
Abstract
Background
Deep-sea hydrothermal vents provide patchy, ephemeral habitats for specialized communities of animals that depend on chemoautotrophic primary production. Unlike eastern Pacific hydrothermal vents, where population structure has been studied at large (thousands of kilometres) and small (hundreds of meters) spatial scales, population structure of western Pacific vents has received limited attention. This study addresses the scale at which genetic differentiation occurs among populations of a western Pacific vent-restricted gastropod, Ifremeria nautilei.
Results
We used mitochondrial and DNA microsatellite markers to infer patterns of gene flow and population subdivision. A nested sampling strategy was employed to compare genetic diversity in discrete patches of Ifremeria nautilei separated by a few meters within a single vent field to distances as great as several thousand kilometres between back-arc basins that encompass the known range of the species. No genetic subdivisions were detected among patches, mounds, or sites within Manus Basin. Although I. nautilei from Lau and North Fiji Basins (~1000 km apart) also exhibited no evidence for genetic subdivision, these populations were genetically distinct from the Manus Basin population.
Conclusions
An unknown process that restricts contemporary gene flow isolates the Manus Basin population of Ifremeria nautilei from widespread populations that occupy the North Fiji and Lau Basins. A robust understanding of the genetic structure of hydrothermal vent populations at multiple spatial scales defines natural conservation units and can help minimize loss of genetic diversity in situations where human activities are proposed and managed.
Similar content being viewed by others


Background
The spatial scales at which individuals within a population interact and the geographic extent of larval dispersal shape the dynamics of marine populations. Dispersal capabilities of some species extend across entire ocean basins [1], but larval propagules of many other species are retained close to their source [2]. Larval development can impose limits on dispersal. Species that brood their offspring (direct development) tend to have more restricted distributions than species with long-lived, planktonic larvae [3], though exceptions exist [4]. Species that aggregate in small patches may interact and reproduce with other individuals in an area encompassing a meter or less [5] and larvae that lack broad dispersal potential may recruit to their natal population [6]. A sampling scheme that fails to account for the localized effects of self-recruiting patches may create an appearance of panmixia, even if substructure exists among patches [7].
Species dependent on deep-sea hydrothermal vents are restricted to patchy, ephemeral habitats that limit the areal extent and occurrence of populations. Hydrothermal vent fields are found on mid-ocean ridges, back-arc spreading centres, and submarine volcanoes [8]. Organisms that thrive at vents are supported by chemoautotrophic microbes that metabolize reduced compounds in the vent effluent [9]. Vent habitats are transient, at temporal scales ranging from days to hundreds of years [10], and constituent species may be subject to frequent local extinction and recolonization events [11, 12]. Survival of vent species therefore depends on fast growth, rapid reproduction, and dispersal abilities that shape the diversity and genetic structure of populations [13, 14].
At mid-ocean ridges, deep-sea hydrothermal vents are distributed along roughly linear axes that may function as dispersal corridors [15–17]. Geographic populations of hydrothermal vent-dependent species can be panmictic across the extent of their range (e.g., the shrimp, Rimicaris exoculata on the Mid-Atlantic Ridge [18–20]) but this is not always the case.
Evidence for isolation-by-distance in vent species has sometimes been ambiguous due to small sample sizes and inconsistency in the resolution of various genetic markers [21]. Considerable evidence exists for geographic subdivision associated with geomorphological features that affect different taxa to varying degrees. For example, the Easter Microplate is associated with isolation of northern and southern East Pacific Rise populations of mussels, but not of polychaete annelids [22, 23]. A 2000-m long "habitat gap" across the Equator is implicated in the isolation of some East Pacific Rise species and variable impedance of gene flow in other species [23, 24]. Similarly, a 350-km long ridge offset, the Blanco Transform Fault, isolates Juan de Fuca and Gorda ridge limpet populations [25]. The same barrier interacts with current regimes and is correlated with southward unidirectional gene flow in the vent polychaete Ridgeia piscesae [17]. Life history and behavioral attributes of various taxa result in these differing responses to shared dispersal barriers [14].
Identification of population structure at various spatial scales depends in part on the choice of genetic markers. For example, amplified fragment length polymorphisms were used to test for fine-scale differentiation among discrete patches of the tubeworm, Riftia pachyptila, separated by as little as 400 m in a venting area along the East Pacific Rise, although sample sizes were small (n < 15 per site [26]). More conservative mitochondrial and nuclear DNA sequences in R. pachyptila revealed panmixia at local scales and isolation-by-distance [27] at greater geographical scales [28, 29].
In contrast to mid-ocean ridge systems, limited attention has been afforded to the population structure of vent organisms from western Pacific back-arc basins. These basins are distributed in a non-linear pattern, reflecting the complex tectonic history of the region [30]. Hydrothermal vents in western Pacific back-arc basins are geographically isolated from vents on the East Pacific Rise [31]. Regional isolation of species was detected among western pacific vents [30]: the Okinawa trough and Izu-Ogasawara Arc have a faunal assemblage distinct from that of other western Pacific hydrothermal vents, and the faunal composition of the Marianna Trough is distinct from that of neighbouring basins [30]. Vent species tend to be shared among Manus, North Fiji, and Lau Basin, but are distinct from species that occur at the Okinawa and Marianna Troughs or the Izu-Ogasawara Arc [30].
Because back-arc basin hydrothermal systems in the western Pacific are located on isolated ridge segments (in contrast to the linear, semi-continuous series of segments on mid-ocean ridges), it has been hypothesized that reduced connectivity among western Pacific back-arc basins may yield more endemic vent fauna within discrete back-arc basins [32]. Some species endemic to these basins appear to be panmictic across multiple basins (e.g., the mussel Bathymodiolus brevior [33]), whereas others are restricted to single basins (e.g., neoverrucid barnacles [34]). Provannid snail species in the genus Alviniconcha represent a cryptic species complex composed of at least three evolutionary lineages, one that occurs at hydrothermal vents in North Fiji Basin, one that is restricted to vents in the Marianna Trough, and one that co-occurs in both Manus and North Fiji Basin [35]. A similar pattern of strong genetic differentiation may exist within other species. To date, comprehensive efforts have not been made to characterize population structure within vent taxa of western Pacific back-arc basins.
Ifremeria nautilei is a provannid gastropod that occurs in Manus, North Fiji, and Lau Basins and depends on sulphur-oxidizing bacterial endosymbionts for nutrition. Sessile adults live in discrete patches near the effluent of diffuse-flow hydrothermal vents [36, 30]. Females possess a specialized brood-pouch in their foot and they release ciliated pre-veliger larvae (Warén's larvae) that are hypothesized to have long-distance dispersal capabilities [37]. Preliminary studies indicated that I. nautilei exhibits distinct mitochondrial haplotypes in Manus and North Fiji Basins [38], but population structure has not been assessed at smaller spatial scales--among vent fields within basins (henceforth sites), among sulphide mounds within vent fields (henceforth mounds), or among discrete patches on vent mounds (henceforth patches).
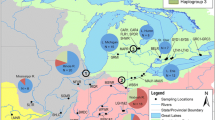
We examined genetic population structure of Ifremeria nautilei from hydrothermal vents in Manus, North Fiji, and Lau Basins at multiple scales, ranging from meters to thousands of kilometres (Figure 1). A nested sampling strategy was employed within Manus Basin to test the null hypothesis that I. nautilei exhibits no population structure among discrete patches at spatial scales of meters to 40 kilometres. The entire Manus Basin population was then compared to North Fiji and Lau basin samples to assess the relationship between increasing spatial scales (1000 kilometres to 3500 kilometres) and genetic differentiation. Genetic markers for differentiation at these scales included partial sequences of mitochondrial cytochrome-c-oxidase subunit I, and an array of nuclear DNA microsatellite loci [39]. By comparing these two types of molecular markers, we can separate evolutionary processes, revealed by COI sequence data and dependent on mutation rates, from ecologic processes, revealed by microsatellite allele frequencies and based on the recombination of alleles with each generation [40]. If the specialized Warén's larvae produced by Ifremeria nautilei are adapted for long-distance dispersal [37], population structure should be minimal over all scales. Alternatively, if I. nautilei disperse in a manner consistent with other sessile invertebrates with specialized habitat needs [41] genetic differentiation may occur at spatial scales less than one kilometre.

Ifremeria nautilei sampling locations in the western Pacific. Grey circles are approximate location of sampling. Dashed lines represent subduction zones. Three sites sampled in Manus Basin are shown in inset.
Results
Summary statistics
Ifremeria nautilei were sampled from Manus, North Fiji, and Lau Basins (275 individuals total; Figure 1, Table 1). Thirty-six partial COI haplotypes (404 bp) were identified from 158 Ifremeria nautilei sampled from Manus Basin; an additional 25 haplotypes were identified from 117 individuals from North Fiji and Lau Basins. Haplotype diversity (H d ) ranged from 0.59 to 0.95 (Table 2) and nucleotide diversity (π) ranged from 0.003 to 0.007. Indices of genetic diversity and tests for selection are reported in Table 2. Fu's F S values were negative, consistent with allelic excess driven by recent population expansion (but may also be indicative of a selective sweep) [42], with two exceptions (Patch 3 and Patch 16).
Nine microsatellite loci were amplified from 40 to 66 individuals per site within Manus Basin (Additional File 1, Table S1). Total alleles per locus ranged from 3 to 23 (mean = 6.6). In permutation tests, allelic richness (Rs) did not vary significantly among patches, mounds, or sites (10,000 permutations, P > 0.05; Additional File 1, Table S1). Three loci (Ifr040, Ifr052, and Ifr078) were monomorphic at the patch level but were polymorphic among patches.
Only eight microsatellite loci were amplified from 20 to 38 individuals per site from North Fiji and Lau Basin (Additional File 1, Table S2). One locus (Ifr086) failed to amplify in any North Fiji or Lau Basin samples. The total number of alleles per locus ranged from 2 to 23 (mean = 6.2). In permutation tests, allelic richness (Rs) did not vary significantly among sites or basins (10,000 permutations, P > 0.05, Additional File 1, Table S2).
Microsatellite marker quality
Tests for HWE deviation were used to assess the quality of sampled microsatellite markers. In Manus Basin, heterozygote deficiency was detected in one locus (Ifr086; Additional File 1, Table S1). Heterozygote excess was detected in only one locus at the site level (Solwara 1, Ifr040; Additional File 1, Table S1). Significant deviation from HWE was not detected at any other patch, mound, or site from Manus Basin (Additional File 1, Table S1).
Two microsatellite loci deviated significantly from Hardy-Weinberg Equilibrium at sites within North Fiji Basin (Ifr068 and Ifr103; Additional File 1, Table S2). Four microsatellite loci were not in equilibrium at the basin level (Ifr068, Ifr078, Ifr093, and Ifr103; Additional File 1, Table S2). Neither directional nor balancing selection was detected among microsatellites at any spatial scale within Manus Basin (LOSITAN, P > 0.05), but one microsatellite locus (Ifr043) was under positive selection (LOSITAN, 25,000 simulations, P < 0.001) at the basin level.
Microsatellite marker identity and excluded markers
Identity tests were used to assess the utility of each microsatellite marker set. Within Manus Basin, probability of identity tests (P ID ) and probability of sibling identity tests (P SIB ) indicated that the nine microsatellite markers identify individuals (P ID = 1.5 × 10-6), including those that shared 50% genetic similarity (P SIB = 3.5 × 10-3). IMa coalescent models require that microsatellite markers adhere to the stepwise mutation model; only four of nine microsatellite markers (Ifr043, Ifr052, Ifr078, and Ifr086) adhered to this model and could be used for IMa analysis. Identity tests for these four markers suggested that they are insufficient for assessment of population structure (P ID = 1.1 × 10-2, P SIB = 0.12).
Within North Fiji and Lau Basins, four of nine microsatellite markers (Ifr043, Ifr068, Ifr086, and Ifr103) failed to amplify, were out of equilibrium, or were under selection. These markers were excluded from all analyses involving North Fiji and Lau Basins. The five remaining microsatellite loci could identify individuals (P ID = 4.0 × 10-4), even those that share 50% genetic similarity (P SIB = 4.4 × 10-2). Only two of those microsatellites (Ifr052 and Ifr078) adhered to the stepwise mutation model and could be used in IMa analyses. Identity tests for these two markers suggested that they are insufficient for assessment of population structure (P ID = 0.13, P SIB = 0.35).
Population structure within Manus Basin
Mitochondrial genealogies revealed two frequent haplotypes at all Manus Basin sites (individuals per haplotype > 30; Figure 2). Less abundant haplotypes radiated from the dominant haplotypes in a star-like pattern (many shallow branches radiating from numerically dominant haplotypes). South Su contained the most private haplotypes (n = 10), followed by Solwara 1 (n = 8) and Solwara 8 (n = 7).

Statistical parsimony network of Ifremeria nautilei haplotypes from Manus, North Fiji, and Lau Basin. Area of circles is proportional to number of individuals that possess each haplotype. Small black circles represent inferred haplotypes not recovered in this data set. Each node represents a one base pair difference between haplotypes. Boxes delineate each putative haplotype group.
No indication of population structure was detected at the patch, mound, or site level within Manus Basin: Analysis of molecular variance (AMOVA) indicated no variation among haplotypes using either mitochondrial or microsatellite markers (P > 0.05); pairwise comparisons of F ST and φ ST revealed no significant genetic differentiation (P > 0.05); hierarchical analysis of F ST and φ ST did not reveal any significant population differentiation among nested samples (HIERFSTAT, P > 0.05; Table 3); assignment tests indicated that all I. nautilei collected in Manus Basin constitute a single population (Structure, K = 1, Figure 3).

Structure inferred two populations of Ifremeria nautilei , finding North Fiji and Lau Basin as one population distinct from Manus Basin. Structure inferred two populations of Ifremeria nautilei, finding North Fiji and Lau Basin as one population distinct from Manus Basin. Manus Basin population indicated in black, North Fiji/Lau population indicated in grey. Solid black line denotes division between samples from Manus and North Fiji Basin. Dashed line indicates division between samples from North Fiji and Lau Basin. Eight microsatellite markers were used to test K (Additional File 1, Table S2). Likelihood plateaued at K = 2.
Population structure among Manus, North Fiji, and Lau Basin
Haplotypes present in Manus, North Fiji, and Lau Basins segregated into three groups (Figure 2). Haplotype group 1 included all samples from Manus Basin while haplotype group 2 contained a mix of individuals from North Fiji and Lau Basin (Figure 2). Haplotype group 3 also contained a mix of individuals from North Fiji and Lau Basin but was not directly connected to haplotype group 2 (Figure 2).
Assignment tests of individual multi-locus genotypes identified two geographical regions hosting distinct populations of Ifremeria nautilei (STRUCTURE, K = 2; Figure 3): one in Manus Basin and a second occupying North Fiji and Lau Basins. STRUCTURE output suggests that one individual from North Fiji Basin may be second-generation migrant from Manus Basin and one individual from Manus Basin may be a second-generation migrant from the North Fiji/Lau Basin population. Hierarchical analyses of F-statistics also detected significant differentiation at this regional level (HIERFSTAT, P < 0.05; Table 3), but no significant differentiation was detected at lower levels [among sites within regions, among sites within the Manus Basin, among mounds within Manus sites, or among patches within Manus mounds (Table 3)]. Pairwise F ST and φ ST values did not show a significant increase in differentiation with geographical distance among samples separated by less than 1100 km (Table 4).
Estimates of migration, effective population size, and divergence time
As reported above, the few microsatellites that adhered to the stepwise mutation model were insufficient to adequately assess gene flow. Only the mitochondrial COI data yielded consistent results in the IMa runs. Coalescent estimates of migration rate (IMa) could not rule out the possibility of equal and bidirectional migration within Manus Basin populations. Due to the high level of connectivity between Manus Basin samples, it is unlikely that estimates of gene flow would converge on the most likely solution. The posterior probabilities display multiple peaks and gradually increasing probabilities and thus results from these analyses should be approached with caution.
Across samples of I. nautilei from all three western Pacific basins, coalescent estimates of migration rate (IMa) suggest that migration between North Fiji and Lau Basin is high (Table 5). Consistent with a single panmictic population, m-values could not be constrained between North Fiji and Lau Basin (Additional File 2, Figure S1). No evidence for migration between Manus and either North Fiji or Lau Basin was detected (IMa), suggesting that I. nautilei from Manus Basin are isolated from North Fiji and Lau Basin (m1 = m2 = 0; Table 5). Posterior probability estimates for gene flow between North Fiji and Lau Basin were inconsistent, due to the fact that Ifremeria nautilei from North Fiji and Lau Basin are part of a single, undifferentiated population. An interpretation of directional migration between these two basins should be approached with caution.
For Manus, North Fiji, and Lau Basin samples of Ifremeria nautilei, estimates of effective population size using either microsatellite linkage disequilibrium or coalescent analysis could not constrain population sizes among basins, suggesting that within each basin, effective population size is functionally infinite. Estimates of splitting time place the oldest divergence between North Fiji and Manus Basin, with a relatively recent split between North Fiji and Lau Basin (Table 5).
Discussion
The complete absence of genetic subdivision in populations of Ifremeria nautilei at distances up to 1000 kilometres suggests this species is able to colonize distant vent habitats and that the ciliated Warén's larvae produced by I. nautilei are adapted for long-distance dispersal, as hypothesized by Reynolds et al. [37]. Despite this dispersal potential, a barrier to gene flow exists between Manus and North Fiji/Lau Basin populations that are separated by 2500 kilometres. Although mitochondrial COI gene sequences and nuclear microsatellite loci are informative at different temporal scales, both markers indicated identical patterns of population structure in I. nautilei, regardless of spatial scale.
Local population structure within Manus Basin
Ifremeria nautilei from the three Manus Basin sites belong to a single, panmictic population based on mitochondrial COI gene sequences and nuclear microsatellite markers. Although patch sizes were generally small, several patches (particularly in Solwara 1) had a sufficient sample size to test and reject the hypothesis that self-recruiting patches of I. nautilei might create the appearance of panmixia within this sample set.
Ifremeria nautilei from South Su had the highest abundance of private haplotypes and private alleles. Under a scenario of colonization with subsequent migration, this pattern could suggest that South Su might serve as a source population that contributes individuals to other sites sampled in Manus Basin. This directional gene flow is consistent with the path of the St. George's Undercurrent, which enters Manus Basin from the southeast and travels northwest, encountering South Su first, then Solwara 1 and Solwara 8, before merging with the Vitiaz Straight Undercurrent to form the New Guinea Coastal Undercurrent [43]. This hypothesis could be tested with development of additional genetic markers that provide more detailed genealogical information than the microsatellite markers used in this study and by sampling and analysis of individuals from sites further west that are known to support Ifremeria nautilei. Rapid population expansion, as suggested by Fu's F S and the star-like mitochondrial genealogies, could account for the emergence of private haplotypes and alleles at each sampled site within Manus Basin. Alternatively, the negative Fu's F S values could be a result of a selective sweep on the mitochondrial genome. Additional sequenced-based nuclear markers would be needed to rule out this possibility.
Basin-scale population structure
Individuals of Ifremeria nautilei sampled from across the known range of the species in the south western Pacific could be subdivided genetically into two populations, one restricted to Manus Basin and one distributed throughout North Fiji and Lau Basins. Under an isolation-by-distance scenario, genetic differentiation is expected to gradually increase with distance [27], but distance alone does not appear to create a significant barrier to gene flow. North Fiji and Lau Basin samples, separated by ~1000 kilometres, were undifferentiated. Coalescent estimates of gene flow suggest that genetic isolation of I. nautilei populations between Manus Basin and North Fiji/Lau Basins may have existed, with occasional migration, for several hundred thousand generations (Table 5), but without an understanding of mutation rates and average generation times in these snails, it is impossible to place these estimates in a geological time frame.
The phylogeographic break between populations from Manus and North Fiji/Lau Ifremeria nautilei is striking, considering the high degree of mixing within each population. Phylogeographic breaks are often associated with oceanographic features (e.g., geomorphology, hydrology) that form effective dispersal barriers for a wide range of taxa (e.g., Cape Hatteras [44], Cape Cod [45], Easter Microplate [14]). A related provannid snail complex, Alviniconcha spp., occurs throughout southwestern Pacific basins [30] and is comprised of several cryptic species [35]. The observed phylogeographic isolation of I. nautilei populations thus does not reflect a pattern that is shared by other southwestern Pacific vent taxa.
The phylogeographic break between Manus and North Fiji/Lau populations of Ifremeria nautilei is not likely the result of a colonization event. Colonization would result in a founder effect, where the founded population contains a subset of alleles from the source population [46]. The single mitochondrial haplotype shared between the Manus and North Fiji/Lau populations is intermediate between dominant haplotypes from the two populations and may be a product of incomplete lineage sorting between formerly connected populations [47]. Haplotype Group 3 (North Fiji/Lau) is more closely related to Haplotype Group 1 (Manus Basin; Figure 2) and it consists of as many missing haplotypes as it does actual haplotypes. This abundance of missing haplotypes, relative to other haplotype groups, could be the result of inadequate sampling, or it may suggest that disproportionately more haplotypes in that lineage have gone extinct. Our interpretation is that the two populations once existed as a single population spanning Manus, North Fiji, and Lau Basins and that this population became isolated through a vicariant process that remains to be determined. The presence of potential second-generation immigrants in each population suggests that isolation might not be complete between the two regions..
While Ifremeria nautilei occurs throughout Manus, North Fiji, and Lau Basins, populations of I. nautilei follow the trend of greater endemism and limited connectivity hypothesized for species endemic to back-arc basin spreading centres [32]. In this context, it is not surprising that a population of I. nautilei is distributed through North Fiji and Lau Basin, as these two basins share genera and species [30]. Water masses tend to be retained within Lau Basin, with some movement of northwestward flowing undercurrents from Lau into North Fiji Basin (Thurnherr, unpublished data, available at http://www.ldeo.columbia.edu/~ant/LAUB-FLEX/). This undercurrent movement is consistent with the weak signal of directional gene flow from Lau into North Fiji Basin (Table 5). Likewise, the barrier to dispersal between North Fiji and Manus Basin that limits gene flow between I. nautilei populations is consistent with a barrier that was hypothesized to restrict species dispersal between these two basins [48]. The barrier could be caused by geomorphological obstacles (i.e. the Vanuatu Archipelago), the lack of depth overlap between sites in Manus Basin and sites in North Fiji and Lau Basin, or by a yet to be determined oceanographic feature.
Conclusions
Theoretical and experimental studies suggest that spatially and temporally unstable environments favor broad dispersal capabilities [49, 50], which in turn should lead to shallow or absent population subdivisions. In dynamic systems such as hydrothermal vents, where habitat availability is unpredictable, survival depends on long-distance dispersal of propagules. No significant genetic differentiation was found among samples of western Pacific Ifremeria nautilei at the patch, mound, or site levels within the Manus Basin. No differentiation was observed between samples of I. nautilei collected from North Fiji and Lau Basins, which are separated by ~1000 kilometres. The Manus Basin population of I. nautilei is isolated from that of the North Fiji and Lau Basins by an unknown process that limits contemporary gene flow.
Reproductive mode and larval type are often poor predictors of population structure in marine environments [3, 40, 41]. Species with broad dispersal potential have been reported with high levels of differentiation at spatial scales of a few kilometres or less [51–53], while species that would otherwise be expected to show fine-scale population structure have been reported to show surprisingly high levels of connectivity throughout their geographic range [54, 55]. The absence of Ifremeria nautilei population structure at all but the broadest spatial scales is consistent with long-distance dispersal and the barrier to gene flow between the North Fiji and Lau Basin population and the Manus Basin population is likely extrinsic and not related to life history characteristics.
Fine-scale spatial sampling and genetic analysis such as that used in this study can inform mitigation and best-management practices for mineral extraction at deep-sea hydrothermal vents. The Solwara 1 site is targeted for deep-sea mineral extraction [56]. A robust understanding of population genetic structure at multiple spatial scales can define natural conservation units that can be used to minimize loss of genetic diversity within and among populations of vent-restricted species [57]. For Ifremeria nautilei, high rates of gene flow among the sampled Manus Basin sites suggests that the Solwara 1 vents are likely to be repopulated from other Manus Basin localities, including South Su and Solwara 8. Monitoring of species recovery and genetic diversity as the Solwara 1 population recovers after extraction operations cease should add insight into the rates at which novel haplotypes and alleles accumulate in this species, providing a means to estimate the ages and sizes of extant populations. The Manus Basin population of I. nautilei comprises a genetically distinct unit that should be managed separately from the North Fiji/Lau population.
Methods
Geographic setting, sample collection, and DNA extraction
Ifremeria nautilei were collected from three hydrothermal vent sites in Manus Basin: Solwara 8, Solwara 1, and South Su (Figure 1). One to four patches of I. nautilei were sampled from three mounds at each site (Table 1). Samples were collected during June-July 2008 with an ST212 trenching ROV modified for biological sampling. Foot tissue was preserved in 95% ethanol. Additional I. nautilei samples were acquired from a cruise that occurred during May-June 2005 with ROV Jason II from North Fiji and Lau Basins (Figure 1). Foot tissue was stored briefly at -20°C and transferred to 95% ethanol prior to DNA extraction. Genomic DNA was isolated by Chelex-Proteinase-K extraction as described in Thaler et al. [39] and extracted DNA was stored at 4°C until amplification.
COIamplification and analysis
The mitochondrial COI (404-bp segment) region was amplified with the species-specific primers COI-3 and COI-6 [38] as follows: 10-100 ng DNA template, 10 × PCR Buffer (20 mM Tris, pH 8.8; 50 mM KCl; 0.01% Triton X-100; 0.02 mg/ml BSA), 2 mM MgCl2, 0.2 mM dNTP's, 0.5 μM each primer, and 1 unit Taq polymerase (Bioline: Taunton, MA) in a 20 μl final volume. Reaction conditions were as follows: 94°C for 1 minute; 30 cycles of 92°C for 40 s, 50°C for 60 s, 72°C for 90 s; final extension of 72°C for 5 min. Amplicons were verified on 1.8% agarose gels. To remove unincorporated nucleotides, 14 μl of PCR product was incubated with 0.2 μl 10 × ExoAP buffer (50 mM Bis-Tris, 1 mM MgCl2, 0.1 mM ZnSO4), 0.05 μl Antarctic Phosphatase (New England Biolabs: Ipswich, MA), 0.05 μl Exonuclease I (New England Biolabs: Ipswich, MA) at 37°C for 60 min followed by 85°C for 15 min to inactivate the enzymes. Bi-directional sequencing reactions were performed using the manufacturer's protocol for Big Dye Terminator Reaction (Applied Biosystems: Foster City, CA). Sequenced PCR product was purified using AMPure magnetic bead system (Agencourt: Morrisville, NC) following manufacturer's protocol, analyzed on an ABI 3730xl DNA Analyzer (Applied Biosystems International), and edited with Sequencher version 4.7 (Gene Codes: Ann Arbor, MI). Consensus sequences were compared against the NCBI GenBank database to confirm species identity [58] and aligned using the MUSCLE alignment algorithm [59]. A sequence for each unique haplotype was deposited in GenBank (North Fiji and Lau haplotypes - accession # JQ074110 to JQ074134; Manus Basin haplotypes - accession # JQ074135 to JQ074170).
Neighbor-joining phylograms of aligned mitochondrial sequences were assembled in MEGA version 4 [60] with an Alviniconcha sp. 2 as an outgroup. Statistical-parsimony networks were assembled in TCS version 1.21 (default settings; [61]). Arlequin version 3.11, [62] was used to estimate haplotype (H), nucleotide diversity (π), Fu's F s , and pairwise φST.
Microsatellite methods
Nine microsatellite markers (Ifr040, Ifr043, Ifr052, Ifr068, Ifr078, Ifr086, Ifr093, Ifr094, and Ifr103) were amplified from Manus, North Fiji, and Lau Basin samples following methods reported in Thaler et al. [39]. To assess marker quality, allelic richness and divergence from Hardy-Weinberg Equilibrium (HWE) were calculated in GENEPOP (version 4.0, [63]). Permutation tests to determine significant variation in allelic richness were conducted in F-stat (version 2.9.3.2; [64]). Departures from HWE toward heterozygote excess or deficiency were assessed for each locus using GENEPOP exact tests. Loci were screened using LOSITAN to test for the potential influence of selection (25,000 simulations; [65]). Microsatellite markers that showed deviations from HWE expectations or found to be under the influence of selection were excluded from subsequent analyses. Identity tests (PID and PSIB) were used to indicate whether a given set of microsatellites contains sufficient information to be useful for assessing population structure [66, 67]. PID and PSIB were calculated for all useful sets of microsatellite markers (Gimlet version 1.3.3; [68]).
Common statistical methods
Arlequin version 3.11 was used to conduct analysis of molecular variance (AMOVA). HIERFSTAT [69, 70] was used to assess hierarchical φST and FST at various nested scales from patch to basin. Microsatellite Analyser (MSA; [71]) was used to identify significant differentiation between patches, mounds, sites, and basins. Alpha levels were adjusts via Sequential Bonferroni correction to account for multiple tests [72]. Structure version 2.3.3 [73] was used to visualize population structure. We used an admixture model with no a priori sample data and with sampling locations as prior distributions. Analyses were conducted with a 100,000 step burn-in, 1,000,000 Markov chain Monte Carlo repetitions, and 3 replicates per level from K = 1 to 12. The most likely K was identified by the average maximal value of Ln P(D) returned by Structure. The program LDNe [74] was used in an attempt to estimate effective population size based on linkage-disequilibrium between microsatellite loci.
Isolation with migration
Migration rate (m), effective population size (θ), and divergence time (τ) between populations were estimated using the coalescent-based isolation-with-migration model implemented in IMa [75]. All estimates were scaled on mutation rates (μ) that are unknown for COI in I. nautilei, so splitting time was calibrated against two hypothetical rates: μ1 = 5 × 10-8 (determined theoretically, see [21]), and μ2 = 1.5 × 10-8 (borrowed from rates in the gastropod, Littorina littorea, see [76]). IMa runs were performed on COI and microsatellite data among three sites within Manus Basin and across all three basins. Only microsatellites that did not deviate from expectations for the stepwise mutation model could be used for IMa analyses [77].
A series of short (< 2,000,000 steps) IMa runs was conducted to optimize model parameters and determine the efficient priors for full runs. Prior probabilities and heating schemes were established at θ = 100, m = 100, and τ = 1.5, in a 40 chain geometric model. Effective sample size, autocorrelations, and trend plots were monitored to evaluate convergence. Three independent runs were compared to ensure that marginal posterior distributions had achieved similar solutions and results were averaged across the three runs. Generated trees were analysed in L-mode for best fit (default settings) and the most likely model was determined using Akaike Information Criterion [78] and 2LRR tests.
References
Scheltema RS: On dispersal and planktonic larvae of benthic invertebrates: An eclectic overview and summary of problems. Bulletin of Marine Science. 1986, 39: 290-322.
Warner RR, Cowen RK: Local retention of production in marine populations: evidence, mechanisms, and consequences. Bulletin of Marine Science. 2002, 70: 245-249.
Kelly RP, Palumbi SR: Genetic Structure among 50 Species of the Northeastern Pacific Rocky Intertidal Community. PLoS ONE. 2010, 5: e8594-10.1371/journal.pone.0008594.
Ayre DJ, Minchinton TE, Perrin C: Does life history predict past and current connectivity for rocky intertidal invertebrates across a marine biogeographic barrier?. Molecular Ecology. 2009, 18: 1887-1903. 10.1111/j.1365-294X.2009.04127.x.
Kinlan BP, Gaines SD, Lester SE: Propagule dispersal and the scales of marine community process. Diversity and Distributions. 2005, 11: 139-148. 10.1111/j.1366-9516.2005.00158.x.
Jones GP, Milicich MJ, Emslie MJ, Lunow C: Self-recruitment in a coral reef fish population. Nature. 1999, 402: 802-804. 10.1038/45538.
Swearer SE, Shima JS, Hellberg ME, Thorrold SR, Jones GP, Robertson DR, Morgan SG, Selkoe KA, Ruiz GM, Warner RR: Evidence of self-recruitment in demersal marine populations. Bulletin of Marine Science. 2002, 70 (Supplement 1): 251-271.
Tunnicliffe V, Fowler CMR: Influence of sea-floor spreading on the global hydrothermal vent fauna. Nature. 1996, 379: 531-533. 10.1038/379531a0.
Cavanaugh CM: Microbial Symbiosis: Patterns of Diversity in the Marine Environment. American Zoologist. 1994, 34: 79-89.
Tunnicliffe V, Juniper SK: Dynamic character of the hydrothermal vent habitat and the nature of sulphide chimney fauna. Progress In Oceanography. 1990, 24: 1-13. 10.1016/0079-6611(90)90015-T.
Tunnicliffe V, Embley RW, Holden JF, Butterfield DA, Massoth GJ, Juniper SK: Biological colonization of new hydrothermal vents following an eruption on Juan de Fuca Ridge. Deep Sea Research Part I: Oceanographic Research Papers. 1997, 44: 1627-1644. 10.1016/S0967-0637(97)00041-1.
Shank TM, Fornari DJ, Von Damm KL, Lilley MD, Haymon RM, Lutz RA: Temporal and spatial patterns of biological community development at nascent deep-sea hydrothermal vents (9°50'N, East Pacific Rise). Deep Sea Research Part II: Topical Studies in Oceanography. 1998, 45: 465-515. 10.1016/S0967-0645(97)00089-1.
Vrijenhoek RC: Gene Flow and Genetic Diversity in Naturally Fragmented Metapopulations of Deep-Sea Hydrothermal Vent Animals. Journal of Heredity. 1997, 88: 285-293.
Vrijenhoek RC: Genetic diversity and connectivity of deep-sea hydrothermal vent metapopulations. Molecular Ecology. 2010, 19: 4391-4411. 10.1111/j.1365-294X.2010.04789.x.
Marsh AG, Mullineaux LS, Young CM, Manahan DT: Larval dispersal potential of the tubeworm Riftia pachyptila at deep-sea hydrothermal vents. Nature. 2001, 411: 77-80. 10.1038/35075063.
Thomson RE, Mihály SF, Rabinovich AB, McDuff RE, Veirs SR, Stahr FR: Constrained circulation at Endeavour ridge facilitates colonization by vent larvae. Nature. 2003, 24: 545-549.
Young CR, Fujio S, Vrijenhoek RC: Directional dispersal between mid-ocean ridges: deep-ocean circulation and gene flow in Ridgeia piscesae. Molecular Ecology. 2008, 17: 1718-1731. 10.1111/j.1365-294X.2008.03609.x.
Creasey S, Rogers AD, Tyler PA: Genetic comparison of two populations of the deep-sea vent shrimp Rimicaris exoculata (Decapoda: Bresiliidae) from the Mid-Atlantic Ridge. Marine Biology. 1996, 125: 473-482.
Shank TM, Lutz RA, Vrijenhoek RC: Molecular systematics of shrimp from deep-sea hydrothermal vents: enigmatic "small orange" shrimp from the Mid-Atlantic Ridge are juvenile Rimicaris exoculata. Marine Molecular Biology and Biotechnology. 1998, 7: 88-96.
Teixera S, Cambon-Bonavita M-A, Serrão E, Desbruyères D, Arnaud-Haond S: Recent population expansion and connectivity in the hydrothermal shrimp Rimicaris exoculata along the Mid-Atlantic Ridge. Journal of Biogeography. 2010, 38: 564-574.
Audzijonytė A, Vrijenhoek RC: When gaps really are gaps: statistical phylogeography of hydrothermal vent invertebrates. Evolution. 2010, 64: 2369-2384.
Won Y, Young CR, Lutz RA, Vrijenhoek RC: Dispersal barriers and isolation among deep-sea mussel populations (Mytilidae: Bathymodiolus) from eastern Pacific hydrothermal vents. Molecular Ecology. 2003, 12: 169-184.
Hurtado LA, Lutz RA, Vrijenhoek RC: Distinct patterns of genetic differentiation among annelids of eastern Pacific hydrothermal vents. Molecular Ecology. 2004, 13: 2603-2615. 10.1111/j.1365-294X.2004.02287.x.
Plouviez S, Shank TM, Faure B, Daguin-Theibaut C, Viard F, Lallier FH, Jollivet D: Comparative phylogeography among hydrothermal vent species along the East Pacific Rise reveals vicariant processes and population expansion in the South. Molecular Ecology. 2009, 18: 3903-3917. 10.1111/j.1365-294X.2009.04325.x.
Johnson SB, Young CR, Jones WJ, Waren A, Vrijenhoek RC: Migration, Isolation, and Speciation of Hydrothermal Vent Limpets (Gastropoda; Lepetodrilidae) Across the Blanco Transform Fault. Biological Bulletin. 2006, 210: 140-157. 10.2307/4134603.
Shank TM, Halanych KM: Toward a mechanistic understanding of larval dispersal: insights from genomic fingerprinting of the deep-sea hydrothermal vent tubeworm Riftia pachyptila. Marine Ecology. 2007, 28: 25-35. 10.1111/j.1439-0485.2007.00146.x.
Wright S: Isolation by distance. Genetics. 1943, 28: 114-138.
Black MB, Lutz RA, Vrijenhoek RC: Gene flow among vestimentiferan tube worm (Riftia pachyptila) populations from hydrothermal vents of the Eastern Pacific. Marine Biology. 1994, 120: 33-39.
Coykendall DK, Johnson SB, Karl S, Lutz RA, Vrijenhoek RC: Genetic diversity and structure of Riftia pachyptila metapopulations (Annelida: Siboglinidae) from deep-sea hydrothermal vents. BMC Evolutionary Biology. 2011, 11: 98-10.1186/1471-2148-11-98.
Desbruyères D, Hashimoto J, Fabri MC: Composition and biogeography of hydrothermal vent communities in western Pacific back-arc basins. Geophysical monograph. 2006, 166: 215-234.
Van Dover CL, German CR, Speer KG, Parson LM, Vrijenhoek RC: Evolution and Biogeography of Deep-Sea Vent and Seep Invertebrates. Science. 2002, 295: 1253-1257. 10.1126/science.1067361.
Van Dover CL: The Ecology of Deep-Sea Hydrothermal Vents. 2000, Princeton, Princeton University Press
Kyuno A, Shintaku M, Fujita Y, Matsumoto H, Utsumi M, Watanabe H, Fujiwara Y, Miyazaki JI: Dispersal and Differentiation of Deep-Sea Mussels of the Genus Bathymodiolus (Mytilidae, Bathymodiolinae). Journal of Marine Biology. 2009, doi:10.1155/2009/625672
Watanabe H, Tsuchida S, Fujikura K, Yamamoto H, Inagaki F, Kyo M, Kojima S: Population history associated with hydrothermal vent activity inferred from genetic structure of neoverrucid barnacles around Japan. Marine Ecology Progress Series. 2005, 288: 233-240.
Kojima S, Segawa R, Fijiwara Y, Fujikura K, Ohta S, Hashimoto J: Phylogeny of Hydrothermal-Vent-Endemic Gastropods Alviniconcha spp. from the Western Pacific Revealed by Mitochondrial DNA Sequences. Biological Bulletin. 2001, 200: 298-304. 10.2307/1543511.
Bouchet P, Warén A: Ifremeria nautilei, nouveau gastéropode d'évents hydrothermaux, probablement associé à des bactéries symbiotiques (Ifremeria nautilei, a new gastropod from hydrothermal vents, probably associated with symbiotic bacteria). Comptes rendus de l'Académie des sciences. Série 3, Sciences de la vie. 1991, 312: 495-501.
Reynolds KC, Watanabe H, Strong EE, Sasaki T, Uematsu K, Miyake H, Kojima S, Suzuki Y, Fujikura K, Kim S, Young CG: New Molluscan Larval Form: Brooding and Development in a Hydrothermal Vent Gastropod, Ifremeria nautilei (Provannidae). Biological Bulletin. 2010, 219: 7-11.
Kojima S, Segawa R, Fujiwara Y, Hashimoto J, Ohta S: Genetic differentiation of populations of a hydrothermal vent-endemic gastropod, Ifremeria nautilei, between the North Fiji Basin and the Manus Basin revealed by nucleotide sequences of mitochondrial DNA. Zoological Science. 2000, 17: 1167-1174. 10.2108/zsj.17.1167.
Thaler A, Zelnio KA, Jones R, Carlsson J, Van Dover CL, Schultz T: Characterization of 12 polymorphic microsatellite loci in Ifremeria nautilei, a chemoautotrophic gastropod from deep-sea hydrothermal vents. Conservation Genetics Resources. 2010, doi:10.1007/s12686-010-9174-9
Hellberg ME: Gene Flow and Isolation among Populations of Marine Animals. Annual Review of Ecology, Evolution, and Systematics. 2009, 40: 291-310. 10.1146/annurev.ecolsys.110308.120223.
Palumbi SR: Marine Reserves and Ocean Neighborhoods: The Spatial Scale of Marine Populations and Their Management. Annual Review of Environment and Resources. 2004, 29: 31-68. 10.1146/annurev.energy.29.062403.102254.
Fu XY: Statistical properties of segregating sites. Theoretical Population Biology. 1995, 48: 172-197. 10.1006/tpbi.1995.1025.
Zenk W, Siedler G, Ishida A, Holfort J, Kashino Y, Kuroda Y, Miyama T, Müller TJ: Pathways and variability of the Antarctic Intermediate Water in the western equatorial Pacific Ocean. Progress In Oceanography. 2005, 67: 245-281. 10.1016/j.pocean.2005.05.003.
Baker P, Austin JD, Bowen BW, Baker SM: Range-wide population structure and history of the northern quahog (Merceneria merceneria) inferred from mitochondrial DNA sequence data. ICES Journal of Marine Science. 2008, 65: 155-163. 10.1093/icesjms/fsn007.
Jennings RM, Shank TM, Mullineaux LS, Halanych KM: Assessment of the Cape Cod Phylogeographic Break Using the Bamboo Worm. Journal of Heredity. 2009, 100: 86-96.
Mayr E: Systematics and the origin of species. 1942, New York, Columbia University Press
Cunningham CW, Collins TM: Beyond area relationships: extinction and recolonization in molecular marine biogeography. Molecular approaches to ecology and evolution. 1998, 297-321.
Hessler RR, Lonsdale PF: Biogeography of Mariana Trough hydrothermal vent communities. Deep Sea Research Part A: Oceanographic Research Papers. 1991, 38: 185-199. 10.1016/0198-0149(91)90079-U.
Cohen D, Levin SA: Dispersal in patchy environments: the effects of temporal and spatial structure. Theoretical Population Biology. 1991, 39: 63-99. 10.1016/0040-5809(91)90041-D.
Friedenberg NA: Experimental evolution of dispersal in spatiotemporally variable microcosms. Ecology Letters. 2003, 6: 953-959. 10.1046/j.1461-0248.2003.00524.x.
Banks SC, Piggot MP, Williamson JE, Bové U, Holbrook NJ, Beheregaray LB: Oceanic variability and coastal topography shape genetic structure in a long-dispersing sea urchin. Ecology. 2007, 88: 3055-3064. 10.1890/07-0091.1.
Bird CE, Holland BS, Bowen BW, Toonen RJ: Contrasting phylogeography in three endemic Hawaiian limpets (Cellana spp.) with similar life histories. Molecular Ecology. 2007, 16: 3173-3186. 10.1111/j.1365-294X.2007.03385.x.
Tatarenkov A, Healey CIM, Avise JC: Microgeographic population structure of green swordail fish: genetic differentiation despite abundant migration. Molecular Ecology. 2010, 19: 257-268. 10.1111/j.1365-294X.2009.04464.x.
Miller KJ, Ayre DJ: Population structure is not a simple function of reproductive mode and larval type: insights from tropical corals. Journal of Animal Ecology. 2008, 77: 713-724. 10.1111/j.1365-2656.2008.01387.x.
Blanquer A, Uriz M: Population genetics at three spatial scales of a rare sponge living in fragmented habitats. BMC Evolutionary Biology. 2010, 10: 13-10.1186/1471-2148-10-13.
Coffey Natural Systems: Environmental Impact Statement: Nautilus Minerals Niugini Limited, Solwara 1 Project, Vol. A. 2008, Coffey Natural Systems, Queensland, Australia, 226-
Van Dover CL: Mining seafloor massive sulphides and biodiversity: what is at risk?. ICES Journal of Marine Science. 2011, 68: 341-348. 10.1093/icesjms/fsq086.
Benson DA, et al: GenBank. Nucleic Acids Research. 2005, 3: D34-D38.
Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Resources. 2004, 32: 1792-1797. 10.1093/nar/gkh340.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Molecular Biology and Evolution. 2007, 24: 1596-1599. 10.1093/molbev/msm092.
Clement M, Posada D, Crandall KA: TCS: a computer program to estimate gene genealogies. Molecular Ecology. 2000, 9: 1657-1659. 10.1046/j.1365-294x.2000.01020.x.
Excoffier L, Laval G, Schneider S: Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evolutionary bioinformatics online. 2005, 1: 47-
Rousset F: Genepop'007: a complete re-implementation of the GENEPOP software for Windows and Linux. Molecular Ecology Resources. 2008, 8: 103-106. 10.1111/j.1471-8286.2007.01931.x.
Goudet J: FSTAT (Version 1.2): A computer program to calculate F-statistics. Journal of Heredity. 1995, 86: 485-486.
Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G: LOSITAN: A workbench to detect molecular adaptation based on a F ST -outlier method. BMC Bioinformatics. 2008, 9: 323-10.1186/1471-2105-9-323.
Woods JG, Paetkau D, Lewis D, McLellan BN, Proctor M, Strobeck C: Genetic tagging of free-ranging black and brown bears. Wildlife Society Bulletin. 1999, 27: 616-627.
Taberlet P, Luikart G: Non-invasive genetic sampling and individual identification. Biological Journal of the Linnean Society. 1999, 68: 41-55. 10.1111/j.1095-8312.1999.tb01157.x.
Valière N: GIMLET: a computer program for analysing genetic individual identification data. Molecular Ecology Notes. 2002, 2: 377-379.
Goudet J: hierfstat, a package for r to compute and test hierarchical F-statistics. Molecular Ecology Notes. 2005, 5: 184-186. 10.1111/j.1471-8286.2004.00828.x.
de Meeûs T, Goudet J: A step-by-step tutorial to use HierFstat to analyze populations hierarchically structured at multiple levels. Infection, Genetics and Evolution. 2007, 7: 731-735. 10.1016/j.meegid.2007.07.005.
Dieringer D, Schlötterer C: microsatellite analyzer (MSA): a platform independent analysis tool for large microsatellite data sets. Molecular Ecology Notes. 2003, 3: 167-169. 10.1046/j.1471-8286.2003.00351.x.
Rice WR: Analyzing Tables of Statistical Tests. Evolution. 1989, 43: 223-225. 10.2307/2409177.
Pritchard JK, Stephens M, Donnelly P: Inference of Population Structure Using Multilocus Genotype Data. Genetics. 2000, 155: 945-959.
Waples RS, Do C: ldne: a program for estimating effective population size from data on linkage disequilibrium. Molecular Ecology Resources. 2008, 8: 753-756. 10.1111/j.1755-0998.2007.02061.x.
Hey J, Nielsen R: Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proceedings of the National Academy of Sciences. 2007, 104: 2785-2790. 10.1073/pnas.0611164104.
Cunningham CW: How to Use Genetic Data to Distinguish Between Natural and Human-Mediated Introduction of Littorina littorea to North America. Biological Invasions. 2008, 10: 1-6. 10.1007/s10530-007-9099-8.
Hey J, Won Y-J, Sivasundar A, Nielsen R, Markert JA: Using nuclear haplotypes with microsatellites to study gene flow between recently separated Cichlid species. Molecular Ecology. 2004, 13: 909-919. 10.1046/j.1365-294X.2003.02031.x.
Kuhner MK: Coalescent genealogy samplers: windows into population history. Trends in Ecology & Evolution. 2009, 24: 86-93. 10.1016/j.tree.2008.09.007.
Acknowledgements
Funding was provided by a research contract from Nautilus Minerals Niugini Limited (CLVD, JC, and TS), Duke University (ADT, TS, JC, CLVD), the Oak Foundation (ADT), grants from the US National Science Foundation (OCE-030554 to CLVD and OCE-0241613 to RCV), the Beaufort Marine Research Award in Fish Population Genetics funded by the Irish Government under the Sea Change Programme (JC). The authors thank Dr. Samantha Smith of Nautilus Minerals, the captain and crew of the M/V Nor Sky, Canyon Offshore ROV team, Rebecca Jones and Pen-Yuan Hsing for assistance with field sampling, and Dr. Sophie Plouviez for discussion. Ifremeria nautilei from Manus Basin are on loan to Duke University from the government of Papua New Guinea.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
CLVD, JC, TFS, and ADT conceived the study; ADT, TFS and RCV collected samples; ADT, KZ, WS, JC, and TFS undertook molecular benchwork and analyses; ADT, CC, RCV, KZ, and CLVD drafted the manuscript. All authors have read and approved the final manuscript.
Electronic supplementary material
12862_2011_1958_MOESM1_ESM.DOC
Additional file 1: Table S1 - Summary statistics for nine microsatellite loci amplified from populations of Ifremeria nautilei within Manus Basin. n = number of individuals, a = number of alleles, Rs = allelic richness, P A = number of private alleles, HE = expected heterozygosity HO = observed heterozygosity (bold = significant deviation from HWE, * = significant heterozygote excess, † = significant heterozygote deficiency, - = null amplification or monomorphic genotype). Patches that contained only 1 allele not shown. Table S2 - Summary statistics for eight microsatellite loci amplified from populations of Ifremeria nautilei from Manus, North Fiji, and Lau Basin. n = number of individuals, a = number of alleles, Rs = allelic richness, P A = number of private alleles, HE = expected heterozygosity HO = observed heterozygosity (bold = significant deviation from HWE, * = significant heterozygote excess, † = significant heterozygote deficiency, - = null amplification or monomorphic genotype). Patches that contained only 1 allele not shown. (DOC 84 KB)
12862_2011_1958_MOESM2_ESM.PNG
{kind=link}
Additional file 2: Figure S1 - Posterior probability densities for migration of Ifremeria nautilei between basins in the western Pacific, based on mitochondrial COI gene region. (PNG 137 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Thaler, A.D., Zelnio, K., Saleu, W. et al. The spatial scale of genetic subdivision in populations of Ifremeria nautilei, a hydrothermal-vent gastropod from the southwest Pacific. BMC Evol Biol 11, 372 (2011). https://doi.org/10.1186/1471-2148-11-372
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-11-372