
Abstract
Background
First identified in fruit flies with temperature-sensitive paralysis phenotypes, the Drosophila melanogaster TipE locus encodes four voltage-gated sodium (NaV) channel auxiliary subunits. This cluster of TipE-like genes on chromosome 3L, and a fifth family member on chromosome 3R, are important for the optional expression and functionality of the Para NaV channel but appear quite distinct from auxiliary subunits in vertebrates. Here, we exploited available arthropod genomic resources to trace the origin of TipE-like genes by mapping their evolutionary histories and examining their genomic architectures.
Results
We identified a remarkably conserved synteny block of TipE-like orthologues with well-maintained local gene arrangements from 21 insect species. Homologues in the water flea, Daphnia pulex, suggest an ancestral pancrustacean repertoire of four TipE-like genes; a subsequent gene duplication may have generated functional redundancy allowing gene losses in the silk moth and mosquitoes. Intronic nesting of the insect TipE gene cluster probably occurred following the divergence from crustaceans, but in the flour beetle and silk moth genomes the clusters apparently escaped from nesting. Across Pancrustacea, TipE gene family members have experienced intronic nesting, escape from nesting, retrotransposition, translocation, and gene loss events while generally maintaining their local gene neighbourhoods. D. melanogaster TipE-like genes exhibit coordinated spatial and temporal regulation of expression distinct from their host gene but well-correlated with their regulatory target, the Para NaV channel, suggesting that functional constraints may preserve the TipE gene cluster. We identified homology between TipE-like NaV channel regulators and vertebrate Slo-beta auxiliary subunits of big-conductance calcium-activated potassium (BKCa) channels, which suggests that ion channel regulatory partners have evolved distinct lineage-specific characteristics.
Conclusions
TipE-like genes form a remarkably conserved genomic cluster across all examined insect genomes. This study reveals likely structural and functional constraints on the genomic evolution of insect TipE gene family members maintained in synteny over hundreds of millions of years of evolution. The likely common origin of these NaV channel regulators with BKCa auxiliary subunits highlights the evolutionary plasticity of ion channel regulatory mechanisms.
Similar content being viewed by others

Background
Voltage-gated sodium (NaV) channels control the passage of sodium ions across the plasma membrane and play a crucial role in the creation and propagation of electrical signals in excitable cells, such as neurons and myocytes. The pore-forming alpha subunits comprise a gene family of up to ten members in vertebrate species [1], which are associated with four types of auxiliary beta-subunits that are important for the biogenesis and gating kinetics of functional alpha-subunits [2]. In contrast, only two NaV channel alpha-subunit genes are found in insects, orthologues of Drosophila melanogaster DSC1 (Drosophila sodium channel 1) and Para (paralytic), and sequence homology searches fail to identify homologues of the vertebrate NaV channel auxiliary beta-subunits. Although DSC1 may rather function as a calcium channel involved in olfactory avoidance responses [1, 3, 4], Para mutants exhibit temperature-sensitive paralysis caused by loss of Na+-based action potentials [5–8]. Thus, insect genomes appear to encode only one functional NaV channel alpha-subunit gene and no orthologues of the vertebrate NaV channel auxiliary beta-subunits.
Studies of temperature-sensitive paralysis phenotypes in D. melanogaster identified a role for the TipE locus (temperature-induced paralysis locus E) in cell excitability, and suggested regulatory interactions with the Para NaV channel [9, 10]. Subsequent identification of the TipE gene facilitated studies that demonstrated its NaV channel regulator activity by stimulating functional expression of Para in Xenopus oocytes, and established that TipE expression during pupal development rescued the paralysis phenotype in adult fruit flies [11]. Moreover, sodium-dependent repetitive firing in Drosophila neurons is regulated by TipE [12], and Para-TipE interactions influence the pharmacology of the Para NaV channel [13]. Despite the lack of insect orthologues of vertebrate NaV channel auxiliary beta-subunits, characterization of the D. melanogaster TipE gene revealed strikingly similar functions: an auxiliary subunit required for the correct assembly and gating of the Para NaV channel. Furthermore, four homologues of TipE (Teh1-4) are encoded in the D. melanogaster genome and their TipE-like functions as auxiliary subunits of Para NaV channels have been characterized through co-expression with Para in Xenopus oocytes [14]. Thus, fruit fly TipE gene family members are important for the optimal expression and functionality of the Para NaV channel but appear quite distinct from auxiliary beta-subunits in vertebrates.
Their key roles in membrane excitability make insect NaV channels attractive targets for the development of insecticides [13, 15, 16]. However, Para mutations lead to knockdown resistance in insects and other arthropods, which reduces the efficacy of many insecticides and intensifies the search for alternative targets [17–19]. Characterizing the evolutionary histories and molecular functions of insect TipE gene family members will not only further the understanding of the mechanisms controlling cell excitability, but may also offer novel opportunities for developing alternative insect control strategies. In this study, we examined the genealogies of TipE gene family members across multiple insect species with sequenced genomes, and revealed a remarkably conserved synteny block of TipE homologues with local gene arrangements that have been well-maintained in many insect genomes over hundreds of millions of years of evolution.
Results and discussion
A genomic cluster of TipE gene family members conserved across Insecta
High levels of genome shuffling observed in many insect species lead to the rapid decay of synteny, leaving only a few identifiable conserved gene clusters [20]. Examples of such well-maintained local gene arrangements include the homeobox (Hox) [21] and wingless (Wnt) [22] gene clusters with key roles in early development. Synteny block analysis across 23 arthropod species with sequenced genomes (see Methods) identified both Hox and Wnt gene clusters, and highlighted a genomic cluster of TipE gene family members as one of the most conserved blocks of orthologues with well-maintained local gene arrangements. This highly conserved synteny block was defined by a core of three sets of orthologous genes from 20 insect species: Drosophila melanogaster and 11 other fruit flies; three mosquitoes: Anopheles gambiae, Aedes aegypti, and Culex quinquefasciatus; the silk moth, Bombyx mori; the flour beetle, Tribolium castaneum; the parasitoid wasp, Nasonia vitripennis; the honey bee, Apis mellifera; and the human body louse, Pediculus humanus. Sequence homology searches of genomic resources of the remaining sequenced arthropod species revealed a similarly conserved gene arrangement in the pea aphid, Acyrthosiphon pisum, as well as four TipE homologues in the crustacean water flea, Daphnia pulex, but no homologous genes in the arachnid deer tick, Ixodes scapularis (possibly due to incomplete genome sequencing). The three D. melanogaster genes are located in a ~15 kb region of chromosome 3L and encode the Para NaV channel auxiliary subunits, Teh2, Teh3, and TipE. Directly neighbouring the Teh2 gene is a fourth homologue, Teh4, which, together with Teh1 on chromosome 3R, make up the full complement of five D. melanogaster TipE gene family members. The conservation of the Teh2-Teh3-TipE synteny block across Insecta, despite generally high rates of genomic shuffling [20] and gene losses [23] over more than ~350 million years of evolution [24], suggests that strong constraints have maintained the local genomic architecture of the TipE locus (Figure 1).

Genomic organization of TipE gene family members in 11 insect species and Daphnia pulex. TipE gene family members exhibit well-maintained local gene neighbourhoods across Insecta. Orthologues of Drosophila melanogaster (Dmela) TipE (red), Teh2 (blue), and Teh3 (green) form a conserved synteny block nested in the last intron of CG18675 orthologues (orange), except in the silk moth (Bombyx mori, Bmori) and the four beetle (Tribolium castaneum, Tcast) where they have escaped from intronic nesting. The synteny block is extended by Teh4 orthologues (purple) in all insects except the mosquitoes (Anopheles gambiae, Agamb; Aedes aegypti, Aaegy; and Culex quinquefasciatus, Cquin) and the silk moth. The gene cluster is conserved in fruit flies with the inserted VhaM9.7-c gene in Dmela also present in Drosophila mojavensis (Dmoja) and ten other drosophilids (not shown). Although Teh1 orthologues (cyan) are often not linked to the TipE locus, the neighbouring location in the human body louse (Pediculus humanus, Phuma) is likely to represent the ancestral arrangement, supported by the genomic proximity of Teh1 in the pea aphid (Acyrthosiphon pisum, Apisu) and parasitoid wasp (Nasonia vitripennis, Nvitr). The honey bee (Apis mellifera, Amell) and flour beetle Teh1 orthologues have undergone translocation events relative to their Teh4-Teh2-Teh3-TipE clusters, but are retained on the same linkage groups. The genome of the crustacean water flea, Daphnia pulex (Dpule), encodes orthologues of TipE, Teh1, and Teh2, but only one Teh3/4-like gene (s8g331). As Teh3 is a single-coding-exon gene in all the insect genomes, it likely arose from the retrotransposition of a Teh4-like gene before the insect radiation. Putative orthologous relations among TipE gene family members are indicated with dashed lines. Start and end positions are indicated for scaffolds, supercontigs, linkage groups, or chromosomes with arrows indicating the direction of the forward strand of the reference assemblies. Complete gene models including untranslated regions and alternative transcripts are displayed for Dmela while only coding-sequence regions are depicted for the other species.
Comparative analysis of the TipE gene family previously identified orthologues of D. melanogaster TipE, Teh1, Teh2, and Teh3 from A. gambiae, B. mori, and A. mellifera, as well as orthologues of all five family members from two other fruit flies, D. pseudoobscura and D. yakuba, and noted their similar genomic clustering [14]. Inspecting the genomic loci of all TipE gene family members in D. pulex and each of the 21 insect genomes facilitated comprehensive gene model curation to confirm and correct their predicted genomic architectures and protein-coding sequences (see Methods). These analyses identified the previously unannotated A. pisum TipE orthologue and the A. aegypti Teh1 orthologue, corrected the previously split gene model of the A. pisum Teh2 orthologue, and improved several other gene model annotations (see Additional file 1, Table S1 for full details on all corrected gene models). The otherwise strictly-conserved arrangement of the Teh2-Teh3-TipE synteny block, with Teh2 and Teh3 encoded on the opposite strand to TipE, is disrupted in the fruit flies by a short gene encoding a Vacuolar H+ ATPase subunit (VhaM9.7-c in D. melanogaster) between Teh3 and TipE, and by a relative inversion of Teh2-Teh3 in the silk moth (Bmori). Teh4 orthologues are located adjacent to Teh2 and extend the core synteny block in all the insects except the three mosquitoes and the silk moth, which appear to have lost the Teh4 gene. In the genome of the human body louse (Phuma), Teh1 is adjacent to TipE (~15 kb apart), while in the pea aphid (Apisu) it is ~63 kb away and separated by two intervening genes; in the parasitoid wasp (Nvitr), it is ~24 kb away with only a single intervening gene. As the compact body louse genome is remarkably free from transposable elements and appears to have experienced less shuffling than other insect genomes [25], the gene cluster in the last common ancestor with all five TipE gene family members is likely to have resembled the genomic arrangement found in the body louse today.
Structural constraints: intronic nesting of the TipE locus
The evidence-based annotations of the D. melanogaster TipE locus provide a comprehensive view of the complex gene architectures and reveal that the coding sequences of this gene cluster - Teh4, Teh2, Teh3, VhaM9.7-c, and TipE - are all nested within the last intron of the neighbouring CG18675 gene (Figure 1). Such nested gene structures in eukaryotes are generally much more frequently gained than lost, suggesting that while intronic insertions may be relatively well-tolerated, deletions are more likely to be constrained due to their possible negative impact on both the nested and the host genes [26]. Single-copy orthologues of CG18675 can be identified in almost all sequenced metazoan genomes, as well as in some flagellate protozoa, green plants, and apicomplexan parasites, but appear to be absent from fungi and nematode worms. These gene sequences are relatively well-conserved, with 49% amino acid identity between CG18675 and the human orthologue (C21orf59, ENSG00000159079), and are matched by the sequence profile, domain of unknown function DUF2870 (IPR021298). Although it has been preserved as a single-copy gene across a broad phyletic range, the nesting of multiple genes within the last intron of CG18675 orthologues appears to have only occurred in insects. A genomic sequence gap obscures the start of the water flea (Dpule) CG18675 orthologue, nevertheless, the C-terminal coding exons align well with other CG18675 orthologues (see Additional file 2), and the last intron contains no nested genes. This suggests that while it is possible that water flea TipE-like genes escaped from intronic nesting, it is more likely that the nesting event occurred in the last common ancestor of insects, after the divergence from crustaceans.
The genomic span of insect CG18675 orthologues with nested TipE gene family members ranges from ~20 kb in the fruit fly (Dmela) and the body louse (Phuma) to ~100 kb in the Aedes mosquito (Aaegy), which, in addition to the insertion of the VhaM9.7-c gene in the drosophilid lineage, highlights the dynamic evolution of this region. Despite these perturbations, the intronic nesting and local gene arrangements of the TipE locus have remained largely intact. In addition to the constraint imposed by intronic nesting, a likely contributing factor is the overlap of the terminal exons of CG18675 and TipE. These exons contain both coding and untranslated regions, however, their overlapping coding regions are out of phase and encode two distinct terminal peptides of 37 (CG18675) and 20 (TipE) amino acids (see Additional file 3, Figure S1). Nesting of the TipE locus and the overlapping terminal exons are observed in the CG18675 orthologues from all the examined insect species except the flour beetle (Tcast) and the silk moth (Bmori). The flour beetle CG18657 orthologue (TC007236) is on a different linkage group to the TipE locus and the silk moth gene (BGIBMGA013920) is on a different scaffold. Both genes show conserved coding sequences that include the last coding exon, indicating that their gene models are complete (see Additional file 2), and their genomic spans of ~2.5 kb (Bmori) and ~1 kb (Tcast) are greatly reduced compared to other CG18675 orthologues. Thus, in these representative species from the Coleoptera and Lepidoptera lineages, the TipE locus appears to have been released from the constraint of intronic nesting. Despite their escape from intronic nesting, Teh2-Teh3 remain adjacent to TipE in the silk moth, and in the flour beetle the local gene arrangements of Teh4, Teh2, Teh3, and TipE are strictly maintained, suggesting possible functional constraints that preserve the architecture of the TipE locus.
Functional constraints: coordinated regulation of TipE gene family members
Intronic nesting may suggest possible mechanisms for coordinated regulation of host and nested genes, however, functional relations between host and their nested genes are usually unclear, and their expression profiles often exhibit inverse or no significant correlations [26, 27]. Insect glucose-methanol-choline (GMC) oxidoreductase genes exhibit a similar nested genomic architecture to that of the TipE locus; most gene family members are encoded in a large intron of the flotillin-2 (Flo-2) gene in D. melanogaster, and its orthologues in other insects [20, 28]. This cluster of GMC oxidoreductases may be coordinately regulated for developmental or physiological roles involved in ecdysteroid metabolism, which may be linked to the function of the host gene product, Flo-2, in lipid rafts [28]. Multiple nesting events of homologous genes within a single host intron could therefore facilitate coordinated regulation of the nested genes. Examining D. melanogaster gene expression data from different tissue samples (FlyAtlas [29]) and developmental series experiments (modENCODE [30]) revealed common and distinct spatial and temporal expression patterns of the TipE locus genes (see Additional file 4). Tissues with the highest transcript levels of TipE gene family members include the brain, thoracicoabdominal ganglion, eye, larval central nervous system, and the head, mirroring the expression of Para and consistent with their roles in excitable cells.
The developmental series data reveal two peaks of gene expression; the first, from the 14-16-hour embryo to the first larval stage (L1), is dominated by Teh4. The start of the second peak is evident from the 12-hour white prepupae, with maximal transcript levels at the 3-day pupae and maintained expression in adult males, but not in adult females. TipE-Teh1 and Teh2-Teh3 are the best correlated pairs of expression profiles, TipE is also well-correlated with Teh2 and Teh4, but to a lesser extent with Teh3 (Spearman correlation test, Table 1). Thus, coordinated regulation appears not only amongst the intron-nested TipE gene family members, but also with the unlinked Teh1 gene. However, these coordinated expression profiles exhibit poor correlations with that of the host gene, CG18657, which displays a small peak during mid-embryogenesis and then increases again from the 12-hour L3 larval stage with greatly-elevated expression in adult males and almost undetectable levels in adult females, consistent with the high transcript levels observed in testis samples. In contrast, the TipE gene family members' expression profiles are much better-matched to that of Para, particularly TipE and Teh1 - these two members also induced the largest increases of expression levels of Para NaV channels in Xenopus oocytes [14]. Thus, D. melanogaster TipE gene family members exhibit coordinated spatial and temporal regulation that also correlates well with the expression of Para, their functional NaV channel partner, but which appears distinct from the host gene, CG18657.
Evolutionary history of the insect TipE gene family
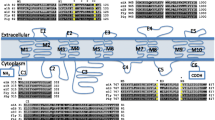
Phylogenetic analysis of TipE gene family members from D. pulex and a representative subset of the 21 insect species highlights the five groups of orthologues (see Figure 2 and Additional file 3, Figure S2). The TipE-Teh1-Teh2 proteins form a distinct clade from Teh3-Teh4, suggesting possible functional divergence between members of these two clades. This is supported by the presence of the epidermal growth factor (EGF)-like domains in the extracellular loop regions of Teh3 and Teh4 proteins that are not found in other TipE gene family members, which may interact with extracellular matrix components [14]. Conserved sequence motif analysis using MEME [31] supports the orthology assignments, highlights the extended extracellular loop regions of the Teh3 and Teh4 orthologues, and shows that the sequence regions common to all TipE gene family members are comprised of the two transmembrane domains and parts of the extracellular loop (see Additional file 3, Figure S3). These regions exhibit high rates of amino acid substitutions, and this variation may explain the lack of confident bootstrap support for several nodes on the TipE gene family phylogeny (see Figure 2 and Additional file 3, Figure S2). The D. pulex genome encodes only one Teh3/4-like gene and Teh3 is found as a single-coding-exon gene in all the examined insect genomes; the alignment of the D. melanogaster genes identifies seven sites with uninterrupted coding sequence in Teh3 but with introns in Teh4, further suggesting that Teh3 arose from the retrotransposition of a Teh4-like gene of the last common ancestor before the insect radiation. In the silk moth and the three mosquitoes, the absence of Teh4 orthologues implies that the Teh3 retrogene, which integrated into the same local genomic environment, could have assumed the functions of the original multi-exon ancestor. The D. pulex TipE orthologue is resolved on the TipE gene family phylogeny (Figure 2), and although Teh1 and Teh2 orthologues are not confidently resolved, they can be identified by comparing their motif architectures (see Additional file 3, Figure S3) with the insect Teh1 and Teh2 orthologues. Thus, it is likely that the ancestral pancrustacean genome encoded four TipE gene family members, the subsequent retrotransposition of Teh4 after the divergence of the crustacean and insect lineages gave rise to Teh3 and created a possible functional redundancy which led to the loss of Teh4 in the silk moth and the mosquitoes.

Phylogenetic tree of TipE gene family members in 11 insect species and Daphnia pulex. The phylogeny of TipE gene family members highlights the five groups of orthologues from the crustacean water flea, Daphnia pulex (Dpule) and 11 representative insect species: two fruit flies, Drosophila melanogaster (Dmela) and Drosophila mojavensis (Dmoja); three mosquitoes, Anopheles gambiae (Agamb), Aedes aegypti (Aaegy), Culex quinquefasciatus (Cquin); the silk moth, Bombyx mori (Bmori); the flour beetle, Tribolium castaneum (Tcast), the honey bee, Apis mellifera (Amell); the parasitoid wasp, Nasonia vitripennis (Nvitr); the pea aphid, Acyrthosiphon pisum (Apisu); and the human body louse, Pediculus humanus (Phuma). The clade of Teh3-Teh4 proteins is distinct from TipE-Teh1-Teh2, linked by dashed lines based on the phylogenetic tree of TipE gene family members in D. pulex and six selected insect species (coloured), rooted using the distantly related vertebrate big-conductance calcium-activated potassium channel beta-4 auxiliary subunits (KCNMB4) (see Additional file 3, Figure S2). The phylogeny resolves the D. pulex TipE orthologue, and although Teh1 and Teh2 orthologues are not confidently resolved, they can be identified by comparing their conserved motif architectures with the insect Teh1 and Teh2 orthologues (see Additional file 3, Figure S3). A single Teh3/4-like gene is found in the D. pulex genome, which probably reflects the ancestral pancrustacean genome, where a retrotransposition event before the insect radiation generated the Teh3 retrogene. This gene duplication may have created a functional redundancy which allowed for the loss of Teh4 orthologues in mosquitoes and the silk moth. The maximum likelihood phylogeny was built from the confidently-aligned regions of the multiple sequence alignment of member proteins (see Methods). Bootstrap support values from 100 samples are given and nodes with less than 60% support are collapsed.
Performing BLAST and PSI-BLAST homology searches of non-redundant sequence databases using TipE gene family members failed to identify any significant matches beyond Pancrustacea. Orthologues of Para are present in all the examined insects and the water flea, as well as in the closest outgroup species, the arachnid deer tick, Ixodes scapularis (see Additional file 1, Table S1 and Additional file 3, Figure S4), however, no TipE homologues were identified in either the deer tick's assembled scaffolds or sequence trace archives. Similarly, TipE-like auxiliary beta-subunits are not found in the genome of the nematode worm, Caenorhabditis elegans, which also lacks any known NaV channel homologues, despite the likely presence of an ancestral sodium channel before the divergence of vertebrates and invertebrates [32]. Scans with Hidden Markov Model (HMM) profiles built from multiple protein sequence alignments of TipE gene family members (see Methods) also produced no significant matches to tick or worm proteins. However, the improved search sensitivity of the HMM profiles revealed distant homology with beta-4 auxiliary subunits of vertebrate big-conductance calcium-activated potassium (BKCa) channels (KCNMB4, a member of the Slo-beta or BK-beta subunit family): the HMM profile built from the 32 TipE gene family members shown in Additional file 3, Figure S2 identifies human, mouse, cow, and frog KCNMB4 proteins with e-values of less than 1e-05. A reciprocal homology search of arthropod proteomes with a vertebrate KCNMB4 HMM profile identifies insect Teh1 orthologues with e-values of less than 1e-03. A notable difference occurs in the extracellular loop region of TipE, Teh3, and Teh4 proteins, which are longer than the loops in Slo-beta proteins and exhibit EGF-like domains in Teh3 and Teh4 [2, 14]. Although the possibility of convergent evolution of these two families of auxiliary subunits cannot be excluded, the similar protein architecture of TipE-like and Slo-beta proteins, with two hydrophobic regions that anchor an extracellular loop containing putative disulphide bridges, further supports their common origin.
Their likely common ancestry with Slo-beta subunits suggests that some TipE gene family members may also play a role in the regulation of BKCa channels. This type of ion channel is found across metazoan species, and is named Slowpoke in D. melanogaster. Although Slowpoke exhibits generally higher expression levels than Para, particularly in the eye, head, and spermatheca, their developmental expression profiles are well-correlated and match those of the TipE gene family members (see Table 1 and Additional file 4). However, rather than suggesting a role for some TipE gene family members in the regulation of Slowpoke, their similar developmental expression profiles may arise simply because both channels function in excitable cells. Indeed, TipE gene family members showed no effects on Slowpoke potassium currents in D. melanogaster cell lines [14], which are instead regulated by an auxiliary subunit with a protein kinase-like domain, Slowpoke binding protein [33]. The likely common ancestor of the TipE-like and Slo-beta auxiliary subunits may therefore have functioned together with the pore-forming alpha-subunits of an ancestral metazoan potassium or sodium channel, subsequently undergoing independent fine-tuning to control calcium-activated potassium currents in vertebrates and voltage-gated sodium currents in the invertebrate lineage.
Conclusion
The TipE gene family of Para NaV channel auxiliary subunits forms a remarkably conserved genomic cluster of orthologues with local gene arrangements maintained across many diverse insect species. This conservation suggests strong constraints that preserve the organisation of the TipE gene cluster, as observed for the well-studied Hox gene clusters that nevertheless appear to tolerate limited gene rearrangements in Drosophila [34]. Comparing the TipE synteny block architecture across 21 insect species and examining expression patterns of D. melanogaster genes provides evidence of both structural and functional constraints that preserve the TipE cluster. The ancestral insect TipE gene cluster is likely to have resembled the genomic arrangement found in the human body louse today; the TipE, Teh2, Teh3, and Teh4 genes nested within the last intron of the CG18657 ancestor, and directly neighbouring Teh1. Although no TipE-like genes were identified in arachnid or nematode genomes, homologues encoded in the D. pulex genome suggest an ancient pancrustacean repertoire of four TipE gene family members. Retrotransposition of Teh4 gave rise to the insect Teh3 retrogene following the divergence of crustaceans and insects, which may have generated functional redundancy that allowed for the loss of Teh4 in the silk moth and the mosquitoes. Local gene arrangements of the TipE locus are well-maintained despite the apparent escape from intronic nesting in the silk moth and the flour beetle genomes, suggesting additional constraints that preserve the genomic architecture. D. melanogaster gene expression data reveal coordinated spatial and temporal regulation of the TipE gene family members that is distinct from the host gene, but which correlates well with the expression of their regulatory target, Para. Such functional constraints are likely to contribute to the preservation of the TipE synteny block across Insecta, to maintain coordinated expression of gene family members with or without the genomic constraint of intronic nesting. Examining the insect TipE gene family reveals several key evolutionary events that characterize the evolution of genes within their genomic contexts: intronic nesting, escape from nesting, overlapping exons, retrotransposition, translocation, gene loss, coordinated regulation, and conserved synteny. Exploration of the evolutionary histories of genes maintained in such synteny blocks therefore offers the opportunity to identify putative structural and functional constraints on gene rearrangements and their relative contributions to genome evolution.
The common ancestry of the TipE-like and Slo-beta auxiliary subunits suggests a remarkable plasticity in the evolution of mechanisms for regulating ion channel activities. An ancestral ion channel auxiliary subunit appears to have evolved to regulate calcium-activated potassium currents in vertebrates and voltage-gated sodium currents in the invertebrate lineage. Thus, although the pore-forming alpha-subunits are generally well-conserved across metazoan species, their regulatory partners appear to exhibit more lineage-specific functions. Consequently, targeting the activity of such auxiliary subunits may prove a successful strategy for the development of insecticides that are not toxic to vertebrates, and a more detailed understanding of their molecular functions in different insect species may facilitate targeted interventions that minimise ecological knock-on effects. As well as regulating functional expression and gating kinetics of ion channels, auxiliary subunits are known to influence interactions with naturally-occurring toxins, such as scorpion venom peptides which inhibit big-conductance calcium channels [35]. Following Nature's lead could therefore offer novel prospects for the development of insect control strategies that target ion channel activities.
Methods
Genomic resources
Official gene sets and assembled genome sequences for 23 arthropod species were retrieved from publically available resources including AphidBase [36], BeetleBase [37], FlyBase [38], Hymenoptera Genome Database [39], SilkDB [40], VectorBase [41], and wFleaBase [42]; see Additional file 1, Table S2 for full details. The examined arthropod species include: Diptera - Drosophila melanogaster (Dmela) [43] and 11 other fruit flies [44]: Drosophila sechellia (Dsech), Drosophila simulans (Dsimu), Drosophila erecta (Derec), Drosophila yakuba (Dyaku), Drosophila ananassae (Danan), Drosophila persimilis (Dpers), Drosophila pseudoobscura (Dpseu), Drosophila willistoni (Dwill), Drosophila mojavensis (Dmoja), Drosophila virilis (Dviri), Drosophila grimshawi (Dgrim), as well as three mosquitoes: Anopheles gambiae (Agamb) [45], Aedes aegypti (Aaegy) [46], Culex quinquefasciatus (Cquin) [47]; Lepidoptera - the silk moth, Bombyx mori (Bmori) [48]; Coleoptera - the flour beetle, Tribolium castaneum (Tcast) [49]; Hymenoptera - the honey bee, Apis mellifera (Amell) [50] and the parasitoid wasp, Nasonia vitripennis (Nvitr) [51]; Hemiptera - the pea aphid, Acyrthosiphon pisum (Apisu) [52]; Phthiraptera - the human body louse, Pediculus humanus (Phuma) [25]; Crustacea - the water flea, Daphnia pulex (Dpule) [53]; and Arachnida - the deer tick, Ixodes scapularis (Iscap).
Synteny block identification
The conserved genomic cluster of TipE gene family members was detected by employing synteny block identification procedures developed from a strategy based on orthologous gene anchors [20, 54]. Initial pairwise species comparisons used single-copy orthologous genes from the OrthoDB [55, 56], requiring at least two pairs of neighbouring orthologues and allowing one intervening orthologue at most. Subsequently, synteny block extension across multiple species was performed by successive projections of pairwise blocks across the species phylogeny.
Gene model curation
Protein sequence comparisons of TipE gene family members and orthologues of CG18675 revealed several annotation errors including missing exons and split gene models. Gene model curation employed additional available information such as expressed sequenced tags (ESTs) and homology-based gene predictions using Fgenesh+ [57] and Wise2 [58] to confirm and correct the annotated gene models. HMM profiles of TipE gene family members and CG18675 orthologues used for gene predictions were built using HMMER [59]. The HMMs were also used to scan the proteomes of additional non-arthropod species including Caenorhabditis elegans, Nematostella vectensis, Hydra magnipapillata, Strongylocentrotus purpuratus, Branchiostoma floridae, Danio rerio, Xenopus tropicalis, Gallus gallus, Bos taurus, Mus musculus and Homo sapiens. Megablast and tBLASTn homology searches against the unassembled sequence trace archives, as well as the assembled genomes, were performed respectively to check for the presence of any additional genes potentially missed by the assembly or annotation procedures.
Phylogenetic analyses
Multiple protein sequence alignments of TipE gene family members and CG18675 orthologues were built using MUSCLE [60], and confidently aligned regions were extracted with Gblocks [61]. Maximum likelihood phylogenetic trees were reconstructed using PhyML [62], with the Jones-Taylor-Thornton (JTT) substitution model, four relative substitution rate categories, estimated proportions of invariable sites and gamma distribution parameters, and 100 bootstrap samples. To support the phylogenetic analyses, conserved motif analysis of the protein sequences of TipE gene family members was performed using MEME [31], with the motif width from 6 to 50 amino acids and a maximum of 10 motifs.
Abbreviations
- BK-beta:
-
big-conductance calcium-activated potassium channel beta subunit
- BKCa channel:
-
big-conductance calcium-activated potassium channel
- DSC1:
-
Drosophila sodium channel 1
- EGF:
-
epidermal growth factor
- Flo-2:
-
flotillin-2
- GMC:
-
glucose-methanol-choline
- HMM:
-
Hidden Markov Model
- JTT:
-
Jones-Taylor-Thornton
- KCNMB4:
-
calcium-activated potassium channel subunit beta-4
- NaV channel:
-
voltage-gated sodium channel
- Para:
-
paralytic
- Slo-beta:
-
Slowpoke-beta
- Teh1-4:
-
TipE homolog 1-4
- TipE:
-
temperature-induced paralytic E
- VhaM9.7-c:
-
Vacuolar H+ ATPase subunit M9.7-c.
References
Widmark J, Sundström G, Ocampo Daza D, Larhammar D: Differential Evolution of Voltage-Gated Sodium Channels in Tetrapods and Teleost Fishes. Molecular Biology and Evolution. 2011, 28: 859-871. 10.1093/molbev/msq257.
Tseng TT, McMahon AM, Johnson VT, Mangubat EZ, Zahm RJ, Pacold ME, Jakobsson E: Sodium channel auxiliary subunits. J Mol Microbiol Biotechnol. 2007, 12: 249-262. 10.1159/000099646.
Kulkarni NH, Yamamoto AH, Robinson KO, Mackay TFC, Anholt RRH: The DSC1 channel, encoded by the smi60E locus, contributes to odor-guided behavior in Drosophila melanogaster. Genetics. 2002, 161: 1507-1516.
Zhou W, Chung I, Liu Z, Goldin AL, Dong K: A voltage-gated calcium-selective channel encoded by a sodium channel-like gene. Neuron. 2004, 42: 101-112. 10.1016/S0896-6273(04)00148-5.
Suzuki DT, Grigliatti T, Williamson R: Temperature-sensitive mutations in Drosophila melanogaster. VII. A mutation (para-ts) causing reversible adult paralysis. Proc Natl Acad Sci USA. 1971, 68: 890-893. 10.1073/pnas.68.5.890.
Siddiqi O, Benzer S: Neurophysiological defects in temperature-sensitive paralytic mutants of Drosophila melanogaster. Proc Natl Acad Sci USA. 1976, 73: 3253-3257. 10.1073/pnas.73.9.3253.
Wu CF, Ganetzky B: Genetic alteration of nerve membrane excitability in temperature-sensitive paralytic mutants of Drosophila melanogaster. Nature. 1980, 286: 814-816. 10.1038/286814a0.
Loughney K, Kreber R, Ganetzky B: Molecular analysis of the para locus, a sodium channel gene in Drosophila. Cell. 1989, 58: 1143-1154. 10.1016/0092-8674(89)90512-6.
Jackson FR, Wilson SD, Hall LM: The tip-E mutation of Drosophila decreases saxitoxin binding and interacts with other mutations affecting nerve membrane excitability. J Neurogenet. 1986, 3: 1-17. 10.3109/01677068609106891.
Ganetzky B: Neurogenetic analysis of Drosophila mutations affecting sodium channels: synergistic effects on viability and nerve conduction in double mutants involving tip-E. J Neurogenet. 1986, 3: 19-31. 10.3109/01677068609106892.
Feng G, Deák P, Chopra M, Hall LM: Cloning and functional analysis of TipE, a novel membrane protein that enhances Drosophila para sodium channel function. Cell. 1995, 82: 1001-1011. 10.1016/0092-8674(95)90279-1.
Hodges DD, Lee D, Preston CF, Boswell K, Hall LM, O'Dowd DK: tipE Regulates Na+-dependent Repetitive Firing in Drosophila Neurons. Molecular and Cellular Neuroscience. 2002, 19: 402-416. 10.1006/mcne.2001.1088.
Warmke JW, Reenan RAG, Wang P, Qian S, Arena JP, Wang J, Wunderler D, Liu K, Kaczorowski GJ, der Ploeg LHTV, Ganetzky B, Cohen CJ: Functional Expression of Drosophila para Sodium Channels. The Journal of General Physiology. 1997, 110: 119-133. 10.1085/jgp.110.2.119.
Derst C, Walther C, Veh RW, Wicher D, Heinemann SH: Four novel sequences in Drosophila melanogaster homologous to the auxiliary Para sodium channel subunit TipE. Biochem Biophys Res Commun. 2006, 339: 939-948. 10.1016/j.bbrc.2005.11.096.
Dong K: Insect sodium channels and insecticide resistance. Invert Neurosci. 2007, 7: 17-30. 10.1007/s10158-006-0036-9.
Davies TGE, Field LM, Usherwood PNR, Williamson MS: DDT, pyrethrins, pyrethroids and insect sodium channels. IUBMB Life. 2007, 59: 151-162. 10.1080/15216540701352042.
Zlotkin E: The insect voltage-gated sodium channel as target of insecticides. Annu Rev Entomol. 1999, 44: 429-455. 10.1146/annurev.ento.44.1.429.
Vais H, Williamson MS, Devonshire AL, Usherwood PN: The molecular interactions of pyrethroid insecticides with insect and mammalian sodium channels. Pest Manag Sci. 2001, 57: 877-888. 10.1002/ps.392.
Soderlund DM, Knipple DC: The molecular biology of knockdown resistance to pyrethroid insecticides. Insect Biochem Mol Biol. 2003, 33: 563-577. 10.1016/S0965-1748(03)00023-7.
Zdobnov EM, Bork P: Quantification of insect genome divergence. Trends Genet. 2007, 23: 16-20. 10.1016/j.tig.2006.10.004.
Garcia-Fernàndez J: The genesis and evolution of homeobox gene clusters. Nat Rev Genet. 2005, 6: 881-892.
Cho SJ, Vallès Y, Giani VC, Seaver EC, Weisblat DA: Evolutionary dynamics of the wnt gene family: a lophotrochozoan perspective. Mol Biol Evol. 2010, 27: 1645-1658. 10.1093/molbev/msq052.
Wyder S, Kriventseva EV, Schröder R, Kadowaki T, Zdobnov EM: Quantification of ortholog losses in insects and vertebrates. Genome Biol. 2007, 8: R242-10.1186/gb-2007-8-11-r242.
Hedges SB, Kumar S: The timetree of life. 2009, Oxford University Press
Kirkness EF, Haas BJ, Sun W, Braig HR, Perotti MA, Clark JM, Lee SH, Robertson HM, Kennedy RC, Elhaik E, Gerlach D, Kriventseva EV, Elsik CG, Graur D, Hill CA, Veenstra JA, Walenz B, Tubío JMC, Ribeiro JMC, Rozas J, Johnston JS, Reese JT, Popadic A, Tojo M, Raoult D, Reed DL, Tomoyasu Y, Krause E, Mittapalli O, Margam VM, et al: Genome sequences of the human body louse and its primary endosymbiont provide insights into the permanent parasitic lifestyle. Proceedings of the National Academy of Sciences. 2010, 107: 12168-12173. 10.1073/pnas.1003379107.
Assis R, Kondrashov AS, Koonin EV, Kondrashov FA: Nested genes and increasing organizational complexity of metazoan genomes. Trends in Genetics. 2008, 24: 475-478. 10.1016/j.tig.2008.08.003.
Kumar A: An Overview of Nested Genes in Eukaryotic Genomes. Eukaryot Cell. 2009, 8: 1321-1329. 10.1128/EC.00143-09.
Iida K, Cox-Foster D, Yang X, Ko WY, Cavener D: Expansion and evolution of insect GMC oxidoreductases. BMC Evolutionary Biology. 2007, 7: 75-10.1186/1471-2148-7-75.
Chintapalli VR, Wang J, Dow JAT: Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet. 2007, 39: 715-720. 10.1038/ng2049.
Celniker SE, Dillon LAL, Gerstein MB, Gunsalus KC, Henikoff S, Karpen GH, Kellis M, Lai EC, Lieb JD, MacAlpine DM, Micklem G, Piano F, Snyder M, Stein L, White KP, Waterston RH: Unlocking the secrets of the genome. Nature. 2009, 459: 927-930. 10.1038/459927a.
Bailey TL, Elkan C: Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994, 2: 28-36.
Yu FH, Catterall WA: Overview of the voltage-gated sodium channel family. Genome Biol. 2003, 4: 207-10.1186/gb-2003-4-3-207.
Ma H, Zhang J, Levitan IB: Slob, a Slowpoke channel-binding protein, modulates synaptic transmission. The Journal of General Physiology. 2011, 137: 225-238. 10.1085/jgp.201010439.
Negre B, Ruiz A: HOM-C evolution in Drosophila: is there a need for Hox gene clustering?. Trends Genet. 2007, 23: 55-59. 10.1016/j.tig.2006.12.001.
Berkefeld H, Fakler B, Schulte U: Ca2+-Activated K+ Channels: From Protein Complexes to Function. Physiological Reviews. 2010, 90: 1437-1459. 10.1152/physrev.00049.2009.
Gauthier JP, Legeai F, Zasadzinski A, Rispe C, Tagu D: AphidBase: a database for aphid genomic resources. Bioinformatics. 2007, 23: 783-784. 10.1093/bioinformatics/btl682.
Kim HS, Murphy T, Xia J, Caragea D, Park Y, Beeman RW, Lorenzen MD, Butcher S, Manak JR, Brown SJ: BeetleBase in 2010: revisions to provide comprehensive genomic information for Tribolium castaneum. Nucleic Acids Research. 2009, 38: D437-D442.
Tweedie S, Ashburner M, Falls K, Leyland P, McQuilton P, Marygold S, Millburn G, Osumi-Sutherland D, Schroeder A, Seal R, Zhang H, The FlyBase Consortium: FlyBase: enhancing Drosophila Gene Ontology annotations. Nucleic Acids Research. 2009, 37: D555-D559. 10.1093/nar/gkn788.
Munoz-Torres MC, Reese JT, Childers CP, Bennett AK, Sundaram JP, Childs KL, Anzola JM, Milshina N, Elsik CG: Hymenoptera Genome Database: integrated community resources for insect species of the order Hymenoptera. Nucleic Acids Research. 2010, 39: D658-D662.
Wang J, Xia Q, He X, Dai M, Ruan J, Chen J, Yu G, Yuan H, Hu Y, Li R, Feng T, Ye C, Lu C, Wang J, Li S, Wong GK-S, Yang H, Wang J, Xiang Z, Zhou Z, Yu J: SilkDB: a knowledgebase for silkworm biology and genomics. Nucleic Acids Res. 2005, 33: D399-402.
Lawson D, Arensburger P, Atkinson P, Besansky NJ, Bruggner RV, Butler R, Campbell KS, Christophides GK, Christley S, Dialynas E, Hammond M, Hill CA, Konopinski N, Lobo NF, MacCallum RM, Madey G, Megy K, Meyer J, Redmond S, Severson DW, Stinson EO, Topalis P, Birney E, Gelbart WM, Kafatos FC, Louis C, Collins FH: VectorBase: a data resource for invertebrate vector genomics. Nucleic Acids Research. 2009, 37: D583-D587. 10.1093/nar/gkn857.
Colbourne J, Singan V, Gilbert D: wFleaBase: the Daphnia genome database. BMC Bioinformatics. 2005, 6: 45-10.1186/1471-2105-6-45.
Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, George RA, Lewis SE, Richards S, Ashburner M, Henderson SN, Sutton GG, Wortman JR, Yandell MD, Zhang Q, Chen LX, Brandon RC, Rogers YH, Blazej RG, Champe M, Pfeiffer BD, Wan KH, Doyle C, Baxter EG, Helt G, Nelson CR, et al: The Genome Sequence of Drosophila melanogaster. Science. 2000, 287: 2185-2195. 10.1126/science.287.5461.2185.
Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA, Kaufman TC, Kellis M, Gelbart W, Iyer VN, Pollard DA, Sackton TB, Larracuente AM, Singh ND, Abad JP, Abt DN, Adryan B, Aguade M, Akashi H, Anderson WW, Aquadro CF, Ardell DH, Arguello R, Artieri CG, Barbash DA, Barker D, Barsanti P, Batterham P, Batzoglou S, Begun D, et al: Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007, 450: 203-218. 10.1038/nature06341.
Holt RA, Subramanian GM, Halpern A, Sutton GG, Charlab R, Nusskern DR, Wincker P, Clark AG, Ribeiro JM, Wides R, Salzberg SL, Loftus B, Yandell M, Majoros WH, Rusch DB, Lai Z, Kraft CL, Abril JF, Anthouard V, Arensburger P, Atkinson PW, Baden H, de Berardinis V, Baldwin D, Benes V, Biedler J, Blass C, Bolanos R, Boscus D, Barnstead M, et al: The genome sequence of the malaria mosquito Anopheles gambiae. Science. 2002, 298: 129-149. 10.1126/science.1076181.
Nene V, Wortman JR, Lawson D, Haas B, Kodira C, Tu ZJ, Loftus B, Xi Z, Megy K, Grabherr M, Ren Q, Zdobnov EM, Lobo NF, Campbell KS, Brown SE, Bonaldo MF, Zhu J, Sinkins SP, Hogenkamp DG, Amedeo P, Arensburger P, Atkinson PW, Bidwell S, Biedler J, Birney E, Bruggner RV, Costas J, Coy MR, Crabtree J, Crawford M, et al: Genome sequence of Aedes aegypti, a major arbovirus vector. Science. 2007, 316: 1718-1723. 10.1126/science.1138878.
Arensburger P, Megy K, Waterhouse RM, Abrudan J, Amedeo P, Antelo B, Bartholomay L, Bidwell S, Caler E, Camara F, Campbell CL, Campbell KS, Casola C, Castro MT, Chandramouliswaran I, Chapman SB, Christley S, Costas J, Eisenstadt E, Feschotte C, Fraser-Liggett C, Guigo R, Haas B, Hammond M, Hansson BS, Hemingway J, Hill SR, Howarth C, Ignell R, Kennedy RC, et al: Sequencing of Culex quinquefasciatus Establishes a Platform for Mosquito Comparative Genomics. Science. 2010, 330: 86-88. 10.1126/science.1191864.
Xia Q, Zhou Z, Lu C, Cheng D, Dai F, Li B, Zhao P, Zha X, Cheng T, Chai C, Pan G, Xu J, Liu C, Lin Y, Qian J, Hou Y, Wu Z, Li G, Pan M, Li C, Shen Y, Lan X, Yuan L, Li T, Xu H, Yang G, Wan Y, Zhu Y, Yu M, Shen W, et al: A draft sequence for the genome of the domesticated silkworm (Bombyx mori). Science. 2004, 306: 1937-1940.
Richards S, Gibbs RA, Weinstock GM, Brown SJ, Denell R, Beeman RW, Gibbs R, Beeman RW, Brown SJ, Bucher G, Friedrich M, Grimmelikhuijzen CJ, Klingler M, Lorenzen M, Richards S, Roth S, Schröder R, Tautz D, Zdobnov EM, Muzny D, Gibbs RA, Weinstock GM, Attaway T, Bell S, Buhay CJ, Chandrabose MN, Chavez D, Clerk-Blankenburg KP, Cree A, Dao M, et al: The genome of the model beetle and pest Tribolium castaneum. Nature. 2008, 452: 949-955. 10.1038/nature06784.
Weinstock GM, Robinson GE, Gibbs RA, Weinstock GM, Robinson GE, Worley KC, Evans JD, Maleszka R, Robertson HM, Weaver DB, Beye M, Bork P, Elsik CG, Evans JD, Hartfelder K, Hunt GJ, Robertson HM, Robinson GE, Maleszka R, Weinstock GM, Worley KC, Zdobnov EM, Hartfelder K, Amdam GV, Bitondi MM, Collins AM, Cristino AS, Evans JD, Lattorff MG, Lobo CH, et al: Insights into social insects from the genome of the honeybee Apis mellifera. Nature. 2006, 443: 931-949. 10.1038/nature05260.
Werren JH, Richards S, Desjardins CA, Niehuis O, Gadau J, Colbourne JK, Beukeboom LW, Desplan C, Elsik CG, Grimmelikhuijzen CJ, Kitts P, Lynch JA, Murphy T, Oliveira DC, Smith CD, van de Zande L, Worley KC, Zdobnov EM, Aerts M, Albert S, Anaya VH, Anzola JM, Barchuk AR, Behura SK, Bera AN, Berenbaum MR, Bertossa RC, Bitondi MM, Bordenstein SR, Bork P, et al: Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science. 2010, 327: 343-348. 10.1126/science.1178028.
Richards S, Gibbs RA, Gerardo NM, Moran N, Nakabachi A, Richards S, Stern D, Tagu D, Wilson AC, Muzny D, Kovar C, Cree A, Chacko J, Chandrabose MN, Dinh HH, Gabisi RA, Hines S, Hume J, Jhangian SN, Joshi V, Lewis LR, Liu YS, Lopez J, Morgan MB, Nguyen NB, Okwuonu GO, Ruiz SJ, Santibanez J, Wright RA, Fowler GR, et al: Genome sequence of the pea aphid Acyrthosiphon pisum. PLoS Biol. 2010, 8: e1000313-10.1371/journal.pbio.1000313.
Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, Oakley TH, Tokishita S, Aerts A, Arnold GJ, Basu MK, Bauer DJ, Cáceres CE, Carmel L, Casola C, Choi JH, Detter JC, Dong Q, Dusheyko S, Eads BD, Fröhlich T, Geiler-Samerotte KA, Gerlach D, Hatcher P, Jogdeo S, Krijgsveld J, Kriventseva EV, Kültz D, Laforsch C, Lindquist E, Lopez J, et al: The ecoresponsive genome of Daphnia pulex. Science. 2011, 331: 555-561. 10.1126/science.1197761.
Zdobnov EM, von Mering C, Letunic I, Torrents D, Suyama M, Copley RR, Christophides GK, Thomasova D, Holt RA, Subramanian GM, Mueller HM, Dimopoulos G, Law JH, Wells MA, Birney E, Charlab R, Halpern AL, Kokoza E, Kraft CL, Lai Z, Lewis S, Louis C, Barillas-Mury C, Nusskern D, Rubin GM, Salzberg SL, Sutton GG, Topalis P, Wides R, Wincker P, et al: Comparative Genome and Proteome Analysis of Anopheles gambiae and Drosophila melanogaster. Science. 2002, 298: 149-159. 10.1126/science.1077061.
Kriventseva EV, Rahman N, Espinosa O, Zdobnov EM: OrthoDB: the hierarchical catalog of eukaryotic orthologs. Nucleic Acids Research. 2008, 36: D271-D275.
Waterhouse RM, Zdobnov EM, Tegenfeldt F, Li J, Kriventseva EV: OrthoDB: the hierarchical catalog of eukaryotic orthologs in 2011. Nucleic Acids Research. 2011, 39: D283-D288. 10.1093/nar/gkq930.
Salamov AA, Solovyev VV: Ab initio Gene Finding in Drosophila Genomic DNA. Genome Research. 2000, 10: 516-522. 10.1101/gr.10.4.516.
Birney E, Clamp M, Durbin R: GeneWise and Genomewise. Genome Research. 2004, 14: 988-995. 10.1101/gr.1865504.
Eddy SR: Profile hidden Markov models. Bioinformatics. 1998, 14: 755-763. 10.1093/bioinformatics/14.9.755.
Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004, 32: 1792-1797. 10.1093/nar/gkh340.
Castresana J: Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Molecular Biology and Evolution. 2000, 17: 540-552.
Guindon S, Gascuel O: A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Systematic Biology. 2003, 52: 696-704. 10.1080/10635150390235520.
Acknowledgements
The authors would like to thank Dr Evgenia Kriventseva, Dr Thomas Junier and all members of the Computational Evolutionary Genomics Group for useful suggestions and discussions. This work was supported by the Swiss National Science Foundation (3100A0-112588).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JL, RMW and EMZ conceived and designed the study, and wrote the manuscript. JL performed the computational data analyses. All authors read and approved the final manuscript.
Electronic supplementary material
12862_2011_1935_MOESM1_ESM.XLS
Additional file 1: Genomic resources and gene information. Table S1: Details of the genes included in this study; Table S2: Details of the genomic resources used in this study. (XLS 48 KB)
12862_2011_1935_MOESM3_ESM.PDF
Additional file 3: Additional Figures. Figure S1: Detailed map of the last exons of CG18675 and TipE in Drosophila melanogaster; Figure S2: Phylogenetic tree of TipE gene family members in six insect species and Daphnia pulex; Figure S3: Motif architectures of TipE-like proteins in six insect species and Daphnia pulex; Figure S4: Phylogenetic tree of Para orthologues in 11 insect species, Daphnia pulex and Ixodes scapularis (PDF 267 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Li, J., Waterhouse, R.M. & Zdobnov, E.M. A remarkably stable TipE gene cluster: evolution of insect Para sodium channel auxiliary subunits. BMC Evol Biol 11, 337 (2011). https://doi.org/10.1186/1471-2148-11-337
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-11-337