein. Trends Genet. 2002, 18 (5): 236-237. 10.1016/S0168-9525(02)02650-1.
Makarova KS, Aravind L, Grishin NV, Rogozin IB, Koonin EV: A DNA repair system specific for thermophilic Archaea and bacteria predicted by genomic context analysis. Nucleic Acids Res. 2002, 30 (2): 482-496. 10.1093/nar/30.2.482.
Makarova KS, Wolf YI, Koonin EV: Potential genomic determinants of hyperthermophily. Trends Genet. 2003, 19 (4): 172-176. 10.1016/S0168-9525(03)00047-7.
McDonald JH, Grasso AM, Rejto LK: Patterns of temperature adaptation in proteins from Methanococcus and Bacillus. Mol Biol Evol. 1999, 16 (12): 1785-1790.
Haney PJ, Badger JH, Buldak GL, Reich CI, Woese CR, Olsen GJ: Thermal adaptation analyzed by comparison of protein sequences from mesophilic and extremely thermophilic Methanococcus species. Proc Natl Acad Sci U S A. 1999, 96 (7): 3578-3583. 10.1073/pnas.96.7.3578.
Thompson MJ, Eisenberg D: Transproteomic evidence of a loop-deletion mechanism for enhancing protein thermostability. J Mol Biol. 1999, 290 (2): 595-604. 10.1006/jmbi.1999.2889.
Sterner R, Liebl W: Thermophilic adaptation of proteins. Crit Rev Biochem Mol Biol. 2001, 36 (1): 39-106. 10.1080/20014091074174.
Chakravarty S, Varadarajan R: Elucidation of factors responsible for enhanced thermal stability of proteins: a structural genomics based study. Biochemistry. 2002, 41 (25): 8152-8161. 10.1021/bi025523t.
Vieille C, Zeikus GJ: Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol Mol Biol Rev. 2001, 65 (1): 1-43. 10.1128/MMBR.65.1.1-43.2001.
Mallick P, Boutz DR, Eisenberg D, Yeates TO: Genomic evidence that the intracellular proteins of archaeal microbes contain disulfide bonds. Proc Natl Acad Sci U S A. 2002, 99 (15): 9679-9684. 10.1073/pnas.142310499.
Hickey DA, Singer GA: Genomic and proteomic adaptations to growth at high temperature. Genome Biol. 2004, 5 (10): 117-10.1186/gb-2004-5-10-117.
Tanaka M, Earl AM, Howell HA, Park MJ, Eisen JA, Peterson SN, Battista JR: Analysis of Deinococcus radiodurans's transcriptional response to ionizing radiation and desiccation reveals novel proteins that contribute to extreme radioresistance. Genetics. 2004, 168 (1): 21-33. 10.1534/genetics.104.029249.
Liu Y, Zhou J, Omelchenko MV, Beliaev AS, Venkateswaran A, Stair J, Wu L, Thompson DK, Xu D, Rogozin IB, Gaidamakova EK, Zhai M, Makarova KS, Koonin EV, Daly MJ: Transcriptome dynamics of Deinococcus radiodurans recovering from ionizing radiation. Proc Natl Acad Sci U S A. 2003, 100 (7): 4191-4196. 10.1073/pnas.0630387100.
Ghosal D, Omelchenko MV, Gaidamakova EK, Matrosova VY, Vasilenko A, Venkateswaran A, Zhai M, Kostandarithes HM, Brim H, Makarova K, Wackett LP, Fredrickson JK, Daly MJ: How radiation kills cells: Survival of Deinococcus radiodurans and Shewanella oneidensis under oxidative stress. FEMS Microbiol Lett. 2005,
Daly MJ, Gaidamakova EK, Matrosova VY, Vasilenko A, Zhai M, Venkateswaran A, Hess M, Omelchenko MV, Kostandarithes HM, Makarova KS, Wackett LP, Fredrickson JK, Ghosal D: Accumulation of Mn(II) in Deinococcus radiodurans facilitates gamma-radiation resistance. Science. 2004, 306 (5698): 1025-1028. 10.1126/science.1103185.
Battista JR: Against all odds: the survival strategies of Deinococcus radiodurans. Annu Rev Microbiol. 1997, 51: 203-224. 10.1146/annurev.micro.51.1.203.
Archibald FS, Fridovich I: . Arch Biochem Biophys. 1982, 214: 452-10.1016/0003-9861(82)90049-2.
Lee Y, Lee C, Yoon J: High temperature dependence of 2,4-dichlorophenoxyacetic acid degradation by Fe3+/H(2)O(2) system. Chemosphere. 2003, 51 (9): 963-971. 10.1016/S0045-6535(03)00043-2.
Snel B, Bork P, Huynen MA: Genome phylogeny based on gene content. Nat Genet. 1999, 21 (1): 108-110. 10.1038/5052.
Wolf YI, Rogozin IB, Grishin NV, Koonin EV: Genome trees and the tree of life. Trends Genet. 2002, 18 (9): 472-479. 10.1016/S0168-9525(02)02744-0.
Wolf YI, Rogozin IB, Grishin NV, Tatusov RL, Koonin EV: Genome trees constructed using five different approaches suggest new major bacterial clades. BMC Evol Biol. 2001, 1 (1): 8-10.1186/1471-2148-1-8.
Korbel JO, Snel B, Huynen MA, Bork P: SHOT: a web server for the construction of genome phylogenies. Trends Genet. 2002, 18 (3): 158-162. 10.1016/S0168-9525(01)02597-5.
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA: The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003, 4 (1): 41-10.1186/1471-2105-4-41.
Phillips RW, Wiegel J, Berry CJ, Fliermans C, Peacock AD, White DC, Shimkets LJ: Kineococcus radiotolerans sp. nov., a radiation-resistant, gram-positive bacterium. Int J Syst Evol Microbiol. 2002, 52 (Pt 3): 933-938. 10.1099/ijs.0.02029-0.
Ferreira AC, Nobre MF, Moore E, Rainey FA, Battista JR, da Costa MS: Characterization and radiation resistance of new isolates of Rubrobacter radiotolerans and Rubrobacter xylanophilus. Extremophiles. 1999, 3 (4): 235-238. 10.1007/s007920050121.
Billi D, Friedmann EI, Hofer KG, Caiola MG, Ocampo-Friedmann R: Ionizing-radiation resistance in the desiccation-tolerant cyanobacterium Chroococcidiopsis. Appl Environ Microbiol. 2000, 66 (4): 1489-1492. 10.1128/AEM.66.4.1489-1492.2000.
Mirkin BG, Fenner TI, Galperin MY, Koonin EV: Algorithms for computing parsimonious evolutionary scenarios for genome evolution, the last universal common ancestor and dominance of horizontal gene transfer in the evolution of prokaryotes. BMC Evol Biol. 2003, 3 (1): 2-10.1186/1471-2148-3-2.
Olendzenski L, Liu L, Zhaxybayeva O, Murphey R, Shin DG, Gogarten JP: Horizontal transfer of archaeal genes into the deinococcaceae: detection by molecular and computer-based approaches. J Mol Evol. 2000, 51 (6): 587-599.
Levin I, Giladi M, Altman-Price N, Ortenberg R, Mevarech M: An alternative pathway for reduced folate biosynthesis in bacteria and halophilic archaea. Mol Microbiol. 2004, 54 (5): 1307-1318. 10.1111/j.1365-2958.2004.04339.x.
Mittenhuber G: Phylogenetic analyses and comparative genomics of vitamin B6 (pyridoxine) and pyridoxal phosphate biosynthesis pathways. J Mol Microbiol Biotechnol. 2001, 3 (1): 1-20.
Jansen R, Embden JD, Gaastra W, Schouls LM: Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 2002, 43 (6): 1565-1575. 10.1046/j.1365-2958.2002.02839.x.
Sauer U, Canonaco F, Heri S, Perrenoud A, Fischer E: The soluble and membrane-bound transhydrogenases UdhA and PntAB have divergent functions in NADPH metabolism of Escherichia coli. J Biol Chem. 2004, 279 (8): 6613-6619. 10.1074/jbc.M311657200.
Venkateswaran A, McFarlan SC, Ghosal D, Minton KW, Vasilenko A, Makarova K, Wackett LP, Daly MJ: Physiologic determinants of radiation resistance in Deinococcus radiodurans. Appl Environ Microbiol. 2000, 66 (6): 2620-2626. 10.1128/AEM.66.6.2620-2626.2000.
Ferry JG: CO dehydrogenase. Annu Rev Microbiol. 1995, 49: 305-333. 10.1146/annurev.mi.49.100195.001513.
Imlay JA: Pathways of oxidative damage. Annu Rev Microbiol. 2003, 57: 395-418. 10.1146/annurev.micro.57.030502.090938.
Zhulin IB, Nikolskaya AN, Galperin MY: Common extracellular sensory domains in transmembrane receptors for diverse signal transduction pathways in bacteria and archaea. J Bacteriol. 2003, 185 (1): 285-294. 10.1128/JB.185.1.285-294.2003.
Melkonyan HS, Chang WC, Shapiro JP, Mahadevappa M, Fitzpatrick PA, Kiefer MC, Tomei LD, Umansky SR: SARPs: a family of secreted apoptosis-related proteins. Proc Natl Acad Sci U S A. 1997, 94 (25): 13636-13641. 10.1073/pnas.94.25.13636.
Davis SJ, Vener AV, Vierstra RD: Bacteriophytochromes: phytochrome-like photoreceptors from nonphotosynthetic eubacteria. Science. 1999, 286 (5449): 2517-2520. 10.1126/science.286.5449.2517.
Battista JR, Park MJ, McLemore AE: Inactivation of two homologues of proteins presumed to be involved in the desiccation tolerance of plants sensitizes Deinococcus radiodurans R1 to desiccation. Cryobiology. 2001, 43 (2): 133-139. 10.1006/cryo.2001.2357.
Chen X, Quinn AM, Wolin SL: Ro ribonucleoproteins contribute to the resistance of Deinococcus radiodurans to ultraviolet irradiation. Genes Dev. 2000, 14 (7): 777-782.
Krogh BO, Shuman S: A poxvirus-like type IB topoisomerase family in bacteria. Proc Natl Acad Sci U S A. 2002, 99 (4): 1853-1858. 10.1073/pnas.032613199.
Koonin EV, Makarova KS: Archaeal genomics: how much have we learned in six years and what's next?. Genome Biol. 2003, in press:
Koonin EV, Makarova KS, Aravind L.: Horizontal gene transfer in prokaryotes - quantification and classification. AnnuRevMicrobiol. 2001
Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Nelson WC, Ketchum KA, McDonald L, Utterback TR, Malek JA, Linher KD, Garrett MM, Stewart AM, Cotton MD, Pratt MS, Phillips CA, Richardson D, Heidelberg J, Sutton GG, Fleischmann RD, Eisen JA, Fraser CM: Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature. 1999, 399 (6734): 323-329. 10.1038/20601.
Aravind L, Tatusov RL, Wolf YI, Walker DR, Koonin EV: Evidence for massive gene exchange between archaeal and bacterial hyperthermophiles. Trends Genet. 1998, 14 (11): 442-444. 10.1016/S0168-9525(98)01553-4.
Worning P, Jensen LJ, Nelson KE, Brunak S, Ussery DW: Structural analysis of DNA sequence: evidence for lateral gene transfer in Thermotoga maritima. Nucleic Acids Res. 2000, 28 (3): 706-709. 10.1093/nar/28.3.706.
Koski LB, Golding GB: The closest BLAST hit is often not the nearest neighbor. J Mol Evol. 2001, 52 (6): 540-542.
Cambillau C, Claverie JM: Structural and genomic correlates of hyperthermostability. J Biol Chem. 2000, 275 (42): 32383-32386. 10.1074/jbc.C000497200.
McDonald JH: Patterns of temperature adaptation in proteins from the bacteria Deinococcus radiodurans and Thermus thermophilus. Mol Biol Evol. 2001, 18 (5): 741-749.
Felsenstein J: Inferring phylogenies. 2004, Sunderland, MA , Sinauer Associates, Inc.
Omelchenko MV, Makarova KS, Wolf YI, Rogozin IB, Koonin EV: Evolution of mosaic operons by horizontal gene transfer and gene displacement in situ. Genome Biol. 2003, 4 (9): R55-10.1186/gb-2003-4-9-r55.
Jordan IK, Makarova KS, Wolf YI, Koonin EV: Gene conversions in genes encoding outer-membrane proteins in H. pylori and C. pneumoniae. Trends Genet. 2001, 17 (1): 7-10. 10.1016/S0168-9525(00)02151-X.
Gury J, Barthelmebs L, Tran NP, Divies C, Cavin JF: Cloning, deletion, and characterization of PadR, the transcriptional repressor of the phenolic acid decarboxylase-encoding padA gene of Lactobacillus plantarum. Appl Environ Microbiol. 2004, 70 (4): 2146-2153. 10.1128/AEM.70.4.2146-2153.2004.
Anantharaman V, Aravind L: MOSC domains: ancient, predicted sulfur-carrier domains, present in diverse metal-sulfur cluster biosynthesis proteins including Molybdenum cofactor sulfurases. FEMS Microbiol Lett. 2002, 207 (1): 55-61. 10.1016/S0378-1097(01)00515-8.
Anantharaman V, Aravind L: New connections in the prokaryotic toxin-antitoxin network: relationship with the eukaryotic nonsense-mediated RNA decay system. Genome Biol. 2003, 4 (12): R81-10.1186/gb-2003-4-12-r81.
Oshima T: [Comparative studies on biochemical properties of an extreme thermophile, Thermus thermophilus HB 8 (author's transl)]. Seikagaku. 1974, 46 (10): 887-907.
Makarova KS, Aravind L, Galperin MY, Grishin NV, Tatusov RL, Wolf YI, Koonin EV: Comparative genomics of the Archaea (Euryarchaeota): evolution of conserved protein families, the stable core, and the variable shell. Genome Res. 1999, 9 (7): 608-628.
Roy R, Menon AL, Adams MW: Aldehyde oxidoreductases from Pyrococcus furiosus. Methods Enzymol. 2001, 331: 132-144.
Roy R, Adams MW: Characterization of a fourth tungsten-containing enzyme from the hyperthermophilic archaeon Pyrococcus furiosus. J Bacteriol. 2002, 184 (24): 6952-6956. 10.1128/JB.184.24.6952-6956.2002.
Xu W, Shen J, Dunn CA, Desai S, Bessman MJ: The Nudix hydrolases of Deinococcus radiodurans. Mol Microbiol. 2001, 39 (2): 286-290. 10.1046/j.1365-2958.2001.02267.x.
Narumi I, Satoh K, Kikuchi M, Funayama T, Yanagisawa T, Kobayashi Y, Watanabe H, Yamamoto K: The LexA protein from Deinococcus radiodurans is not involved in RecA induction following gamma irradiation. J Bacteriol. 2001, 183 (23): 6951-6956. 10.1128/JB.183.23.6951-6956.2001.
Martins A, Shuman S: An RNA ligase from Deinococcus radiodurans. J Biol Chem. 2004, 279 (49): 50654-50661. 10.1074/jbc.M407657200.
Narumi I, Satoh K, Cui S, Funayama T, Kitayama S, Watanabe H: PprA: a novel protein from Deinococcus radiodurans that stimulates DNA ligation. Mol Microbiol. 2004, 54 (1): 278-285. 10.1111/j.1365-2958.2004.04272.x.
Harris DR, Tanaka M, Saveliev SV, Jolivet E, Earl AM, Cox MM, Battista JR: Preserving genome integrity: the DdrA protein of Deinococcus radiodurans R1. PLoS Biol. 2004, 2 (10): e304-10.1371/journal.pbio.0020304.
Earl AM, Mohundro MM, Mian IS, Battista JR: The IrrE protein of Deinococcus radiodurans R1 is a novel regulator of recA expression. J Bacteriol. 2002, 184 (22): 6216-6224. 10.1128/JB.184.22.6216-6224.2002.
Hua Y, Narumi I, Gao G, Tian B, Satoh K, Kitayama S, Shen B: PprI: a general switch responsible for extreme radioresistance of Deinococcus radiodurans. Biochem Biophys Res Commun. 2003, 306 (2): 354-360. 10.1016/S0006-291X(03)00965-3.
Ludwig ML, Metzger AL, Pattridge KA, Stallings WC: Manganese superoxide dismutase from Thermus thermophilus. A structural model refined at 1.8 A resolution. J Mol Biol. 1991, 219 (2): 335-358. 10.1016/0022-2836(91)90569-R.
Moskovitz J: Methionine sulfoxide reductases: ubiquitous enzymes involved in antioxidant defense, protein regulation, and prevention of aging-associated diseases. Biochim Biophys Acta. 2005, 1703 (2): 213-219.
Morgan PE, Dean RT, Davies MJ: Protective mechanisms against peptide and protein peroxides generated by singlet oxygen. Free Radic Biol Med. 2004, 36 (4): 484-496. 10.1016/j.freeradbiomed.2003.11.021.
Moskovitz J, Weissbach H, Brot N: Cloning the expression of a mammalian gene involved in the reduction of methionine sulfoxide residues in proteins. Proc Natl Acad Sci U S A. 1996, 93 (5): 2095-2099. 10.1073/pnas.93.5.2095.
Grimaud R, Ezraty B, Mitchell JK, Lafitte D, Briand C, Derrick PJ, Barras F: Repair of oxidized proteins. Identification of a new methionine sulfoxide reductase. J Biol Chem. 2001, 276 (52): 48915-48920. 10.1074/jbc.M105509200.
van Vliet AH, Ketley JM, Park SF, Penn CW: The role of iron in Campylobacter gene regulation, metabolism and oxidative stress defense. FEMS Microbiol Rev. 2002, 26 (2): 173-186. 10.1016/S0168-6445(02)00095-5.
Grove A, Wilkinson SP: Differential DNA Binding and Protection by Dimeric and Dodecameric forms of the Ferritin Homolog Dps from Deinococcus radiodurans. J Mol Biol. 2005, 347 (3): 495-508. 10.1016/j.jmb.2005.01.055.
Grant RA, Filman DJ, Finkel SE, Kolter R, Hogle JM: The crystal structure of Dps, a ferritin homolog that binds and protects DNA. Nat Struct Biol. 1998, 5 (4): 294-303. 10.1038/nsb0498-294.
Mattimore V, Battista JR: Radioresistance of Deinococcus radiodurans: functions necessary to survive ionizing radiation are also necessary to survive prolonged desiccation. J Bacteriol. 1996, 178 (3): 633-637.
Piatkowski D, Schneider K, Salamini F, Bartels D: Characterization of five abscisic acid-responsive cDNA clones isolated from the desiccation-tolerant plant Craterostigma plantagineum and their relationship to other water-stress genes. Plant Physiol. 1990, 94 (1682-88):
JGI, Deinococcus geothermalis draft genome. [http://genome.jgi-psf.org/draft_microbes/deige/deige.home.html]. [http://genome.jgi-psf.org/draft_microbes/deige/deige.home.html]
Grady R, Hayes F: Axe-Txe, a broad-spectrum proteic toxin-antitoxin system specified by a multidrug-resistant, clinical isolate of Enterococcus faecium. Mol Microbiol. 2003, 47 (5): 1419-1432. 10.1046/j.1365-2958.2003.03387.x.
Cooper TF, Heinemann JA: Postsegregational killing does not increase plasmid stability but acts to mediate the exclusion of competing plasmids. Proc Natl Acad Sci U S A. 2000, 97 (23): 12643-12648. 10.1073/pnas.220077897.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25 (17): 3389-3402. 10.1093/nar/25.17.3389.
Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV: The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001, 29 (1): 22-28. 10.1093/nar/29.1.22.
Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Marchler GH, Mullokandov M, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Yamashita RA, Yin JJ, Zhang D, Bryant SH: CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res. 2005, 33 Database Issue: D192-6.
Lespinet O, Wolf YI, Koonin EV, Aravind L: The role of lineage-specific gene family expansion in the evolution of eukaryotes. Genome Res. 2002, 12 (7): 1048-1059. 10.1101/gr.174302.
Edgar RC: MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004, 5 (1): 113-10.1186/1471-2105-5-113.
Hasegawa M, Kishino H, Saitou N: On the maximum likelihood method in molecular phylogenetics. J Mol Evol. 1991, 32 (5): 443-445. 10.1007/BF02101285.
Felsenstein J: Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods. Methods Enzymol. 1996, 266: 418-427.
Acknowledgements
This research was partially supported by the Office of Science (Biological and Environmental Research), U. S. Department of Energy (DOE) grant number DE-FG02-04ER63918 awarded to MJD. We are grateful to James K. Fredrickson and Heather M. Kostandarithes at Pacific Northwest National Laboratory for Fe and Mn ICP-MS analyses of T. thermophilus.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
MVO performed the majority of genomic comparisons, phylogenetic analysis and helped to draft the manuscript. YIW and KSM performed some genomic comparisons and statistical analysis of the data. EKG, VYM, AV and MZ performed the experiments on the comparison of the Thermus and Deinococcus response to irradiation and desiccation and the analysis of Mn/Fe ratio of Thermus. MJD, EVK, and KSM participated in design of experiments and project coordination, and contributed to the writing of the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12862_2005_169_MOESM1_ESM.doc
Additional File 1: (contains supplementary text and supplementary tables 1S, 2S, 3S, 4S and supplementary figures 1S, 2S, 3S) (DOC 659 KB)
12862_2005_169_MOESM3_ESM.xls
Additional File 3: contains DR and TT protein comparison with COGs predicted to be associated with the thermophilic phenotype. (XLS 43 KB)
12862_2005_169_MOESM4_ESM.xls
Additional File 4: contains the results of the reconstruction of the gene repertoire of the common ancestor of TT and DR. (XLS 392 KB)
12862_2005_169_MOESM5_ESM.xls
Additional File 5: contains information on taxonomic assignments for the best hits of the TT and DR proteins. (XLS 982 KB)
12862_2005_169_MOESM6_ESM.zip
Additional File 6: contains compressed file for all the trees (in PHYLIP format) for 122 proteins from the reconstructed gene set of the DR-TT common ancestor that showed affinity to thermophiles (see also Additional file 1: "Phylogenetic analysis of the genes of the reconstructed DR-TT common ancestor"). (ZIP 46 KB)
12862_2005_169_MOESM7_ESM.zip
Additional File 7: contains compressed file for all the trees (in PHYLIP format) for proteins from DR-TT core that had best hits to thermophiles. (ZIP 55 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Omelchenko, M.V., Wolf, Y.I., Gaidamakova, E.K. et al. Comparative genomics of Thermus thermophilus and Deinococcus radiodurans: divergent routes of adaptation to thermophily and radiation resistance. BMC Evol Biol 5, 57 (2005). https://doi.org/10.1186/1471-2148-5-57
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-5-57