
Abstract
Background
Cryptic population structure can be an indicator of incipient speciation or historical processes. We investigated a previously documented deep break in the mitochondrial haplotypes of Heliconius erato chestertonii to explore the possibility of cryptic speciation, and also the possible presence of endosymbiont bacteria that might drive mitochondrial population structure.
Results
Among a sample of 315 individuals from 16 populations of western Colombia, two principal mtDNA clades were detected with 2.15% divergence and we confirmed this structure was weakly associated with geography. The first mtDNA clade included 87% of individuals from northern populations and was the sister group of H. erato members of Andes western, while the second clade contained most individuals from southern populations (78%), which shared haplotypes with an Ecuadorian race of H. erato. In contrast, analysis using AFLP markers showed H. e. chestertonii to be a genetically homogeneous species with no association between mitochondrial divergence and AFLP structure. The lack of congruence between molecular markers suggests that cryptic speciation is not a plausible explanation for the deep mitochondrial divergence in H. e chestertonii. We also carried out the first tests for the presence of endosymbiontic bacteria in Heliconius, and identified two distinct lineages of Wolbachia within H. e. chestertonii. However, neither of the principal mitochondrial clades of H. e. chestertonii was directly associated with the patterns of infection.
Conclusions
We conclude that historical demographic processes are the most likely explanation for the high mitochondrial differentiation in H. e. chestertonii, perhaps due to gene flow between Cauca valley H. e. chestertonii and west Pacific slope populations of H. erato.
Similar content being viewed by others

Background
Sequences derived from the mitochondrial genome are commonly used both in species delimitation and historical phylogeography. For example, deep divergence in mitochondrial DNA sequences (mtDNA) between related individuals is often taken as evidence for the existence of cryptic species [1–4]. The discovery of cryptic species-level variation has important implications for characterising biodiversity and for studies of speciation. Nonetheless, it is now well recognised that inference of evolutionary processes and species boundaries from mitochondrial sequences alone can be problematic [5, 6]. For example, similar patterns of divergence can be due to host-parasite interactions, whereby selection leads different molecular markers to show different histories [7]. Mitochondrial lineages may also be retained through admixture between divergent species or populations [8], or perhaps due to unusual population structures [9, 10]. Indeed, in diverse tropical radiations, mtDNA 'barcoding' may perform rather poorly as a species identification tool [6]. It is therefore of general interest to pursue individual cases of deep mtDNA divergence in order to determine how often such divergence is indeed an indicator of cryptic species-level variation.
Heliconius butterflies are an excellent ecological and genetic system for studying speciation. These unpalatable butterflies are recognized for their diversity of wing color patterns associated with mimicry [11, 12]. The classical example of this adaptive radiation occurs between the comimetic species H. erato and H. melpomene. These butterflies co-occur in Central and South America and show convergent changes in their color pattern. They are represented by more than 20 different geographic forms which are considered subspecies [13]. Neutral molecular markers show geographic structure among subspecies of H. erato and H. melpomene, and most named races fall within a particular geographic clade [13–16]. However, in H. erato two forms: H. e. hydara and H. e. chestertonii are polyphyletic in the mitochondrial phylogeny [13, 16]. Individuals of H. e. chestertonii fall into two distinct mtDNA clades that show over 2% divergence, with no clear biogeographic explanation [17]. H. e. chestertonii is found in the western Colombian Andes on disturbed, dry habitats and forms a hybrid zone with the geographically closest subspecies: H. e. venus. Although H. e. chestertonii is a member of the erato clade, it has an unusual wing color pattern compared with the characteristic red/yellow/black pattern of H. erato. H. e. chestertonii has an iridescent and melanic forewing while the hindwing displays a broad yellow band. The co-mimic for this wing color pattern is H. cydno weymeri f. gustavi, a member of the H. melpomene clade.
Rapid evolution in the early stages of the speciation process is expected to lead to incongruence between morphological and molecular markers [14, 18, 19]. However, in H. e. chestertonii the deep divergence in the mitochondrial haplotypes is not readily explained through ancestral polymorphism or hybridization. Such mtDNA division has not been observed in any other Heliconius taxon to date. One possible explanation might be that structure is due to the presence of endosymbiotic bacteria [19–21]. Wolbachia are intracellular bacteria and infect numerous species of arthropods and nematodes [22]. Interactions between these microorganisms and their eukaryotic hosts often has consequences for host reproduction, leading in some cases to breaks between populations or species [23, 24]. Wolbachia are inherited maternally, so their evolutionary fate is tightly linked to that of the mitochondrion [25]. Furthermore, hybridization between H. e. chestertonii and its nearby relative, H. e. venus, produces partially infertile eggs [26]. Hybrid sterility of this form can also be generated by endosymbionts where parental populations are infected by different strains.
However, an alternative hypothesis for the mtDNA break within the continuous distribution of H. e. chestertonii is cryptic reproductive isolation, unrelated to endosymbionts. Given the emphasis on wing color pattern in Heliconius speciation, a potential case of cryptic speciation would be of considerable interest. To investigate these possibilities we first extend previous mtDNA sampling to better document the distribution of mtDNA lineages. In order to test for cryptic speciation, we have then complemented these data with nuclear Amplified Fragment Length Polymorphisms (AFLP) markers, to provide a comparison with nuclear biparentally inherited markers. Finally, we have tested for a variety of endosymbiotic bacteria to investigate whether mtDNA structure could be a result of patterns of infection among populations.
Results
Mitochondrial relationships
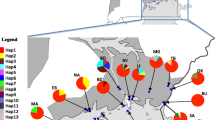
Our expanded sampling of mitochondrial sequences confirms the deep divergence within H. e. chestertonii, with a similar topology obtained with either Bayesian or parsimony analyses (Figure 1). As expected, both taxa studied here are members of the E1 clade, containing H. erato species from Costa Rica, Panama, Colombia and west Ecuador [16]. H. e. venus forms a monophyletic group together with some members of H. e. hydara, while individuals of H. e. chestertonii showed a complex relationship with other subspecies and is a polyphyletic taxon. Three principal clades are observed. The first primarily includes individuals collected in populations north of the hybrid zone in Calima River Valley (Figure 1). These haplotypes form a sister group to the other subspecies of the western clade E1: H. e. petiverana, H. e venus and H. e. hydara (Figure 1). The second principal clade includes the west Ecuadorean race H. e. cyrbia and primarily individuals of H. e. chestertonii collected in southern populations (Figure 1). The third clade, with only three northern individuals, is sister to all races of H. erato in the western E1 clade and south clade of H. e. chestertonii. The different clades involving members of H. e. chestertonii has strong bootstrap support (100%) and a high posterior probability (1) (Figure 1). A haplotype network analysis revealed the same relationships among populations (Additional file 1). Three main star-like species/geographical clusters, H. e. venus, H. e. chestertonii-North and H. e. chestertonii-South. A topology test showed that monophyly of H. e. chestertonii could be strongly rejected (P < 0.0001 Shimodaira-Hasegawa test).

Sampling sites of Heliconius erato chestertonii and H. e . venus and bayesian tree for the Clade E1 of Heliconius erato. Symbols correspond to population of origin for the individuals of H. e. chestertonii, south (gray) or north (black) and H. e. venus (white). The tree is based on mitochondrial genes of Cytochrome oxidase subunits I and II, leucine-tRNA and has the same topology as the Parsimony analysis. Posterior probability and bootstrap support are indicated on branches of the principal clusters. Individuals of the different subspecies are identified by bars (right).
Genetic analysis of mitochondrial DNA (mtDNA) within populations, showed that H. e. chestertonii is more polymorphic than H. e. venus (θ W = 0.01 and θ W = 0.003 respectively, Table 1). We then subdivided H. e. chestertonii to compare polymorphism within (1) northern and southern populations and (2) the two principal clades obtained in the phylogenetic tree. The northern and southern populations had similar levels of DNA polymorphism (θ W = 0.01 and 0.009 respectively) with no fixed difference between groups and 35 shared mutations (Table 1). Nonetheless, the comparison between clades showed 17 fixed differences and just three shared mutations (Table 1). The Analysis of Molecular Variance (AMOVA) showed that the maximum variability is within populations (63.65%) and only 18.22% of the total variability was due to geography, and a further 18.13% explained by species (groups in the AMOVA). When the species were analyzed separately, we found higher genetic structure among localities of H. e. chestertonii than between H. e. venus populations (F ST = 0.2775, P < 0.01; F ST = 0.1197, P > 0.5; Additional file 2).
AFLP Analysis
Of the 327 loci examined, we found that 318 were polymorphic, representing 97% of loci. Genetic diversity statistics estimated from AFLP data showed that populations are differentiated within and among species (Additional file 3, P < 0.001). Both H. e. venus and H. e. chestertonii had values of genetic diversity of 27.6%, while the hybrid zone populations (Calima River Valley) were more diverse than in any allopatric locality (H. e. chestertonii (CV) = 29% and H. e. venus (CV) = 30%).
The AFLP-based bayesian population assignment test identified two clusters which correspond to H. e. chestertonii and H. e. venus (Figure 2a, L = -20596.26). We compared the mean likelihood values for all runs with the statistic ΔK and confirmed that K = 2 is the best estimate for this data. Additionally, the PCA analysis gave a consistent result, with 56% of the variance in the data explained by differences between species (Additional file 4). In addition, all the populations used in this study showed significant genetic structure for AFLP markers (Figure 3 and Additional file 2). The AMOVA showed that 48% of the total variation in the AFLPs was explained within populations and the 42% among groups, which correspond to species in our analysis. When we excluded H. e. venus and repeated the AMOVA grouping data for northern and southern populations of H. e. chestertonii, only 9% of the variation was accounted for by geography. In order to test for congruent differentiation between AFLP and mtDNA markers, we ran a Structure analysis using only those individuals with data for mtDNA and assuming K = 3 (putatively representing H. e. venus and the two H. e. chestertonii clades). This analysis showed no evidence for any population structure associated with the mtDNA clades (Figure 2b).

Structure Analysis. a. Graphical representation of results obtained from Structure. Upper black and gray bars represent the phenotype of each individual, H. e. chestertonii and H. e. venus respectively. The colours represent the Bayesian clusters when the analysis was carried out with K = 2 (upper, L = -20596.26) and K = 3 (lower, L = -20852.973) and correspond to H. e. venus (yelow) and H. e. chestertonii (violet and pink). The lower bars and letters show the population origin of individuals (for description of locations see Table 3). b. The results for K = 3 including only the individuals used in mtDNA analysis (L = -12353). Lower black and gray bars represent individuals in the southern and northern clades.

Matrix of pairwise F ST . Upper and lower matrices show the F ST values for mtDNA and AFLP markers respectively, between each population. H. e. chestertonii (black) and H. e. venus (red) localities are provided in Table 3. In the Calima River Valley hybrid zone, individuals were separated by phenotype: H. e. chestertonii (CV) or H. e. venus (CV*).
Interactions with endosymbionts
PCR analysis of 307 wild caught H. e. chestertonii did not identify any positive results for Ricketsia and Spiroplasma infection. All screens were run with a positive control of DNA from known infected individuals of other insects, so this failure is unlikely to be due to technical PCR error, although we cannot completely rule out the presence of a divergent strain of one of these bacterial taxa. In contrast, 7% of individuals screened (10 females/11males) were infected with Wolbachia. There are two major clades within Wolbachia, both of which are known to infect insects. Interestingly, the sequences derived from our samples fell into both clades, with 19 in clade-B and the remaining two in clade-A (Table 2). The presence of this endosymbiont is almost exclusive to H. e. chestertonii, with just one female H. e. venus from the hybrid zone infected. Most of these butterflies were collected in northern populations of H. e. chestertonii (85%). However not all individuals of the same population were infected and some localities did not show the presence of Wolbachia.
Discussion
It is generally accepted that genetic differentiation between subpopulations can lead to the formation of new reproductively isolated species over time [27]. Phylogeographic analysis can be useful in identifying both cryptic species and incipient subpopulations on the way to becoming new species [2, 3]. The analysis of this cryptic population structure can show how genetic, behavioral and ecological processes have acted during the earliest stages of speciation [27]. Studies that include independent sources of evidence, such as morphological comparisons, reproductive biology and phylogenetics, are necessary to understand the history of diverging lineages and to resolve species identification [28].
In this study, we first confirmed the unusual degree of mitochondrial divergence within H. e. chestertonii. Our analysis was based on a broad sampling of H. e. chestertonii which enabled the confirmation of two principal groups of mitochondrial haplotypes (Figure 1 and Additional file 1). Our broader sampling showed that these clades are not completely associated with the geographic distribution of the populations, as had been suspected previously (Figure 2b) [17]. The principal haplotype groups (Figure 1 and Additional file 1) were on average 2.15% divergent, similar to that estimated in previous studies [13, 15, 17]. This is considerably more divergent than within any other race of H. erato, apart from the trans-Andean biogeographic break which occurs within the distribution of the race H. e. hydara as described above. However, here there is no clear biogeographic context for this population structure. Nonetheless, the haplotypes do show some geographic structure, with 87% of northern individuals being Clade 1 and 78% of southern individuals Clade 2.
Cryptic speciation
If cryptic speciation was occurring, whereby two species are considered as one based on wing pattern morphology, we would expect the mtDNA haplotypes to be associated with a detectable level of genetic differentiation at nuclear markers. Such cryptic species have been identified in a diversity of organisms such as bryophytes, fungi, elasmobranches and arthropods [1, 4, 27, 29–32]. Within Heliconius, a case of cryptic speciation has been recently discovered in two closely related species: H. timareta and H. melpomene [33]. However, in H. e. chestertonii we have found that despite deep divergence in mtDNA between two principal clades (2.15%), the AFLP analysis shows H. e. chestertonii to be a genetically homogeneous species (Figure 2).
Endosymbionts infection
Discordance between maternally inherited genetic markers and those transmitted biparentally, can often be explained by the spread of endosymbionts such as Wolbachia, Spiroplasma, Rickettsia, Arsenophonus, Cardinium, and others [34–37]. In most of the cases, these parasites are transmitted together with the mitochondrial genome through the egg cytoplasm, so associations over time can be detected when the mitochondrial genome is analyzed [38]. Some of these endosymbiont microorganisms can lead to reproductive alterations in their arthropod hosts and will lead to divergence between populations [19, 39]. This is the first published study in which the presence of endosymbionts is tested in Heliconius, and we have identified two distinct lineages of Wolbachia within H. e. chestertonii. However, neither of the principal mitochondrial clades of H. e. chestertonii is directly associated with the infection (Table 2). We did not find evidence of presence of other endosymbionts in populations of H. e. chestertonii. We should add a caveat to our results, which is that our PCR assay might not have detected all possible infected individuals. Indeed, the stage of development of the host can lead to over or underestimates of the real density of Wolbachia within populations [22]. This study was limited to adult butterflies, and future analysis might include other stages such as egg, larvae and pupae. Nonetheless, the density of Wolbachia estimated within H. e. chestertonii populations (7%) would be considered "very low" according to a recent classification [40]. In the future it would be interesting to further investigate the phenotypic effects of this infection. It is also of course possible that another endosymbiont is present, which was not sampled with the PCR assays described here. However, for the moment there is no evidence that the mitochondrial structure in H. e. chestertonii is a result of endosymbiont infection. In summary, we have provided no evidence that mtDNA structure in H. e. chestertonii is due to either cryptic speciation or endosymbiotic bacteria. This leaves historical processes within the species as the most likely cause for the pattern.
Historical processes
Climatic changes during glacial and interglacial periods can lead to contractions, expansions and fragmentations of populations [41–44]. The 2% divergence between lineages within H. e. chestertonii suggests divergence within approximately the last million years. Even without such vicariance, isolation by distance can lead to genetic structure within species. Intriguingly, recent work on H. cydno has shown that this species similarly has a marked genetic break in the center of its Cauca Valley range (Arias and Salazar, pers. comm.). In both species, southern Cauca populations are more closely related to subspecies on the Pacific coast than those in the North. A plausible scenario is that the Cauca Valley has been subject to a double colonization first from the central Andean valleys and subsequently from the Pacific populations in the west. Nonetheless, we are not aware of any geological evidence that would support this hypothesis.
Conclusions
Our genetic analysis shows that northern and southern populations of H. e. chestertonii are genetically differentiated, but with only frequency differences in mtDNA clades, and no corresponding genetic structure at nuclear markers, despite the deep divergence (2.15%) between the two principal mtDNA clades. Our results support the assertion that mtDNA evidence alone should be used with caution in delimiting species boundaries. In this case, divergent haplotype groups within populations could not be explained by either cryptic speciation or endosymbiont infections.
Methods
Butterflies collections
Between 1997 and 2008, 315 specimens of Heliconius erato chestertonii and H. e. venus were collected at 15 different localities throughout southwest Colombia and one locality in Panama (Figure 1, Table 3). These two members of H. erato form a bimodal hybrid zone mantained by strong premating isolation in one of these Colombian localities [17, 26]. The body of each butterfly was separated from the wings and preserved in DMSO 96% at -80°C, while wings were kept in glassine envelopes. Tissue and wings are stored in the Instituto de Genética, Universidad de los Andes, Colombia or in CDJs laboratory, University of Cambridge, UK (Table 3).
DNA Butterflies extraction
Genomic DNA was extracted from one-third of thorax or the end of the abdomen using DNeasy Blood & Tissue kit (Qiagen) following the manufacturer's tissue-extraction protocol. The thorax extractions were used for mitochondrial and AFLPs analysis and abdomen extractions for endosymbiont assays (Table 3).
Sequencing and phylogenetic analysis of mitochondrial markers
At least six individuals of each population, three of each sex when possible (Table 3), were used to amplify a total of 1551 bp of a mitochondrial region, which covered the subunits I and II of Cytocrome Oxidase (CO I_II) and leucine-tRNA. We used published primers and PCR conditions from Beltran et al. 2002 for our amplifications. Subsequently, all the PCR products were purified for sequencing with Exonuclease I and Shrimp Alkaline Phosphatase enzymes (Fermentas) and sent to Macrogen Sequencing Service (Macrogen, Korea). Sequence editing was performed using Geneious Ver. 5.4 [45]. Sequences were aligned using Clustal W Ver. 2.0 [46] and checked for reading-frame errors in protein-coding regions with MacClade Ver. 4.08 [47]. Sequences were deposited in GenBank (Access numbers: JF912810-JF912880). To complement our analysis, we included publicly available sequences of H. erato [13, 16, 17] and sequences of H. hecalesia and H. clysonimus were used as outgroups (GenBank access numbers in Additional file 5). Phylogenetic analyses were performed using two different approaches: Maximum-Parsimony (MP) and Bayesian inference (BI).
Maximum-parsimony (MP) methods were implemented in PAUP 4.0b8 [48], using a heuristic search and tree bisection-reconnection (TBR) branch swapping option. A majority rule consensus tree was computed whenever multiple equally parsimonious trees were obtained. Parsimony bootstrap support values were estimated through 1000 bootstrap replicates. A Bayesian analysis was conducted with GTR + I + G nucleotide substitution model, which was the best-fit model obtained in JModel Test Ver. 0.1.1 [49] based on a Hierarchical Likelihood Ratio Test (HLRT). Bayesian inference (BI) was carried out using four simultaneous chains for ten million generations of Markov Chain Montecarlo (MCMC), sampling every 100 generations. The consensus tree and posterior probability of the nodes was estimated with Mr Bayes Ver. 3.1 [50, 51].
Topology test and population genetics analysis
The Shimodaira-Hasegawa (SH) log-likelihood test, as implemented in PAUP 4.0b8 [48, 52], was used to test the monophyly of H. e. chestertonii. To test this a priori hypothesis, BI analysis was first performed using the same parameters as described earlier but this time by enforcing the monophyly of the complex as a topological constraint. The SH test was then used to compare trees obtained from both constrained and unconstrained analysis.
We described DNA polymorphism in populations of H. e. chestertonii and H. e. venus (Table 1) with DnaSP Ver. 5.10.01 [53]. The measures employed were: 1) number of segregating sites (S), 2) number of haplotypes (h), 3) average pairwise number of differences between sequences (k) and genetic diversity (θ). An analysis of molecular variance (AMOVA) and F ST values were obtained in ARLEQUIN Ver. 3.5 to determine genetic structure between populations from mtDNA markers [54]. To investigate relationships between populations a haplotype network was constructed using NETWORK Ver. 4.5.1.6 [55].
AFLPs, cluster analysis and genetic distance
Genomic DNA from 185 individuals from all the populations, was used for DNA fingerprinting with AFLP markers (Table 3). The DNA quality was quantified by spectrophotometry and only samples with an A260/A280 ratio between 1.8 and 2.0 were used. For AFLP generation, we applied the general method of Vos et al. [56] with minor modifications. The AFLP® Core Reagent Kit (Invitrogen) was used for the digestion of 125 ng of DNA for samples with EcoRI and MseI restriction endonucleases and the ligation with EcoRI/MseI adaptors, following the manufacturers protocol. The best primer combinations (high variation and fragments between 50 to 400 bp) for pre- and selective amplifications were selected by comparing the final sequencing of eight different primer mixtures for 48 individuals (Additional file 6). Sixteen samples chosen at random were run in duplicate during the screening process to ensure reproducibility of selected markers. As result, primers carrying a single selective nucleotide at the 3-end: EcoRI+A (5-GAC TGC GTA CCA ATT CA-3) and MseI+C (5-GAT GAG TCC TGA GTA AC-3) were chosen for the pre-amplification cycle. The conditions in this first PCR were: denaturation at 94°C for 60s, followed by 20 cycles with denaturation at 94°C for 30s, annealing at 56°C for 60s and extension at 72°C for 120s. Subsequently, selective amplifications were performed using four pairs of primers that contained three selective bases and EcoRI primers were labelled with the fluorescent dyes: EcoRI (FAM)+ACA/MseI+CGT, EcoRI (VIC)+ACC/MseI+CGT, EcoRI (PET)+ACT/MseI+CGT and EcoRI (NED)+ACG/MseI+CAC. For these amplifications we applied a touchdown PCR of 12 cycles with denaturation at 94°C for 30s, an annealing temperature with 0.7°C stepwise reduction from 65°C to 56°C for 60s and extension at 72°C for 120s, after which 23 additional cycles were run with fixed annealing temperature of 56°C for 30s. AFLP reaction products were labelled with an internal size standard (GeneScan™-500 LIZ™) and run on an ABI 3730 automated DNA sequencer (Applied Biosystems). Fragment data were collected and analysed with GeneMapper Ver. 4.0 (Applied Biosystems). All bands were visually confirmed and only unambiguous loci between 50 and 400 bp were included in the analysis. Each individual genotype was manually assessed. A binary data matrix of presence (1) or absence (0) of bands of each size by sample was generated.
We used the presence/absence matrix of AFLP fragments to differentiate all individuals and populations using a Bayesian clustering algorithm implemented in the program Structure Ver. 2.3.3 [57, 58]. The number of clusters (K) was determined by comparing the likelihood ratios for K values between 1 and 6. Previous runs including more than 6 clusters were done and the likelihood values were lower and are not shown in this analysis. Each likelihood value was estimated with runs that involved a burn-in of between 104 and 107 MCMC generations with 10 interactions. An admixture model under the assumption of Hardy-Weinberg equilibrium was implemented in the runs. The best number of clusters was confirmed with the ad hoc statistic ΔK [59]. Additionally we implemented a principal component analysis (PCA) using the software Genetix Ver. 4.05. [60] to confirm the results and variation of our data. Measures of heterozygosity and variation were estimated with AFLP-SURV Ver.1.0 [61] and with an AMOVA in Arlequin [54].
Determination of the presence of endosymbionts
A total of 301 individuals (at least 10 from each population), were used to search for the presence of endosymbionts (Table 2 and 3). We used PCR to test for the presence of Wolbachia, Ricketsia and Spiroplasma in H. e. chestertonii and H. e. venus. Where the presence of Wolbachia was detected, additional PCR was carried out to determine the Wolbachia clade (A or B). Amplifications were carried out with specific primers that amplified the wsp gene of Wolbachia (A and B-clade) and 16S rDNA for the other two bacteria (Table 4). We designed three novel specific primers from our Heliconius wsp gene sequences in addition to those proposed by Zhou et al. 1998 (Table 4). Different combinations of these primers allowed us to determine the presence of Wolbachia (diagnostic combination: H81F/H691R), Wolbachia A-clade (H308F/H691R) and Wolbachia B-clade (H81F/wsp522R). Positive controls for these PCR reactions were DNA from known infected insects supplied by Emily Hornett (Wolbachia) and Francis Jiggins (Ricketsia and Spiroplasma) in the University of Cambridge, UK. We followed the amplification conditions as described previously [35, 62–65].
References
Sandoval-Castillo J, Rocha-Olivares A: Deep mitochondrial divergence in baja california populations of an aquilopelagic elasmobranch: the golden cownose ray. J Hered. 2011, 102 (3): 269-274.
Baker JM, Funch P, Giribet G: Cryptic speciation in the recently discovered American cycliophoran Symbion americanus; genetic structure and population expansion. Mar Biol. 2007, 151: 2183-2193.
Vogler C, Benzie J, Lessios H, Barber P, Worheide G: A threat to coral reefs multiplied? Four species of crown-of-thorns starfish. Biol Lett. 2008, 4 (6): 696-699.
Shaw AJ: Biogeographic patterns and cryptic speciation in bryophytes. Journal of Biogeography. 2001, 28: 253-261.
Meyer CP, Paulay G: DNA barcoding: error rates based on comprehensive sampling. PLoS Biol. 2005, 3 (12): e422-
Elias M, Hill RI, Willmott KR, Dasmahapatra KK, Brower AV, Mallet J, Jiggins CD: Limited performance of DNA barcoding in a diverse community of tropical butterflies. Proc R Soc B. 2007, 274 (1627): 2881-2889.
Paterson S, Piertney SB: Frontiers in host-parasite ecology and evolution. Mol Ecol. 2011, 20 (5): 869-871.
Roy L, Buronfosse T: Using mitochondrial and nuclear sequence data for disentangling population structure in complex pest species: a case study with Dermanyssus gallinae. PLoS One. 2011, 6 (7): e22305-
Haase M, Misof B, Wirth T, Baminger H, Baur B: Mitochondrial differentiation in a polymorphic land snail: evidence for Pleistocene survival within the boundaries of permafrost. J Evol Biol. 2003, 16 (3): 415-428.
Thomaz D, Guiller A, Clarke B: Extreme divergence of mitochondrial DNA within species of pulmonate land snails. Proc Biol Sci. 1996, 263 (1368): 363-368.
Brown KS: The biology of Heliconius and related genera. Annual Review of Entomology. 1981, 26: 427-456.
Mallet J, Gilbert LE: Why are there so many mimicry rings? Correlations between habitat, behaviour and mimicry in Heliconius butterflies. Biological Journal of the LInnean Society. 1995, 55: 59-180.
Brower AVZ: Phylogeny of Heliconius butterflies inferred from mitochondrial DNA sequences (Lepidoptera: Nymphalidae). Mol Phylogenet Evol. 1994, 3: 159-174.
Beltran M, Jiggins CD, Bull V, Linares M, Mallet J, McMillan WO, Bermingham E: Phylogenetic discordance at the species boundary: comparative gene genealogies among rapidly radiating Heliconius butterflies. Mol Biol Evol. 2002, 19 (12): 2176-2190.
Brower AVZ: Parallel race formation and the evolution of mimicry in Heliconius butterflies: a phylogenetic hypothesis from mitochondrial DNA sequences. Evolution. 1996, 195-221. 50
Quek SP, Counterman BA, Albuquerque de Moura P, Cardoso MZ, Marshall CR, McMillan WO, Kronforst MR: Dissecting comimetic radiations in Heliconius reveals divergent histories of convergent butterflies. Proc Natl Acad Sci USA. 2010, 107 (16): 7365-7370.
Arias CF, Munoz AG, Jiggins CD, Mavarez J, Bermingham E, Linares M: A hybrid zone provides evidence for incipient ecological speciation in Heliconius butterflies. Mol Ecol. 2008, 17 (21): 4699-4712.
Mavarez J, Salazar CA, Bermingham E, Salcedo C, Jiggins CD, Linares M: Speciation by hybridization in Heliconius butterflies. Nature. 2006, 441 (7095): 868-871.
Werren JH, Baldo L, Clark ME: Wolbachia: master manipulators of invertebrate biology. Nature Reviews Microbiology. 2008, 6: 741-751.
Jiggins FM, Hurst GD: Microbiology. Rapid insect evolution by symbiont transfer. Science. 2011, 332 (6026): 185-186.
Watts T, Haselkorn TS, Moran NA, Markow TA: Variable incidence of Spiroplasma infections in natural populations of Drosophila species. PLoS One. 2009, 4 (5): e5703-
Arthofer W, Riegler M, Schneider D, Krammer M, Miller WJ, Stauffer C: Hidden Wolbachia diversity in field populations of the European cherry fruit fly, Rhagoletis cerasi (Diptera, Tephritidae). Mol Ecol. 2009, 18 (18): 3816-3830.
Bordenstein SR, O'Hara FP, Werren JH: Wolbachia-induced incompatibility precedes other hybrid incompatibilities in Nasonia. Nature. 2001, 409 (6821): 707-710.
Zabal-Aguirre M, Arroyo F, Bella JL: Distribution of Wolbachia infection in Chorthippus parallelus populations within and beyond a Pyrenean hybrid zone. Heredity. 2010, 104 (2): 174-184.
Wade MJ: Evolution: Infectious speciation. Nature. 2001, 409: 675-677.
Munoz AG, Salazar C, Castano J, Jiggins CD, Linares M: Multiple sources of reproductive isolation in a bimodal butterfly hybrid zone. J Evol Biol. 2010, 23 (6): 1312-1320.
Marshall DC, Hill KB, Cooley JR, Simon C: Hybridization, Mitochondrial DNA Phylogeography, and Prediction of the Early Stages of Reproductive Isolation: Lessons from New Zealand Cicadas (Genus Kikihia). Syst Biol. 2011
Douhan GW, Smith ME, Huyrn KL, Westbrook A, Beerli P, Fisher AJ: Multigene analysis suggests ecological speciation in the fungal pathogen Claviceps purpurea. Mol Ecol. 2008, 17 (9): 2276-2286.
Boyer SL, Baker JM, Giribet G: Deep genetic divergences in Aoraki denticulata (Arachnida, Opiliones, Cyphophthalmi): a widespread 'mite harvestman' defies DNA taxonomy. Mol Ecol. 2007, 16 (23): 4999-5016.
Mathews LM: Cryptic biodiversity and phylogeographical patterns in a snapping shrimp species complex. Mol Ecol. 2006, 15 (13): 4049-4063.
Matute DR, McEwen JG, Puccia R, Montes BA, San-Blas G, Bagagli E, Rauscher JT, Restrepo A, Morais F, Niño-Vega G, et al: Cryptic Speciation and Recombination in the Fungus Paracoccidioides brasiliensis as Revealed by Gene Genealogies. Molecular Biology and Evolution. 2006, 23 (1): 65-73.
McBride CS, Arguello JR, O'Meara BC: Five Drosophila genomes reveal nonneutral evolution and the signature of host specialization in the chemoreceptor superfamily. Genetics. 2007, 177 (3): 1395-1416.
Giraldo N, Salazar C, Jiggins CD, Bermingham E, Linares M: Two sisters in the same dress: Heliconius cryptic species. BMC Evol Biol. 2008, 8: 324-
Dyer KA, Jaenike J: Evolutionarily stable infection by a male-killing endosymbiont in Drosophila innubila: molecular evidence from the host and parasite genomes. Genetics. 2004, 168 (3): 1443-1455.
Hurst GD, Jiggins FM: Male-killing bacteria in insects: mechanisms, incidence, and implications. Emerg Infect Dis. 2000, 6 (4): 329-336.
Jiggins FM: Male-killing Wolbachia and mitochondrial DNA: selective sweeps, hybrid introgression and parasite population dynamics. Genetics. 2003, 164 (1): 5-12.
McMeniman CJ, Lane RV, Cass BN, Fong AW, Sidhu M, Wang YF, O'Neill SL: Stable introduction of a life-shortening Wolbachia infection into the mosquito Aedes aegypti. Science. 2009, 323 (5910): 141-144.
Charlat S, Le Chat L, Mercot H: Characterization of non-cytoplasmic incompatibility inducing Wolbachia in two continental African populations of Drosophila simulans. Heredity. 2003, 90 (1): 49-55.
Goto S, Anbutsu H, Fukatsu T: Asymmetrical interactions between Wolbachia and Spiroplasma endosymbionts coexisting in the same insect host. Appl Environ Microbiol. 2006, 72 (7): 4805-4810.
Hilgenboecker K, Hammerstein P, Schlattmann P, Telschow A, Werren JH: How many species are infected with Wolbachia?--A statistical analysis of current data. FEMS Microbiol Lett. 2008, 281 (2): 215-220.
Arora N, Nater A, van Schaik CP, Willems EP, van Noordwijk MA, Goossens B, Morf N, Bastian M, Knott C, Morrogh-Bernard H, et al: Effects of Pleistocene glaciations and rivers on the population structure of Bornean orangutans (Pongo pygmaeus). Proc Natl Acad Sci USA. 2010, 107 (50): 21376-21381.
Derycke S, Remerie T, Backeljau T, Vierstraete A, Vanfleteren J, Vincx M, Moens T: Phylogeography of the Rhabditis (Pellioditis) marina species complex: evidence for long-distance dispersal, and for range expansions and restricted gene flow in the northeast Atlantic. Mol Ecol. 2008, 17 (14): 3306-3322.
Brumfield RT, Edwards SV: Evolution into and out of the Andes: a Bayesian analysis of historical diversification in Thamnophilus antshrikes. Evolution. 2007, 61 (2): 346-367.
Hughes C, Eastwood R: Island radiation on a continental scale: exceptional rates of plant diversification after uplift of the Andes. Proc Natl Acad Sci USA. 2006, 103 (27): 10334-10339.
Drummond AJ, Ashton B, Buxton S, Cheung M, Cooper A, Duran C, Field M, Heled J, Kearse M, Markowitz S, et al: Geneious Ver 5.4. 2011, [http://www.geneious.com]
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al: Clustal W and Clustal X version 2.0. Bioinformatics. 2007, 23: 2947-2948.
Maddison DR, Maddison WP: MacClade 4: Analysis of phylogeny and character evolution. Edited by: Associates S. 2000, Sunderland, Massachusetts, 4.08
Swofford DL: PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Edited by: Associates S. 2003, Sunderland, Massachusetts, 4.0
Posada D: jModelTest: Phylogenetic Model Averaging. Mol Biol Evol. 2008, 25: 1253-1256.
Huelsenbeck JP, Ronquist F: MRBAYES: Bayesian inference of phylogeny. Bioinformatics. 2001, 17: 754-755.
Ronquist F, Huelsenbeck JP: MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19: 1572-1574.
Shimodaira H, Hasegawa M: Multiple Comparisons of Log-Likelihoods with Applications to Phylogenetic Inference. Mol Biol Evol. 1999, 16 (8): 1114-1116.
Librado P, Rozas J: DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009, 25: 1451-1452.
Excoffier L, Lischer HEL: Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources. 2010, 10: 564-567.
Bandelt H-J, Forster P, Röhl A: Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999, 16: 37-48.
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, et al: AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 1995, 23 (21): 4407-4414.
Hubisz MJ, Falush D, Stephens M, Pritchard JK: Inferring weak population structure with the assistance of sample group information. Molecular Ecology Resources. 2009, 9 (5): 1322-1332.
Pritchard JK, Stephens M, Donnelly P: Inference of Population Structure Using Multilocus Genotype Data. Genetics. 2000, 155: 945-959.
Evanno G, Regnaut S, Goudet J: Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005, 14 (8): 2611-2620.
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F: GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Edited by: Laboratoire Génome P. 2004, Interactions, CNRS UMR 5171, Université de Montpellier II. Montpellier (France)
Vekemans X, Beauwens T, Lemaire M, Roldan-Ruiz I: Data from amplified fragment length polymorphism (AFLP) markers show indication of size homoplasy and of a relationship between degree of homoplasy and fragment size. Molecular Ecology. 2002, 11: 139-151.
Graf von der Schulenburg JH, Habig M, Sloggett JJ, Webberley KM, Bertrand D, Hurst GDD, Majerus MEN: Incidence of Male-Killing Rickettsia spp. (a- Proteobacteria) in the Ten-Spot Ladybird Beetle Adalia decempunctata L. (Coleoptera: Coccinellidae). Appl Environ Microbiol. 2001, 67 (1): 270-277.
Hurst GD, Jiggins FM: Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc Biol Sci. 2005, 272 (1572): 1525-1534.
van Kuppeveld FJ, van der Logt JT, Angulo AF, van Zoest MJ, Quint WG, Niesters HG, Galama JM, Melchers WJ: Genus- and species-specific identification of mycoplasmas by 16S rRNA amplification. Appl Environ Microbiol. 1993, 59 (2): 655-
Zhou W, Rousset F, O'Neill S: Phylogeny and PCR-based classification of Wolbachia strains using wsp gene sequences. Proc R Soc Lond B. 1998, 265: 509-515.
Acknowledgements
AGM was supported by COLCIENCIAS, Facultad de Ciencias and Vicerrectoria de Investigaciones, Universidad de los Andes. ML and AGM were funded by Banco de la República (Project 2582), and private donations (Continautos S.A., Proficol El Carmen S.A. and Didacol S.A). CDJ was fund by Funding from a Leverhulme Research Leadership grant.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
AGM obtained some specimens from Cauca Valley, carried out laboratory work, genetic and statistical analyses. SWB, ML and CDJ participated in the design and coordination of the study. AGM, SWB and CDJ drafted the manuscript. ML obtained most of the specimens from Cauca Valley used in this study. All authors read and approved the final manuscript
Electronic supplementary material
12862_2011_1977_MOESM1_ESM.PDF
Additional file 1: Network of mtDNA haplotypes. The median joining network of mtDNA haplotypes is congruent with the three clades in phylogenetic analysis (see Results). The colours represent H. e. venus (white), H. e. chestertonii samples of the north (black) and south of Cauca Valley (gray). (PDF 638 KB)
12862_2011_1977_MOESM2_ESM.PDF
Additional file 2: Matrix with F ST values for mtDNA markers (lower triangle) and AFLP markers (upper triangle). Abbreviations correspond to localities. In the hybrid zone, individuals were separated by phenotype: H. e. chestertonii (CV) or H. e. venus (CV_V). Asterisk shows comparisons with significant values (P < 0.05). (PDF 47 KB)
12862_2011_1977_MOESM3_ESM.PDF
Additional file 3: Genetic diversity from AFLP markers. Estimates of total gene diversity (H T ) and within group (H S ). Index of fixation (F ST ) was calculated grouping the populations within species (** P < 0.01). (PDF 61 KB)
12862_2011_1977_MOESM4_ESM.PDF
Additional file 4: Principal Components Analysis of AFLP markers. The circles represent individuals of H. e. chestertonii and asterisks H. e. venus. Black circles showed individuals from south populations and gray circles those from the north. (PDF 303 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Muñoz, A.G., Baxter, S.W., Linares, M. et al. Deep mitochondrial divergence within a Heliconiusbutterfly species is not explained by cryptic speciation or endosymbiotic bacteria. BMC Evol Biol 11, 358 (2011). https://doi.org/10.1186/1471-2148-11-358
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-11-358