
Abstract
Background
The genus Uropsilus comprises a group of terrestrial, montane mammals endemic to the Hengduan and adjacent mountains. These animals are the most primitive living talpids. The taxonomy has been primarily based on cursory morphological comparisons and the evolutionary affinities are little known. To provide insight into the systematics of this group, we estimated the first multi-locus phylogeny and conducted species delimitation, including taxon sampling throughout their distribution range.
Results
We obtained two mitochondrial genes (~1, 985 bp) and eight nuclear genes (~4, 345 bp) from 56 specimens. Ten distinct evolutionary lineages were recovered from the three recognized species, eight of which were recognized as species/putative species. Five of these putative species were found to be masquerading as the gracile shrew mole. The divergence time estimation results indicated that climate change since the last Miocene and the uplift of the Himalayas may have resulted in the diversification and speciation of Uropsilus.
Conclusions
The cryptic diversity found in this study indicated that the number of species is strongly underestimated under the current taxonomy. Two synonyms of gracilis (atronates and nivatus) should be given full species status, and the taxonomic status of another three potential species should be evaluated using extensive taxon sampling, comprehensive morphological, and morphometric approaches. Consequently, the conservation status of Uropsilus spp. should also be re-evaluated, as most of the species/potential species have very limited distribution.
Similar content being viewed by others

Background
The Asian shrew-like moles (Uropsilus spp.) are the only living genus in Uropsilinae and encompass the most primitive subfamily of Talpidae [1, 2]. In contrast to semi-fossorial shrew moles, fossorial moles, and aquatic desmans, the members of Uropsilus possess a shrew-like body shape, conspicuous external ears, a slender tail as long as the head-body length, and compressed claws without specialized characters for burrowing. All of these characters indicate an ambulatory lifestyle [3]. Although the ancestor species of Uropsilinae were widely distributed throughout Eurasia, the distribution of the relict genus Uropsilus is limited to southwest China and north Myanmar, in middle to high montane forests. The basal position of Uropsilinae in the family is supported by morphological characters [4, 5] and DNA sequences [2, 6, 7], although the evolutionary relationships within this genus remain poorly understood. Despite a recent description of a new species (Uropsilus aequodonenia) using morphometric and molecular approaches [8], the classifications of living uropsilines has been based exclusively on morphological and morphometric comparisons, and the taxonomy has changed several times (Table 1): three genera (Nasillus, Rhynchonax, and Uropsilus ), five species (U. aequodonenia, N. investigator, R. andersoni, N. gracilis, and U. soricipes) and two subspecies (R. a. atronates and R. a. nivatus) have been named (Table 1). Five taxa are currently recognized as full species of the genus Uropsilus (aequodonenia, andersoni, gracilis, investigator, and soricipes), whereas the other two (atronates and nivatus) are recognized as synonyms of U. gracilis[1, 9]. However, only a few CYT B sequences are available for four of the recognized species, and most of the taxon sampling was limited to western Sichuan [8]; furthermore, there are no available sequences for U. investigator. Thus, it would be interesting to examine whether the current taxonomy can be recovered by molecular approaches and whether morphological cryptic species exist within the widely distributed species [10].
The mountains of southwest China and adjacent areas harbor an extremely high biodiversity [17]. Two nonexclusive hypotheses, including a pre-Pleistocene diversification model and a Pleistocene refugia model have been proposed to explain the expansion of endemic species [18]. Under the pre-Pleistocene diversification model, the uplifting of the Qinghai-Tibetan Plateau (QTP) in association with climate changes boosted allopatric speciation (e.g., [19–21]). Conversely, Quaternary glacial-interglacial cycles caused repeated shifts in distribution ranges [22], which may also have motivated diversification and speciation in isolated mountain chains [23–25]. Given that Uropsilinae might have evolved for approx. 30 million years [26], this genus could be a promising model to test these two hypotheses.
In this study, we sampled Asian shrew-like moles throughout their distribution range, sequenced both mitochondrial and nuclear loci, and adopted molecular phylogenetic and species delimitation approaches to represent a systematic framework of the genus Uropsilus. Our goals are the following: (i) to examine the proposed taxonomy based on morphology/morphometrics; (ii) to assess the evolutionary relationships among living taxa; and (iii) to test alternative scenarios of species/diversification patterns within the genus.
Results
Sequence characteristics
We obtained 6,330 bp sequences for each voucher specimen, including 1,985 bp mitochondrial (CYT B [1,140 bp] and 12S rRNA [845 bp]) and 4,345 bp nuclear (ADORA3 [321 bp], ATP7A [675 bp], BDNF [555 bp], BMI1 [313 bp], CREM [389 bp], PLCB4 [331 bp], RAG1 [1,010 bp], and RAG2 [751 bp]) sequences. A total of 552 sequences were deposited in GenBank with Accession nos. from KF777818 to KF778377 (Additional file 1: Table S1). No premature stop codon was observed in the coding regions of the protein coding genes examined. The mitochondrial genes showed relatively higher genetic polymorphisms than the nuclear genes (Additional file 2: Table S2). Note that the BMI1 gene had no variable site and was thus not used in the species delimitation or network tree reconstruction.
Gene trees and divergence times
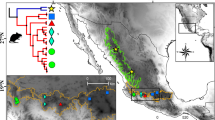
The Bayesian and Maximum likelihood (ML) phylogenetic reconstructions recovered very similar topologies; only the ML gene trees are shown (Figure 1). In addition to U. aequodonenia and U. andersoni, 10 distinct lineages were recovered in the mitochondrial gene tree (i.e., clades A-J; Figure 1a), and the monophyly of all these lineages was strongly supported (i.e., maximum-likelihood bootstrap proportions [BS] ≥ 85, Bayesian posterior probability [PP] ≥ 0.95). Two of these clades represented U. investigator (A and B), two represented U. soricipes (I and J), and the others represented U. gracilis (C-H). The relationships among the clades were overall strongly supported, except at two nodes. U. aequodonenia and U. andersoni were supported as sister species (BS = 100, PP = 1.0) and are sister to clade C (BS = 100, PP = 1.0). U. gracilis was supported as polyphyletic. The nuclear gene tree differed from the mitochondrial gene tree in not recovering the monophyly of clades E, F, and G, even though E + F + G + H was still supported as monophyletic (BS = 74, PP = 1.0; Figure 1b). Moreover, clade J was not supported as the sister group of clade I, but is sister to clade E + F + G + H + I (BS = 96, PP = 1.0). The mitochondrial-nuclear combined gene tree was very similar to the mitochondrial gene tree (Figure 1c) but supported clade C as the sister taxon of the other clades, except clade A + B (BS = 100, PP = 0.58). The partitioned branch support scores showed that both the mitochondrial and nuclear datasets support all but two relationships each (PBS < 1; Additional file 3: Figure S1).

Concatenated gene trees of genus Uropsilus based on Bayesian and ML analyses. ML gene trees of Uropsilus based on: (a) two mitochondrial genes, (b) eight nuclear genes, and (c) a combined mitochondrial-nuclear dataset. The node numbers represent the maximum-likelihood bootstraps [BS] and Bayesian posterior probabilities [PP]. A-J represents clade derived from the mitochondrial tree estimation.
The ultrametric tree derived from the divergence time estimation is shown in Figure 2. It is noteworthy that all relationships were strongly supported. The most recent common ancestor of Uropsilus occurred in the latest Miocene (6.18 Ma, 95% CI: 8.65-4.27), and the following divergences occurred from 4.35 Ma to 1.37 Ma (6.08-0.91).

Maximum credibility ultrametric tree of Uropsilus based on mitochondrial-nuclear concatenated data. The numbers above branches are the Bayesian posterior probabilities and the numbers under the branches represent the median divergence times. The node bars indicate the 95% interval confidence for the divergence time estimates. The five vertical lines represent the major historical events in the diversification of Uropsilus: 1) 5.93 Ma, Messinian salinity crisis [27]; 2) 3.6 Ma, strongest uplift of the QTP [28, 29]; 3) 2.4 Ma, modern Asian monsoon system began and Pleistocene glaciation started [30, 31]; 4) 1.8 Ma, uplift of the QTP [32]; 5) 1.1 Ma, uplift of QTP [28, 32].
Species delimitation and species trees
The split recognized clades diverged older than 0.61 Ma as putative species; therefore, all 10 monophyletic clades in the ML tree were recognized as putative species (Figure 3a). The K2P distance between clades E and F was calculated to be 0.013 and 0.039 between I and J. All of the other pairwise distances between the clades are ranged from 0.085 to 0.203 (Additional file 4: Table S4). The *BEAST analysis recovered the same topology as the mitochondrial-nuclear concatenated gene tree (Figure 3b), which was used as the guide species tree for the BPP analyses. In each Bayesian delimitation analysis, the posteriors of all parameters had a high ESS (> 1000); the results are provided in Additional file 5: Table S4. When using both mitochondrial and nuclear genes, BPP recognized all 10 clades as putative species (PP ≥ 0.99; Figure 3a; Additional file 5: Table S4). However, when using nuclear genes alone, the posterior supports for clades A and B and, I and J as distinct species were much lower (PP ≤ 0.47; Figure 3a; Additional file 5: Table S4). Following a conservative approach, we recognized A + B and G + H as single species each. We again reconstructed a species tree using the 8-species scenario, and the topology did not change (Figure 3c). In addition, the 7 nuclear gene network trees showed that U. investigator did not share any haplotypes with other species/putative species when the other 7 species/putative species shared haplotypes in 3 to 4 genes (Figure 4).

Results of species delimitation using splits and BPP, and species trees reconstructed using the *BEAST model. Node numbers indicate Bayesian posterior probabilities supporting each clade as two putative species when using nuclear genes alone and using all genes, respectively (a); and Bayesian posterior probabilities estimated in *BEAST analyses (b and c).

Nuclear gene network trees and distribution of each putative species.
Discussion
Mitochondrial vs. nuclear genes
Although incongruences between mitochondrial and nuclear or between different nuclear genes have been observed in many studies (e.g., [33]), the mitochondrial gene trees and PBS analyses suggested that in our case mitochondrial genes still represent good genetic markers. When both mitochondrial and nuclear genes alone failed to fully resolve the relationships, combined mitochondrial and nuclear genes using both concatenated and coalescent approaches recovered identical topologies and were robustly supported. It follows that a combination of both rapidly evolving mitochondrial and conservative nuclear genes appears to be the best approach to resolve ancient but closely spaced divergences.
Species delimitation and taxonomic reappraisal
The accuracy of molecular species delimitation has been discussed several times [34, 35]. BPP has been recognized as the most accurate approach when compared to other coalescent-based methods like ABC and SpeDeSTEM [34]. Recently, Miralles and Vences found that the number of putative species strikingly ranged from 9 to 34, depending on the method implemented [35]. Although BPP was not recognized as the best approach in their study, the results could be affected by small sample size of each putative species and the small number of loci (see [36] for detail). Despite the uncertainty of the number of putative species recognized, molecular data represent only a crude estimate rather than a final conclusion and should be diagnosed using comprehensive morphological characters.
Splits and BPP recognized 10 and 8 putative species, respectively. It is of note that clades A + B and G + H were recognized as one species each in the BPP analyses, even the pairwise K2P distances of CYT B were higher than 0.100. This value is higher than the distance between sister species U. aequodonenia and U. andersoni (0.085) and the average genetic distance between sister species of mammals (0.081; [37]). This result may be due to the small sample size of clade A (n = 2) and potential gene flow (see [36]) because A and B are sympatrically distributed and G and H were not supported as monophyletic by nuclear genes (Figure 1b). On the other hand, U. sp. 2, U. sp. 3 were recognized as putative species even though the K2P distance between them and their sister taxa were very low (K2P = 0.013-0.039), which might due to mitochondrial introgression or different lineage sorting scenarios. We treated U. spp. 2 and 3 as putative species here, as the BPP analyses suggested there was no recent gene flow. Regardless of the ambiguity of species delimitation, the high pairwise genetic distances and the polyphyly of previously known U. gracilis strongly suggested that number of species of Uropsilus has been underestimated.
With respect to the previous taxonomy of Uropsilus, dental formulas have been used as a key for species diagnosis. Four dental formulas have been observed, and U. gracilis and U. investigator share the same dentition (Additional file 3: Figure S1; [12]). Our results revealed that six polyphyletic taxa (species/putative species) share the same dental formula (i2/1,c1/1,p4/4,m3/3 = 38; Additional file 3: Figure S1). Within the six species/putative species, clade E was sampled from the type locality of U. gracilis, and can be safely assigned to this species (Figure 4). The type locality of U. investigator is distributed to the west of the Mekong River, geographically close to the clades A and B, thus we assigned clade A + B to U. investigator. Clades G + H and C consist of populations from the type localities of atronates and nivatus, respectively, and these two taxa should most likely be recognized as valid species. There is no available name for spp. 1–3, and their taxonomic status should be evaluated based on extensive sampling and comprehensive morphological/morphometric comparisons.
Divergence pattern of Uropsilus
The results of our divergence time estimation suggested that neither the pre-Pleistocene speciation nor the Pleistocene “speciation pump” hypothesis could exclusively explain the speciation/diversification pattern. Indeed, the diversification of Uropsilus may have been affected by periodical orogenic processes and climate change. Prevailing trends toward cooling and desiccation in the late Miocene (i.e., Messinian salinity crisis; [27, 38]) and the Pliocene/Pleistocene boundary [39] have been well documented, and leading to the diversification of humid-dwelling taxa (e.g., [40, 41]). Therefore, global cooling may be responsible for the splits of the Asian shrew-like moles at 6.18 Ma and 2.40 Ma. Similarly, geological studies have supported rapid uplifts of QTP at 3.6 Ma, 1.8 and 1.1 Ma [28, 30], which may also have resulted in the diversifications of the genus.
The distribution of different species/putative species showed a strong geographic pattern, which could be partly due to the extremely complex topography, understory habitats, and low dispersal ability of the animals [42, 43]. Nonetheless, at least four species/putative species, including U. aequodonenia, U. andersoni, U. soricipes, and U. sp.1, are distributed in the western Sichuan mountains, indicating a very complex geographic history. Extensive sampling is required to uncover the vicariant, migration, and speciation history patterns.
Cryptic diversity and conservation implications
Identifying cryptic diversity is essential for the accurate assessment of genetic diversity and conservation planning [10, 44]. Cryptic divergence and strong geographic structures have been observed in other endemic taxa [45, 46], indicating that topography has strong effect on diversification, particularly with regard to small and sedentary animals. The current conservation statuses of Uropsilus spp. are all considered as “Least Concern” or “Data Deficient” (IUCN Red List Category; [47]), due to their presumed wide distribution and/or large population size [48]. However, our results indicated that unrecognized species exist within Uropsilus, and most of the species/putative species have very limited distribution. Indeed, because of global warming and continuous habitat loss, the species diversity of the genus may be actually threatened. Therefore, re-evaluation of the endangered categories relying on a new systematic background is warranted; at present, these putative species should be considered as evolutionary significant units and taken into consideration for conservation planning.
Conclusions
In the present study, we obtained sequences of Uropsilus throughout their distribution in the mountains of southwest China. We reconstructed a robust phylogeny for this most primitive talpid genus and found cryptic diversity. Five putative species were determined in addition to the five recognized species. We suggested that atronates and nivatus should be recognized as full species, and a comprehensive morphological diagnosis is warranted for three unidentified species. Moreover, the conservation statues should re-evaluated, as most of the species/putative have limited distribution. Finally, the divergence of the genus may be affected by climatic changes and tectonic activities, providing clues for the expansion of endemic fauna.
Methods
Sample collection and ethics statement
A total of 56 individuals were collected, representing 3 of the 5 named species from 13 localities throughout the distribution area. These localities include the type localities of the gracilis (Mt. Jinfo, Chongqing), soricipes (Baoxing, Sichuan), synonyms atronates (Mucheng, Yunnan), and nivatus (Mt. Yulong, Yunnan) species (Figure 5, Table 2). All of the animal samples were obtained following the regulations of China for the implementation of the protection of terrestrial wild animals (State Council Decree [1992] No. 13) and approved by the Ethics Committee of Kunming Institute of Zoology, Chinese Academy of Sciences, China (no specific permit number). Voucher specimens were deposited in the Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, China.

Distribution, type locality, and sampling sites of Uropsilus spp. The coloured ranges indicate for the natural distribution of four Uropsilus species. Species distribution maps are modified from the the IUCN Red List of Threatened Species. Version 2013.1. (http://www.iucnredlist.org). Downloaded on 09 June 2013.
Laboratory protocols
All samples were frozen in ethanol at−70°C prior to DNA extraction. Total genomic DNA was extracted by a standard phenol/chloroform method [49] or using the DNeasy Tissue kit (Qiagen, Gemamy) from either liver or muscle tissues. Two mitochondrial (CYT B, 12S rRNA) and eight nuclear (adenosine A3 receptor [ADORA3], ATP7A, brain-derived neurotrophic factor [BDNF], polycomb ring finger oncogene [BMI1], cAMP responsive element modulator [CREM], phospholipase C beta 4 [PLCB4], recombination activating protein 1 [RAG1], and recombination activating protein 1 [RAG2]), genes including the coding sequences (CDSs) and 3′ untranslated regions (3′ UTRs) were applied. All these genes have been widely used in mammalian phylogenetic studies, including the mammal Tree of Life project (e.g., [50]). The gene fragments were amplified with rtaq DNA Polymerase (TaKaRa, China). Touchdown PCR was used to increase the specificity, sensitivity and yield [51]. The primers and high and low annealing temperatures are provided in Table 3. All PCR products were purified using the UNIQ-10 spin column DNA gel extraction kit (Sangon, China). Sequencing was performed using the BigDye Terminator Cycle kit v3.1 (Invitrogen, USA) and an ABI 3730xl sequencer (Applied Biosystems, USA).
Sequence assembling and alignment
Sequences were assembled and edited using DNASTAR Lasergene version 7.1. All genes were aligned in MUSCLE [53] and further examined by eye in MEGA5 [54]. In addition, the CYT B sequences determined in a recent study representing Uropsilus aequodonenia, U. andersoni, U. gracilis and U. soricipes were downloaded from GenBank [8]. Sequences of Talpa altaica of the subfamily Talpinae and Sorex araneus of the family Soricidae were chosen as outgroup taxa (Additional file 1: Table S1).
Phylogenetic analyses and divergence time estimation
To reconstruct the phylogenetic relationships of Uropsilus, ML analyses were performed using RAxML v7.3.2 [55], and Bayesian inference (BI) was performed using BEAST v1.7.5 [56]. The phylogenetic analyses were conducted on the following four datasets: 1) a mitochondrial combined gene dataset; 2) a nuclear gene combined dataset; 3) an all gene combined dataset; 4) the same as dataset 3 but with the CYT B sequences downloaded from GenBank removed (Additional file 6). We used a partitioning strategy to incorporate the variation in evolutionary processes among different sites [57]. The best-fit partitioning scheme and the appropriate model of DNA evolution for each partition were determined in PartitionFinder v1.0 [58]. The alignment was partitioned by gene and by codon position. Twelve models of molecular evolution (K80, HKY, TrNef, TrN, SYM, GTR, K80 + G, HKY + G, TrNef + G, TrN + G, SYM + G, GTR + G) were compared and ranked by the Bayesian Information Criterion (BIC) [59]. The best partitioning scheme and substitution models are given in Additional file 7. The ML analyses were performed using the CIPRES Science Gateway [60]. We selected the GTR + gamma model for each partition and the rapid Bootstrapping algorithm (Stamatakis A, Hoover P, Rougemont J: A Rapid Bootstrap Algorithm for the RAxML, Web-Servers, unpublished) and ran 500 bootstrap replicates. The Bayesian phylogenetic trees were calculated in BEAST v1.7.5 [56]. We employed relaxed uncorrelated exponential clock models that allowed the rate in each branch to evolve independently [61]. The combined mitochondrial fragment and each nuclear gene were given specific exponential clock models, and a Continuous-time Markov chains (CTMCs) model was employed as a prior for each clock model [62]. Each analysis used a random staring tree, a birth-death tree prior and the program’s default prior distribution of the model parameters. Each analysis was run for 50 million generations with a sampling interval of 5,000 was conducted. Trace v1.5 [63] was used to confirm the effective sample sizes (ESSs) as greater than 200 and the first 30% of the generations were treated as burn-in. The BEAST analyses were repeated four times. To assess the support for the mitochondrial and nuclear genes at each node, partitioned branch support scores (PBSs; [64]) were calculated using TreeRot v2.0 [65] and PAUP 4.0b10 [66]. We performed this analysis using dataset 4. We further constructed network trees for each nuclear gene using NETWORK v4.611; the nuclear genes were unphased with DnaSP v5.10 [67] using the algorithms provided by PHASE [68], and the unphased haplotypes were used to construct median-joining haplotype networks [69]. We ran the MP calculation post-processing option to delete all of the superfluous median vectors and chose one of the shortest trees.
The molecular divergence time was estimated using BEAST v1.7.5. Because mitochondrial genes evolve much faster than nuclear genes [70], missed nuclear genes will lead to inaccurate estimates of branch lengths and divergence times. Therefore, we used dataset 4 for the molecular dating analyses with U. aequodonenia and U. andersoni excluded. For the fossil-calibrated age constraints, lognormal and exponential distributions were used to account for uncertainty in fossil calibrations [71]. Three calibrations were used. (i) The split of the most recent common ancestor (MRCA) shared by Talpidae and Erinaceidae + Soricidae was at approximately 73 (78–68) million years ago (Ma) [72]. We set up the prior using a lognormal distribution (offset = 63, mean = 10.4, standard deviation = 0.24), such that the mean age was at 73 Ma and the 95% upper boundary was at 78 Ma. (ii) The oldest known Talpini was from the early Oligocene (Palaeogene mammal unit MP 21) [73] at approximately 33.9-32.6 Ma [74]; thus, we set up the prior using an exponential distribution (offset = 32.6, mean = 10.8; 32.6*0.333) [75]. (iii) We employed the oldest known U. soricipes from 2.4-2.0 million-year-old strata (early Pleistocene) in Hubei Province, China, and set up the prior using an exponential distribution, (offset = 2.0, mean = 0.67 2.0*0.333) [76, 77].
Species delimitation and species tree estimation
We conducted species delimitation analyses using the “splits” v1.0-14 package (SPecies LImits by Threshold Statistics) for the R statistical environment and the programe BPP v2.2. When using splits, the time-calibrated concatenated gene tree was used as the input tree, whereas the number of putative species was identified using a generalized mixed Yule-coalescent model (GMYC) [78]. We further used BPP v2.2 to conduct a Bayesian species delimitation [79, 80]. This software provides the most accurate molecular species delimitation to date [34], and the mixing of the Markov chain is improved in the new version v2.2 [81]. The computational simulation demonstrated that the correct species models could be inferred with high posterior probabilities when 5 or 10 sequences from each population were sampled [36]. A previous study found that a mis-specified guide tree can result in strong support for more species [82]. Therefore, we estimated the species tree using the *BEAST model implemented in BEAST (see below), and assigned the 56 specimens to 10 putative species based on the results of splits (see Results). The analyses were performed using both the mitochondrial-nuclear combined data (dataset 4) and the nuclear genes alone (dataset 2). Gamma prior G (6, 6,000) was used on the population size parameters (θs), and the age of the root in the species tree (τ0) was assigned gamma prior G (4, 1,000). Multiple runs were performed using both the species delimitation algorithm 0 and algorithm 1. “Locusrate = 1” specifying the random-rates model of Burgess and Yang [83], or “Heredity = 1” allowing θ to vary among loci, were also used but not at the same time. The analyses for each data set were repeated 12 times. Each rjMCMC was run for 1 million generations and sampled every 10 generations after discarding 10,000 generations as pre-burn-in.
The species trees were estimated using the *BEAST model [84] in BEAST v1.7.5. Because the *BEAST model uses very different assumptions from a concatenated gene tree estimation, we did not use the partitioning scheme derived from Partitionfinder. Instead, we gave 12S rRNA, each codon position of CYT B gene and each nuclear gene a different substitution model. The best-fit models were calculated using jModeltest [85] and are provided in Additional file 7. Because *BEAST requires that all species have at least one sequence at each locus, we used dataset 4 for the *BEAST analyses. The 56 specimens were assigned to 8 or 10 putative species based on the results of splits and BPP (see Results). We used the same priors as described above. Each analysis was run for 100 million generations and sampled every 10,000 generations.
Data accessibility
DNA sequences: accession numbers are provided in Additional file 1: Table S1.
References
Hoffmann RS, Lunde D: Soricomorpha. A guide to the mammals of china. Edited by: Smith AT, Xie Y. 2008, Princeton, New Jersey: Princeton University Press, 213-216.
Shinohara A, Campbell KL, Suzuki H: Molecular phylogenetic relationships of moles, shrew moles, and desmans from the new and old worlds. Mol Phylogenet Evol. 2003, 27 (2): 247-258. 10.1016/S1055-7903(02)00416-5.
Allen GM: The mammals of China and Mongolia. Part 1. 1938, New York: American Museum of Natural History
Sánchez-Villagra MR, Horovitz I, Motokawa M: A comprehensive morphological analysis of talpid moles (Mammalia) phylogenetic relationships. Cladistics. 2006, 22 (1): 59-88. 10.1111/j.1096-0031.2006.00087.x.
Motokawa M: Phylogenetic relationships within the family Talpidae (Mammalia: Insectivora). J Zool. 2004, 263 (2): 147-157. 10.1017/S0952836904004972.
Douady CJ, Douzery EJP: Molecular estimation of eulipotyphlan divergence times and the evolution of “Insectivora”. Mol Phylogenet Evol. 2003, 28 (2): 285-296. 10.1016/S1055-7903(03)00119-2.
Shinohara A, Suzuki H, Tsuchiya K, Zhang YP, Luo J, Jiang XL, Wang YX, Campbell KL: Evolution and biogeography of talpid moles from continental east Asia and the Japanese islands inferred from mitochondrial and nuclear gene sequences. Zool Sci. 2004, 21 (12): 1177-1185. 10.2108/zsj.21.1177.
Liu Y, Liu SY, Sun ZY, Guo P, Fan ZX, Murphy RW: A new species of Uropsilus ( Talpidae: Uropsilinae) from Sichuan China. Acta Theriol Sin. 2013, 33 (2): 113-
Hutterer R: Order Soricomorpha. Mammal species of the world: a taxonomic and geographic reference. Edited by: Wilson DE, Reeder DAM. 2005, Baltimore: John Hopkins University Press, 220-311.
Bickford D, Lohman DJ, Sodhi NS, Ng PKL, Meier R, Winker K, Ingram KK, Das I: Cryptic species as a window on diversity and conservation. Trends Ecol Evol. 2007, 22 (3): 148-155. 10.1016/j.tree.2006.11.004.
Ellerman JR, Morrison-Scott TCS: Checklist of Palaearctic and Indian mammals 1758–1946. 1951, London: British Museum (Natural History)
Hoffmann RS: A review of the shrew moles (Genus Uropsilus) of China and Burma. J Mamm Soc Japan. 1984, 10 (2): 69-80.
Nowak RM, Paradiso JL: Walker’s mammals of the world. 1999, Baltimore, MD: Johns Hopkins University Press
Cranbrook TE: Notes on the habits and vertical distribution of some insectivores from the Burma-Tibetan frontier. Proc Linnean Soc London. 1960–61, 173: 121-127.
Corbet GB, Hill JE: A world list of mammalian species. 1980, London and Ithaca, NY: British Museum (Natural History) and Cornell University Press
Honacki JH, Kinman KE, Koeppl JW: Mammal species of the world: a taxonomic and geographic reference. 1982, New York: Allen Press and the Association of Systematics Collections
Myers N, Mittermeier R, Mittermeier C, Da-Fonseca G, Kent J: Biodiversity hotspots for conservation priorities. Nature. 2000, 403 (6772): 853-10.1038/35002501.
Zhang RZ: Geological events and mammalian distribution in China. Acta Zool Sin. 2002, 48 (2): 141-153.
Che J, Zhou WW, Hu JS, Yan F, Papenfuss TJ, Wake DB, Zhang YP: Spiny frogs (Paini) illuminate the history of the Himalayan region and Southeast Asia. Proc Natl Acad Sci U S A. 2010, 107 (31): 13765-13770. 10.1073/pnas.1008415107.
Liu J, Wang Y, Wang A, Hideaki O, Abbott R: Radiation and diversification within the Ligularia-Cremanthodium-Parasenecio complex (Asteraceae) triggered by uplift of the Qinghai-Tibetan Plateau. Mol Phylogenet Evol. 2006, 38 (1): 31-10.1016/j.ympev.2005.09.010.
Yu N, Zheng C, Zhang YP, Li WH: Molecular systematics of pikas (genus ochotona) inferred from mitochondrial DNA sequences. Mol Phylogenet Evol. 2000, 16 (1): 85-95. 10.1006/mpev.2000.0776.
Hewitt G: Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond B Biol Sci. 2004, 359 (1442): 183-195. 10.1098/rstb.2003.1388.
Hewitt G: The genetic legacy of the Quaternary ice ages. Nature. 2000, 405 (6789): 907-913. 10.1038/35016000.
McCormack J, Bowen B, Smith T: Integrating paleoecology and genetics of bird populations in two sky island archipelagos. BMC Biol. 2008, 6 (1): 28-10.1186/1741-7007-6-28.
McCormack JE, Huang H, Knowles LL: Sky islands. Encyclopedia of Islands. 2009, 4: 841-843.
McKenna MC, Bell SK, Simpson GG: Classification of mammals above the species level. 1997, New York: Columbia University Press
Krijgsman W, Hilgen F, Raffi I, Sierro F, Wilson D: Chronology, causes and progression of the Messinian salinity crisis. Nature. 1999, 400 (6745): 652-655. 10.1038/23231.
Li J, Fang X: Uplift of the Tibetan plateau and environmental changes. Chinese Sci Bull. 1999, 44: 2117-2124. 10.1007/BF03182692.
Song Y, Fang X, Torii M, Ishikawa N, Li J, An Z: Late Neogene rock magnetic record of climatic variation from Chinese eolian sediments related to uplift of the Tibetan Plateau. J Asian Earth Sci. 2007, 30 (2): 324-332. 10.1016/j.jseaes.2006.10.004.
Xiao JL, An ZS: Three large shifts in East Asian monsoon circulation indicated by loess–paleosol sequences in China and late Cenozoic deposits in Japan. Palaeogeogr Palaeoclimatol Palaeoecol. 1999, 154: 179-189. 10.1016/S0031-0182(99)00110-8.
Tungsheng L, Zhongli D: Stepwise coupling of monsoon circulations to global ice volume variations during the late Cenozoic. Global Planet Change. 1993, 7 (1–3): 119-130.
Meng X, Xia P, Zheng J, Wang X: Evolution of the East Asian monsoon and its response to uplift of the Tibetan plateau since 1.8 Ma recorded by major elements in sediments of the south china Sea. Chinese Sci Bull. 2011, 56 (6): 547-551. 10.1007/s11434-010-4258-1.
Pages M, Bazin E, Galan M, Chaval Y, Claude J, Herbreteau V, Michaux J, Piry S, Morand S, Cosson JF: Cytonuclear discordance among Southeast Asian black rats (Rattus rattus complex). Mol Ecol. 2013, 22 (4): 1019-1034. 10.1111/mec.12149.
Camargo A, Morando M, Avila LJ, Sites JW: Species delimitation with abc and other coalescent-based methods: a test of accuracy with simulations and an empirical example with lizards of the Liolaemus Darwinii complex (Squamata: Liolaemidae). Evolution. 2012, 66 (9): 2834-2849. 10.1111/j.1558-5646.2012.01640.x.
Miralles A, Vences M: New metrics for comparison of taxonomies reveal striking discrepancies among species delimitation methods in madascincus lizards. PLoS ONE. 2013, 8 (7): e68242-10.1371/journal.pone.0068242.
Zhang C, Zhang DX, Zhu TQ, Yang ZH: Evaluation of a Bayesian coalescent method of species delimitation. Syst Biol. 2011, 60 (6): 747-761. 10.1093/sysbio/syr071.
Bradley RD, Baker RJ: A test of the genetic species concept: cytochrome-b sequences and mammals. J Mammal. 2001, 82 (4): 960-973. 10.1644/1545-1542(2001)082<0960:ATOTGS>2.0.CO;2.
Xu X, Fang X: Rock magnetic record of Cenozoic lake sediments from the Linxia basin and aridification of the Asian inland. Front Earth Sci. 2008, 2: 217-224. 10.1007/s11707-008-0040-y.
Webb T, Bartlein P: Global changes during the last 3 million years: climatic controls and biotic responses. Annu Rev Ecol Syst. 1992, 23: 141-173.
He K, Li YJ, Brandley MC, Lin LK, Wang YX, Zhang YP, Jiang XL: A multi-locus phylogeny of Nectogalini shrews and influences of the paleoclimate on speciation and evolution. Mol Phylogenet Evol. 2010, 56 (2): 734-746. 10.1016/j.ympev.2010.03.039.
Plana V, Gascoigne A, Forrest LL, Harris D, Pennington RT: Pleistocene and pre-Pleistocene Begonia speciation in Africa. Mol Phylogenet Evol. 2004, 31 (2): 449-461. 10.1016/j.ympev.2003.08.023.
Zhou W, Wen Y, Fu J, Xu Y, Jin J, Ding L, Min M, Che J, Zhang Y: Speciation in the Rana chensinensis species complex and its relationship to the uplift of the Qinghai-Tibetan Plateau. Mol Ecol. 2012, 21 (4): 960-973. 10.1111/j.1365-294X.2011.05411.x.
Fu J, Zeng X: How many species are in the genus batrachuperus? a phylogeographical analysis of the stream salamanders (family hynobiidae) from southwestern china. Mol Ecol. 2008, 17 (6): 1469-1488. 10.1111/j.1365-294X.2007.03681.x.
Murray SW, Campbell P, Kingston T, Zubaid A, Francis CM, Kunz TH: Molecular phylogeny of hipposiderid bats from Southeast Asia and evidence of cryptic diversity. Mol Phylogenet Evol. 2012, 62 (2): 597-611. 10.1016/j.ympev.2011.10.021.
Chen SD, Liu SY, Liu Y, He K, Chen WC, Zhang XY, Fan ZX, Tu FY, Jia XD, Yue BS: Molecular phylogeny of asiatic short-tailed shrews, genus Blarinella Thomas, 1911 (Mammalia: Soricomorpha: Soricidae) and its taxonomic implications. Zootaxa. 1911, 2012 (3250): 43-53.
Li S, He K, Yu FH, Yang QS: Molecular phylogeny and biogeography of Petaurista inferred from the cytochrome b gene, with implications for the taxonomic status of P. caniceps, P. marica and P. sybilla. PLoS One. 2013, 8 (7): e70461-10.1371/journal.pone.0070461.
Baillie JE, Hilton-Taylor C, Stuart SN: 2004 IUCN red list of threatened species: a global species assessment. 2004, Gland, Switzerland and Cambridge, UK: IUCN
The IUCN Red list of threatened species. Version. 2012, http://www.iucnredlist.org ], .2
Sambrook J, Fritsch EF, Maniatis T: Molecular cloning: a laboratory manual. 1989, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 2
Meredith RW, Janečka JE, Gatesy J, Ryder OA, Fisher CA, Teeling EC, Goodbla A, Eizirik E, Simão TLL, Stadler T, et al: Impacts of the cretaceous terrestrial revolution and KPg extinction on mammal diversification. Science. 2011, 334 (6055): 521-524. 10.1126/science.1211028.
Don RH, Cox PT, Wainwright BJ, Baker K, Mattick JS: Touchdown PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res. 1991, 19 (14): 4008-4008. 10.1093/nar/19.14.4008.
Teeling EC, Scally M, Kao DJ, Romagnoli ML, Springer MS, Stanhope MJ: Molecular evidence regarding the origin of echolocation and flight in bats. Nature. 2000, 403 (6766): 188-192. 10.1038/35003188.
Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32 (5): 1792-1797. 10.1093/nar/gkh340.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28 (10): 2731-2739. 10.1093/molbev/msr121.
Stamatakis A, Hoover P, Rougemont J: A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol. 2008, 57 (5): 758-771. 10.1080/10635150802429642.
Drummond AJ, Suchard MA, Xie D, Rambaut A: Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012, 29 (8): 1969-1973. 10.1093/molbev/mss075.
Brandley MC, Schmitz A, Reeder TW: Partitioned Bayesian analyses, partition choice, and the phylogenetic relationships of scincid lizards. Syst Biol. 2005, 54 (3): 373-390. 10.1080/10635150590946808.
Lanfear R, Calcott B, Ho SYW, Guindon S: PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012, 29 (6): 1695-1701. 10.1093/molbev/mss020.
Luo A, Qiao H, Zhang Y, Shi W, Ho SYW, Xu W, Zhang A, Zhu C: Performance of criteria for selecting evolutionary models in phylogenetics: a comprehensive study based on simulated datasets. BMC Evol Biol. 2010, 10 (1): 242-10.1186/1471-2148-10-242.
Miller MA, Pfeiffer W, Schwartz T: Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Gateway computing environments workshop (GCE): 2010. 2010, : IEEE, 1-8.
Drummond AJ, Ho SYW, Phillips MJ, Rambaut A: Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4 (5): e88-10.1371/journal.pbio.0040088.
Ferreira MA, Suchard MA: Bayesian analysis of elapsed times in continuous-time Markov chains. Can J Stat. 2008, 36 (3): 355-368. 10.1002/cjs.5550360302.
Rambaut A, Drummond A: Tracer v1.4.1: MCMC trace analysis tool. 2007, Institute of Evolutionary Biology: University of Edinburgh
Baker RH, DeSalle R: Multiple sources of character information and the phylogeny of Hawaiian drosophilids. Syst Biol. 1997, 46 (4): 654-673. 10.1093/sysbio/46.4.654.
Sorenson MD: TreeRot, version 2. 1999, Boston, Massachusetts: Boston University
Swofford DL: PAUP*: phylogenetic analysis using parsimony, version 4.0 b10. 2002, Sunderland, Massachusetts, USA: Sinauer Associates
Librado P, Rozas J: DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009, 25 (11): 1451-1452. 10.1093/bioinformatics/btp187.
Stephens M, Donnelly P: A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003, 73 (5): 1162-1169. 10.1086/379378.
Bandelt HJ, Forster P, Rohl A: Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999, 16 (1): 37-48. 10.1093/oxfordjournals.molbev.a026036.
Zheng Y, Peng R, Kuro-o M, Zeng X: Exploring patterns and extent of bias in estimating divergence time from mitochondrial DNA sequence data in a particular lineage: a case study of salamanders (order caudata). Mol Biol Evol. 2011, 28 (9): 2521-2535. 10.1093/molbev/msr072.
Ho SYM: Calibrating molecular estimates of substitution rates and divergence times in birds. J Avian Biol. 2007, 38 (4): 409-414.
Roca AL, Kahila Bar-Gal G, Eizirik E, Helgen KM, Maria R, Springer MS, O’Brien JS, Murphy WJ: Mesozoic origin for West Indian insectivores. Nature. 2004, 429 (6992): 649-651. 10.1038/nature02597.
Ziegler R: Moles (talpidae, mammalia) from early Oligocene karstic fissure fillings in south Germany. Geophys J Roy Astron Soc. 2012, 45: 501-513.
İslamoğlu Y, Harzhauser M, Gross M, Jiménez-Moreno G, Coric S, Kroh A, Rögl F, van der-Made J: From Tethys to eastern paratethys: Oligocene depositional environments, paleoecology and paleobiogeography of the Thrace basin (NW turkey). Int J Earth Sci. 2010, 99 (1): 183-200. 10.1007/s00531-008-0378-0.
Heath TA: A hierarchical Bayesian model for calibrating estimates of species divergence times. Syst Biol. 2012, 61 (5): 793-809. 10.1093/sysbio/sys032.
Agustí J, Cabrera L, Garcés M, Krijgsman W, Oms O, Parés J: A calibrated mammal scale for the Neogene of Western Europe. State of the art. Earth-Sci Rev. 2001, 52 (4): 247-260. 10.1016/S0012-8252(00)00025-8.
Huang W, Fang Q: The wushan Man site. 1991, Beijing: Ocean Press
Pons J, Barraclough TG, Gomez-Zurita J, Cardoso A, Duran DP, Hazell S, Kamoun S, Sumlin WD, Vogler AP: Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst Biol. 2006, 55 (4): 595-609. 10.1080/10635150600852011.
Rannala B, Yang Z: Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics. 2003, 164 (4): 1645-1656.
Yang Z, Rannala B: Bayesian species delimitation using multilocus sequence data. Proc Natl Acad Sci U S A. 2010, 107 (20): 9264-9269. 10.1073/pnas.0913022107.
Rannala B, Yang ZH: Improved reversible jump algorithms for Bayesian species delimitation. Genetics. 2013, 194 (1): 245-253. 10.1534/genetics.112.149039.
Leache AD, Fujita MK: Bayesian species delimitation in West African forest geckos (Hemidactylus fasciatus). Philos Trans R Soc London, Ser B. 2010, 277 (1697): 3071-3077.
Burgess R, Yang Z: Estimation of hominoid ancestral population sizes under Bayesian coalescent models incorporating mutation rate variation and sequencing errors. Mol Biol Evol. 2008, 25 (9): 1979-1994. 10.1093/molbev/msn148.
Heled J, Drummond AJ: Bayesian inference of species trees from multilocus data. Mol Biol Evol. 2010, 27 (3): 570-580. 10.1093/molbev/msp274.
Darriba D, Taboada GL, Doallo R, Posada D: jModelTest 2: more models, new heuristics and parallel computing. Nat Meth. 2012, 9 (8): 772-772.
Acknowledgements
We appreciate the constructive comments and suggestions from the editor Dr. Miguel Vences and the anonymous reviewer. We thank Chi Zhang for discussing the BPP analyses in detail. We thank Nai-Qing Hu, Wei-Wei Zhou, Wan-Sheng Jiang, Feng Dong, Lan-Ping Zheng and Min-Sheng Peng for their meaningful suggestions on the laboratory work and earlier versions of this manuscript. This work was supported by grants from the National Natural Science Foundation of China (No. 31301869 and 31272276), Knowledge Innovation Program of the Chinese Academy of Sciences (KSZD-EW-2-011), and the Fund of State Key Laboratory of Genetics Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences (GREKF11-03).
This work is dedicated to Dr. Robert S. Hoffmann, who greatly contributed to the science of mammalogy in China.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TW and KH contributed to the laboratory work, data analyses, and manuscript writing; XLJ conceived the study and commented on the manuscript; all authors have read and approved the final manuscript.
Tao Wan, Kai He contributed equally to this work.
Electronic supplementary material
{kind=link}
12862_2013_2466_MOESM5_ESM.xls
Additional file 5: Table S4: Results of the BPP analyses based on both mitochondrial-nuclear combined data set and nuclear genes alone.(XLS 32 KB)
12862_2013_2466_MOESM7_ESM.doc
Additional file 7: Partitioning schemes and molecular evolution model used in the gene tree and species tree estimations.(DOC 48 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wan, T., He, K. & Jiang, XL. Multilocus phylogeny and cryptic diversity in Asian shrew-like moles (Uropsilus, Talpidae): implications for taxonomy and conservation. BMC Evol Biol 13, 232 (2013). https://doi.org/10.1186/1471-2148-13-232
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-13-232