
Abstract
Advanced glycation end products (AGEs), a group of compounds that are formed by non-enzymatic reactions between carbonyl groups of reducing sugars and free amino groups of proteins, lipids or nucleic acids, can be obtained exogenously from diet or formed endogenously within the body. AGEs accumulate intracellularly and extracellularly in all tissues and body fluids and can cross-link with other proteins and thus affect their normal functions. Furthermore, AGEs can interact with specific cell surface receptors and hence alter cell intracellular signaling, gene expression, the production of reactive oxygen species and the activation of several inflammatory pathways. High levels of AGEs in diet as well as in tissues and the circulation are pathogenic to a wide range of diseases. With respect to mobility, AGEs accumulate in bones, joints and skeletal muscles, playing important roles in the development of osteoporosis, osteoarthritis, and sarcopenia with aging. This report covered the related pathological mechanisms and the potential pharmaceutical and dietary intervention strategies in reducing systemic AGEs. More prospective studies are needed to determine whether elevated serum AGEs and/or skin autofluorescence predict a decline in measures of mobility. In addition, human intervention studies are required to investigate the beneficial effects of exogenous AGEs inhibitors on mobility outcomes.
Similar content being viewed by others
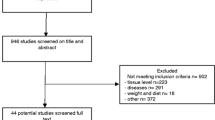


Background
Overview of advanced glycation end products (AGEs)
What are they?
Advanced glycation end products (AGEs) are a heterogeneous group of compounds that are formed by non-enzymatic reactions between the carbonyl groups of reducing sugars and the free amino groups of proteins, lipids or nucleic acids. AGEs are produced in the Maillard reaction, which can cause browning, fluorescence and protein cross-linking, as well as the formation of flavour and aroma compounds [1]. AGEs can be formed within the body and can also originate from exogenous sources such as diet and smoking. Increased levels of AGEs are generated during the heat processing of food and the browning continues during storage. Various AGEs precursors are present and are formed during the initial, intermediate and final stages of the Maillard reaction and, depending on their composition and their molecular size, different AGEs compounds are created through a series of reactions involving enolisation, dehydration, cyclisation, fragmentation and oxidation. Such compounds include pyrraline, Nɛ-carboxymethyllysine (CML), Nɛ-carboxyethyllysine (CEL), pentosidine, argpyrimidine, derivatives of methylglyoxal (MG), hydroimidazolones derived from MG, glyoxal (GO), 3-deoxyglucosone (3-DG), arginine-derived Nδ-ornithine and bis(lysyl)imidazolium derivatives, such as methylglyoxal-lysine dimer (MOLD) and glyoxal-lysine-dimer (GOLD) [1, 2]. Among these, CML, pentosidine, and furosine are considered as the common AGEs in foods and human plasma [3, 4], in which CML has been found as the most abundant AGEs in human plasma [5].
AGEs can be divided into three categories according to their ability to create cross-links on proteins and to show fluorescence: (1) fluorescent cross-linking AGEs such as pentosidine and crossline; (2) non-fluorescent cross-linking AGEs such as imidazolium dilysine cross-links, alkyl formyl glycosyl pyrrole (AFGP) cross-links and arginine-lysine imidazole (ALI) cross-links; (3) non-cross-linking AGEs such as pyrraline, CML and CEL [6]. Because of the complexity and heterogeneity of AGEs formation in vivo, the structures of cross-linked AGEs have not been completely determined.
Some researchers also classify AGEs as being toxic or non-toxic. Compounds such as CML, CEL and pyrraline are considered to be non-toxic AGEs. Toxic AGEs are usually derived from glyceraldehyde or glycolaldehyde. However, the structural identity of toxic AGEs remains unknown [7, 8]. In fact, recent studies have indicated the pathogenic role of some non-cross-linking AGEs (non-toxic) such as CML. For example, through an AGEs receptor, CML may affect cell signalling, may stress cells and may trigger cell injury, leading to pathological endothelial cell dysfunction and apoptosis of macrophages [9, 10].
Exogenous formation of AGEs
AGEs are naturally occurring chemicals in raw animal-origin foods, and cooking propagates and accelerates the generation of more AGEs within them. Studies have shown that dry heating results in the formation of more than ten to hundred times of new AGEs in foods as compared to the uncooked state [1]. For the food industry AGEs are greatly desirable owing to the profound effect of AGEs on safety and convenience as well as to enhance food flavour, colour and appearance, and thus increase food consumption [11, 12]. Dry heat, irradiation or ionization in modern food processing considerably promotes the formation of new AGEs [12,13,14]. Contents of AGEs in food are mainly calculated from measurements of a single marker (CML or MG derivatives), which is regarded as a major limitation considering levels of individual AGEs significantly vary in different foods [1, 4]. On the whole, AGEs contents in foods analysed for MG derivatives are associated with corresponding levels of CML. Animal-origin foods, as well as foods with a high level of fat and protein, contain relatively high contents of AGEs. On the other hand, low values of AGEs were found in uncooked and even cooked carbohydrate-rich foods such as fruits, vegetables, milk and whole grains [1]. The order of dietary AGEs levels in foods is found to be beef>cheeses>poultry>pork>fish>eggs [1].
AGEs enter the circulation together with other nutrients in food. Currently, there are limited data on absorption and bioavailability of AGEs [15]. Both animal studies and human studies confirmed that dietary AGEs are partially absorbed in the intestine (10–30%) [15,16,17]. The absorption rate differs between low molecular weight (LMW) AGEs and high molecular weight (HMW) AGEs. LMW AGEs may be relatively quickly absorbed, biotransformed, and excreted whereas HMW AGEs are absorbed more slowly and less efficiently due to insufficient degradation by gastrointestinal enzymes. About two thirds of the absorbed AGEs remain in the body for 3 days [17,18,19,20], resulting in increased oxidation stress, AGEs and potentially organ damage. The bioavailability of AGEs is largely influenced by factors such as diet, structures and gut environment etc. Global AGEs distribution in tissues were observed in animal studies, which have shown that more than half of the absorbed AGEs were bound in liver and kidney after 72 h, the rest could be found in heart, lung and spleen [15, 20].
Recent animal and human studies with an oral intake of an AGE-rich meal, labelled AGEs or specific AGEs have clearly demonstrated that dietary AGEs represent an important source for circulating AGEs and contribute to the in vivo AGEs pool under physiological conditions [17, 20,21,22,23,24].
Studies on dietary AGEs intakes in the general population are however scarce. The estimated average dietary AGEs intake in adults has been shown to be 15,000 AGEs kU/day [1, 25], which is considered to be high and inductive for inflammation. Diets rich in grilled or roasted meats, fats, and highly processed foods could achieve a level of AGEs higher than 20,000 kU/day [1]. The influence of dietary AGEs on the formation of endogenous AGEs is discussed in Section “Factors that Affect AGEs Formation in vivo” of this report.
Besides dietary AGEs, smoking is another source of exogenous AGEs. Scarce information, however, is found on sources of AGEs in smokers [26]. Cerami et al. (1997) reported that the water extracts of tobacco leaves contain reactive glycation products (glycotoxins) and formation of AGEs in vivo and in vitro was promoted by tobacco smoke, the process of which was concentration and time dependent [27]. The highly reactive glycotoxins can induce the formation of AGEs formation in hours whereas glucose or glucose-6- phosphate induced AGEs formation takes days to weeks [27]. Glycotoxins from cigarette enter the body via lung alveoli and then are transported to blood stream or lung cells where the formation of AGEs occurs by interacting with other glycation products [26, 28].
Endogenous formation of AGEs
The formation of AGEs endogenously is a part of the normal consequence of metabolism. However, this can be pathogenic if high levels of AGEs accumulate in tissues and the circulation. The formation of AGEs can be accelerated under certain conditions, such as hyperglycaemia, hyperlipidaemia and increased oxidative stress (OS). In fact, with aging and different diseases, elevated amounts of AGEs have been found in vivo [6, 29,30,31,32].
Generally, AGEs are formed physiologically in all tissues and body fluids, both intracellularly and extracellularly, when the carbonyl groups of reducing sugars react non-enzymatically with the free amino groups on proteins [33]. As this reaction occurs at a lower temperature and is less complex, compared with food production, there is less diversity of the AGEs compounds.
The research on AGEs in the human body has progressed dramatically during the last 20 years. One of the early examples is the identification of haemoglobin A1c (HbA1c), which is an Amadori rearrangement product, and results from the combination of glucose with the N-terminal valine residue of a haemoglobin B chain. It is measured clinically as an index of hyperglycaemia [34, 35].
At least four types of process in the formation of AGEs under physiological conditions have been identified (Fig. 1) [15, 36].
-
1)
Monosaccharide autoxidation (auto-oxidative glycosylation) or the degradation of saccharides unattached to a protein. This is an auto-oxidative pathway in which sugars give rise to reactive products by autoxidation.
-
2)
Unstable Schiff base fragmentation, which is typically followed by the generation of a stable Amadori product.
-
3)
Fructosamine (ketosamine) degradation.
-
4)
The direct reaction of α,β-dicarbonyl compounds (oxoaldehydes) formed from the reducing carbohydrates and lipid peroxidation.

Formation of AGEs in vivo. Adapted from Gugliucci and Menini (2014) [104]
During formation of AGEs, the important sites of glycation are lysyl side chains, N-terminal amino groups and arginyl guanidine groups of proteins, guanyl bases of nucleotides and amino groups of phosphatidylethanolamine and phosphatidylserine [15]. Under physiological conditions, glucose is the most studied carbonyl precursor because it is the most commonly seen reducing sugar. The reaction between glucose and proteins in vivo is relatively slow. During the early stages of the Maillard reaction, Schiff bases are formed between reactive sugars and ɛ-amino groups. These can lead to an irreversible intramolecular rearrangement, which forms more stable Amadori products. The Amadori products undergo further structural changes through dehydration, oxidation and degradation to form highly stable AGEs, such as CML [37]. However, some highly reactive dicarbonyl compounds, such as GO, MG and 3-DG, are capable of rapid AGEs formation. They are generated by various pathways including glucose autoxidation, lipid peroxidation and interruption of glycolysis by reactive oxygen species (ROS). A large quantity of these dicarbonyl compounds induces so-called “carbonyl stress”, because they are highly reactive with both intracellular and extracellular proteins [7]. For example, 3-DG, which can be formed by the non-oxidative rearrangement and hydrolysis of Amadori products, reacts rapidly with protein amino groups to form AGEs such as imidazolone, pyrraline and CML [38, 39]. MG can be produced by the autoxidation of carbohydrates and lipid peroxidation in most glucose-metabolising cells, and reacts with the lysine residues on proteins to form CEL [40].
Factors that affect AGEs formation in vivo
As mentioned in Section “Exogenous Formation of AGEs”., the dietary AGEs load contributes to circulating AGEs and the AGEs pool in vivo, and hence dietary AGEs can affect endogenous AGEs formation through their effects on AGEs metabolism. Generally, AGEs are generated within the body in homeostasis. The rate of AGEs formation in vivo depends on a number of factors, including the nature and concentration of the substrate groups, the glycating agents, the half-life of the proteins, the availability of catalytic compounds, the OS or redox balance and the presence of inhibitors such as aminoguanidine and pyridoxamine.
The abilities of sugars to react with amino groups differ. The smaller sugar molecules with fewer carbon atoms are more reactive. The reactivity increases when there are more reactive open chains and furanose ring structures. For example, fructose is about 10-fold more reactive than glucose. The low reactivity of glucose, which is the predominant sugar in vivo, works as a native protection mechanism against the intracellular accumulation of AGEs and their precursors. As a result, the Maillard reaction is slow under normal metabolism [41]. However, under some sub-clinical conditions, such as hyperglycaemia and increased OS, elevated AGEs formation is triggered by increasing levels of reactive carbonyl intermediates, such as GO, MG and 3-DG. In addition, ROS are generated during the formation of AGEs, including stages such as the autoxidation of glucose, Schiff bases and Amadori adducts. When the level of ROS is elevated under unbalanced OS, a vicious cycle of AGE/ROS promotes more oxidation of lipids and glucose and accelerates the formation of AGEs in vivo [42].
The antioxidant systems in the body, including reducing agents, antioxidant enzymes and the detoxification system, can limit the level of AGEs precursors and reduce the generation of ROS. For example, the enzymes in the glyoxalase system, which carry out detoxification, can prevent the MG-mediated glycation and can convert most of the MG into harmless molecules such as lactate [43]. In the blood and body fluids, some early detoxification proteins, such as defensins, lactoferrin and lysozyme, are able to bind AGEs before their cellular uptake or cross-linking to other molecules [44, 45].
The most important mechanisms involved in the degradation of endogenous AGEs are extracellular proteolysis and the AGEs-receptor 1 (AGER1)-mediated intracellular uptake and degradation within cells [46]. For example, the degradation of AGEs by certain cells such as macrophages generates soluble AGEs, which can be excreted by the kidney [47].
In addition, as renal clearance works as an important factor for the level of circulating AGEs, it affects the accumulation of AGEs and the formation of endogenous AGEs. In fact, renal AGEs clearance can be affected by aging and the accumulation of AGEs in the kidney with renal impairment [48]. It can also be mediated by some functional compounds such as lysozyme, which may accelerate renal AGEs clearance [49]. In conclusion, AGEs are metabolised by innate defence and/or intracellular degradation after receptor-dependent uptake [45]. These protecting systems balance the endogenous formation of AGEs (Fig. 2) [40].

The cycle of endogenous and exogenous AGEs. Adapted from Stirban et al.(2013) [47]
Direct impact of AGEs on plasma and extracellular proteins
The direct toxic effects of AGEs result from altering the structure and function of plasma and extracellular proteins by glycation and cross-linking. The glycation of proteins affects their normal function by disrupting their molecular conformation, interfering with their receptor function and altering their enzymatic activity. In addition, AGEs cross-link not only with proteins but also with other intracellular and extracellular molecules such as lipids and nucleic acids, which leads to structural and functional changes [6]. The alterations to the enzymatic proteolysis and degradation rate of some proteins such as elastin and collagen could lead to their accumulation in the extracellular matrix, which may result in changes in cellular adhesion and cell growth [50, 51]. This may decrease the elastic properties of both arterial and ventricular walls, causing the reductions in vascular and myocardial compliance that are characteristic of aging and diabetes [40].
Some long-lived structural proteins are more prone to AGEs accumulation because of their slow turnover. As these proteins provide the framework for most of the parenchymal organs, either in their fibrous form or in their basement membrane, their glycation and accumulation of AGEs could result directly in pathological outcomes, including renal function impairment, diabetic complications and affected bone health. For example, AGEs change the properties of collagen such as decreasing its solubility and changing its rigidity [52]. In the extracellular matrix region of the kidney, in which AGEs accumulate in collagen, these AGEs could cause changes in elasticity, ionic charge, thickness and turnover of basement membrane components, and hence could affect renal function [48]. Furthermore, the accumulation of AGEs in bone can affect bone strength and can lead to skeletal fragility by decreasing bone toughness and increasing stiffness [53, 54]. Non-enzymatic glycation of collagen may also exert a negative effect on bone remodelling and interfere with osteoblast differentiation [6, 55, 56].
Impact of AGEs on inflammation, oxidative stress and insulin resistance
Apart from the direct impact of AGEs on proteins and the extracellular matrix, AGEs can also interact with specific cell surface receptors and hence alter cell intracellular signalling, gene expression, the production of ROS and the activation of several inflammatory pathways, including the release of pro-inflammatory cytokines, growth factors and adhesion molecules via activation of the nuclear factor kappa B (NF-κB) pathway (Table 1) [57].
The most well-studied AGEs receptor is the receptor of advanced glycation end products (RAGE), which is the main up-regulator of cell activation in response to the AGEs load. RAGE is a multi-ligand receptor, belongs to the immunoglobulin superfamily and has a highly charged, cytoplasmic domain. It recognises a range of ligands including AGEs, leukocyte integrin Mac-1, modified low density lipoprotein (LDL), DNA, RNA and S100 calcium-binding protein B (S100B) [58]. AGE–RAGE interaction triggers various intracellular signalling cascades, followed by the transcription of a range of genes involved in different biological systems, which perpetuates the inflammatory/pro-inflammatory signals [59]. Specifically, this axis stimulates Janus kinase/signal transducers and activators of transcription (JAK/STAT), p38 mitogen-activated protein kinase (p38 MAPK), extracellular signal-regulated protein kinases 1 and 2 (ERK 1/2) and c-Jun N-terminal kinase (JNK), which leads to the activation of transcription factors NF-κB and interferon-stimulated response elements (ISRE). This causes increased expression of cytokines, growth factors and adhesion molecules. Furthermore, AGE–RAGE interaction also stimulates the generation of ROS via the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase pathway [47, 57, 59, 60].
In contrast, there are a number of AGEs receptors, such as the advanced glycation end product receptor (AGER) family and the scavenger receptor (SR) family, that mediate endocytosis, leading to the intracellular uptake and degradation of AGEs by their fusion with lysosomes [61,62,63,64]. Furthermore, AGEs peptides can be transferred to the renal system, whereas the receptors are recycled and available for further endocytosis processes [57, 65].
The expression level and the activation of AGEs receptors depend on the cell or tissue type (Table 1) and are regulated in response to the AGEs load, other metabolic changes, conditions such as hyperlipidaemia, aging and diabetes mellitus [66]. For example, in Fig. 3a, in response to conditions with a low AGEs burden, the expression of RAGE is down-regulated whereas the expression of AGER1 is up-regulated. As the RAGE signalling pathway leads to the activation of transcription factors NF-κB, activator protein 1 (AP-1) and forkhead box protein O subclass (FOXO), the down-regulated RAGE reduces the transcription of genes related to OS and inflammation. The up-regulation of AGER1 also inhibits these transcription factors through the sirtuin-1 (SIRT1) pathway. In addition, the increased expression of AGER1 may accelerate the intracellular degradation of AGEs, which results in an overall lower degree of OS and inflammation caused by AGEs. In Fig. 3b, when there is an AGEs burden, RAGE is up-regulated, leading to increased OS and inflammation. The prolonged high AGEs burden leads to down-regulation of AGER1, which, therefore, cannot exert strong inhibitory effects on RAGE signalling or reduce the levels of AGEs by their degradation [15]. Recent studies demonstrate that the interaction between AGEs and RAGE can cause cell migration and adhesion. For example, the activation of RAGE on monocytes can induce the migration of these cells into the sub-endothelial space [67]. Via interaction with integrin CD11b and their increased expression on endothelial cells, RAGEs can work as receptors for recruiting monocytes and neutrophils, and promoting the adhesion of leukocytes to the vessel wall [68, 69].

Scheme of the interaction of AGEs with RAGE and AGER1 under conditions with different AGE loads, a) a low AGEs burden, and b) an overload of AGEs. AGER1, AGE receptor 1; AP-1, activator protein 1; Cdc42-Rac, cell division control protein 42 homolog-Rac; ERK 1/2, extracellular signal-regulated protein kinases 1 and 2; FOXO, forkhead box protein O subclass; JAK/STAT, Janus kinase/signal transducers and activators of transcription; JNK, c-Jun N-terminal kinases; NADPH, nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor kappa B; p38 MAPK, p38 mitogen-activated protein kinases; RAGE, receptor for AGEs; SIRT1, sirtuin-1; TIRAP-MyD88, toll-interleukin 1 receptor domain containing adaptor protein and myeloid differentiation primary response protein 88. Adapted from Poulsen et al.(2013) [15]
Oxidative Stress-induced cell signal transduction disturbances results in increased OS and disrupted antioxidant defense system, which is implicated in the development and persistence of Insulin Resistance (IR) [70]. Molecular mechanisms of IR involves an array of pathways including cell-specific redox regulation of protein kinases C (PKCs) and changes in the insulin signal pathways phosphatidylinositol 3-kinase (PI3K) and MAPK, suppressing protective survival systems AGER1 and SIRT1 [70,71,72,73,74,75,76,77]. High AGEs in muscle, insulin sensitive tissue, induce IR and activate NFκB by oxidative activation of PKC that phosphorylates regulatory serine residues on insulin receptor substrate-1 (IRS1) in the PI3K pathway [70, 71].
Methods for measuring AGEs and cut-off values for different populations
Several analytical methods are available for measurement of circulating or tissue-bound AGEs, e.g., enzyme-linked immunosorbent assays (ELISA) [22], fluorescence spectroscopy [78], and mass spectrometry (MS)-based high-performance liquid chromatography (HPLC)/gas chromatography (GC) [79]. MS-based methods are often used in laboratories for the diagnosis and monitoring of age-related chronic diseases due to high sensitivity, reproducibility and accuracy. LC coupled with tandem mass spectrometric (LC–MS/MS) has been used to accurately quantify glycation adducts in plasma, urine, and dialysate samples in patients with uremia [80].
Tissue-bound AGEs are usually measured in the skin due to easy accessibility by skin autofluorescence (SAF), a simple and non-invasive technique [78], which has been validated against the gold standard method skin biopsies [81]. Validation studies indicated significant association of SAF with AGEs content in skin biopsies. Meta-analysis of three validation studies has shown that skin AGEs content attributed up to 76% of the variance in the SAF levels, suggesting that SAF can act as a biomarker of cumulative skin AGEs [78, 82,83,84]. Koetsier et al. (2010) conducted a cross-sectional study and provided reference values of SAF for healthy Caucasian control subjects over a broad age range [85].
SAF has been demonstrated to be potentially better predictor for the development of chronic complications and mortality in diabetes over time (5–10 years) than glycated haemoglobin A1c which reflects short-term glycemic status (3–6 months) [81, 82, 86, 87]. Smit et al. (2013) suggested that decision tree method could be used for early diabetes screening in risk groups [88]. According to previously published reference values, cut-off values for SAF would be ≥80th age percentile for age group < 50 years or ≥ 70th age percentile for age group ≥50 years [85, 88,89,90]. In addition, SAF values (> 2.0 AU over 5 years) has been revealed to be a significant marker for the induction and development of vascular complications that can predict CV risk and death [82, 87].
Effects of AGEs on mobility
Effects of AGEs on chronic diseases
In healthy individuals, there are associations between serum AGEs levels and the risk factors for developing chronic diseases. Generally, the circulating AGEs levels are positively correlated with age, oxidative stress and insulin resistance [21, 25, 33]. Higher levels of AGEs have been found in healthy individuals with high dietary AGEs intakes than in individuals who eat foods containing fewer AGEs [91]. In recent studies, the serum concentrations of AGEs have been positively associated with a wide range of diseases, such as obesity, insulin resistance, diabetes, metabolic dysfunction, renal diseases, cardiovascular diseases (CVD), osteoporosis, rheumatoid arthritis, cognitive impairments and cancer [9, 31, 48, 66, 92,93,94,95,96,97,98,99,100,101,102,103,104]. There are direct pathological contributions of AGEs because of their protein cross-linking and accumulation. For instance, in kidney diseases, this could reduce the renal clearance of AGEs and could also increase endogenous AGEs formation [17, 105]. The increased levels of AGEs in vivo can be seen not only as an outcome of the development of these diseases and their relevant complications, but also as a cause of the pathogenesis of some diseases, such as diabetes [12]. This is generally related to the effects of AGEs on protein dysfunction, oxidative stress and inflammation.
Dietary AGEs have been shown to be correlated with serum levels of AGEs, inflammation markers, metabolic dysfunction and life expectancy [5, 21, 24, 25], suggesting that dietary AGEs are pathogenic, and predisposing the body to the development of CVD, diabetes and other chronic diseases, possibly via inducing systemic oxidative stress [21]. Animal studies demonstrated that AGEs supplementation promotes oxidative stress [21, 45, 106]. Similar results were observed in humans, implicating a correlation between dietary AGEs and oxidative stress, and thus increased risk of CVD, renal diseases and diabetes [25, 45]. Modern diet contains high levels of AGEs, resulting in excessive influx of AGEs into the circulation system, and thus enhancing the basal oxidant stress and inflammation [99, 107, 108]. As a result, β-cell functions are prone to be compromised, leading to insulin dysfunction and other diabetic complications. Studies on mice have demonstrated that reduction in dietary AGEs decreased oxidative stress and thus prevents or ameliorates type 1 and type 2 diabetes [109,110,111].
AGEs and mobility
Mobility underlies the ability to perform the basic activities of daily living that are necessary for independence and is a core indicator of health and function in aging [112]. In recent years, there has been increasing evidence that poor mobility outcomes are linked to in vivo AGEs levels [53, 113,114,115,116,117].
AGEs and bone health
Loss of bone mineral and/or bone mass is considered to be the major cause of age-related bone fractures. In fact, every year, more than 8.9 million fractures worldwide are caused by osteoporosis. As there is a lack of the initial symptoms of osteoporosis, many cases are diagnosed only after the first clinical fracture has occurred [118].
Osteoporosis is a progressive skeletal disease, the aetiology of which is attributed to various factors such as endocrine, metabolic and mechanical factors. It is characterised by a systemic impairment of bone mass, density and strength and structural deterioration of the bone microarchitecture, which leads to enhanced bone fragility and an increased risk of fractures. Osteoporosis can occur at any age, but is predominantly found in certain populations such as the elderly and with diseases such as diabetes [119].
Bone is a permanently remodelling organ; it is continually renewed in a complex process of formation by osteoblasts and resorption by osteoclasts. AGEs accumulated in the bone matrix may result in suppressed bone formation as evidenced by an animal study, which showed that significant reductions in mineral apposition rate, mineralized surface per bone surface and bone formation rate were observed in rats with an autograft implant containing AGEs [120]. Bone matrix homeostasis is heavily influenced by nutritional and hormonal factors. Recent studies have proposed non-enzymatic protein glycation as a new factor that affects bone remodelling. The glycation pathway has been implicated as a strong contributor to age-related diseases such as osteoporosis [121]. In support of this concept, increased serum levels of AGEs, such as pentosidine and CML, are found in individuals with osteoporosis [114]. Recent findings also provide important evidence that bone proteins are affected by AGEs modification. Furthermore, these AGEs can influence both osteoclasts and osteoblasts [55, 56, 115].
Type I collagen, contributing to 85% of the bone matrix, is the most abundant protein of bone and has an exceptionally long lifetime, making them susceptible to modification by AGEs [122]. Adverse changes in the collagen network of the bone matrix occur as people age and such changes may lead to deterioration in bone quality. For example, a significantly higher level of pentosidine has been found in the collagen in both cortical and trabecular bone for individuals aged over 65 years [121]. When collagen is cross-linked with AGEs, it has decreased solubility and becomes highly resistant to proteolytic degradation, which consequently leads to stiff collagen with disrupted function. The increased level of AGEs cross-links between collagen molecules is one of the dominant factors affecting the integrity of the collagen network in bone. Furthermore, it has been demonstrated that AGE-modified collagen boosts the intracellular release of ROS, interferes with the adhesion of osteoblastic cells to the matrix and inhibits osteoblastic differentiation and proliferation [123,124,125].
It is known that AGEs affect osteoblast differentiation and proliferation by binding to their receptors [126]. Similarly (Fig. 3), in these cells, the binding of AGEs to RAGE activates NF-κB, resulting in increased expression of cytokines, growth factors and cell adhesion molecules. This initiates inflammatory processes and elicits oxidative stress, leading to abnormal osteoblast function and bone remodelling disorder [115]. For example, there is reduced synthesis of Type I collagen and osteocalcin by human osteoblast-like cells after they have been treated with AGE-modified bovine serum albumin (BSA) [127]. In human osteoblast primary cell culture, there is a dose-dependent effect of the AGE pentosidine on osteoblast function [56]. In primary osteoblasts derived from fetal rat, AGE-collagen suppressed mature bone nodule formation, one of the osteoblastic parameters [122]. These studies have shown that AGEs impaired both bone matrix production and mineralization of osteoblasts.
Although contribution of AGEs to osteoblasts differentiation and function has been well documented in vitro [125, 128,129,130,131], their roles in osteoclast activity and differentiation remain mostly elusive. Different AGEs receptors including RAGE were found expressed in both osteoclast progenitors and mature osteoclasts. AGEs might interact with specific cell-surface receptors to interfere with the process of osteoclastic differentiation and activity [131]. In vitro resorption assay on AGE-modified mineralized matrices revealed that AGEs impaired the structural integrity of bone matrix proteins and the osteoclastic differentiation process, resulting in decreased osteoclast-induced bone resorption. Osteoclastogenesis was inhibited in vitro in the presence of AGEs, most likely by impairing the process of osteoclast progenitors into pre-osteoclastic cells, possibly mainly by a RAGE-dependent manner.
AGE-induced chronic inflammation in bone can be seen as a pathogenetic factor in osteoporosis [116]. It can significantly affect bone turnover, influencing the intrinsic balance of bone mineralisation and resorption [132]. By stimulating the expression of pro-inflammatory cytokines, such as interleukin (IL)-1, IL-6, tumour necrosis factor (TNF)-α and leukaemia inhibitory factor (LIF), AGEs may work as pro-osteoporotic mediators, regulating both osteoblasts and osteoclasts [133, 134]. For example, IL-1, IL-6 and TNF-α work as stimulators for bone resorption by promoting osteoclast activity and inhibiting bone formation [116]. Some modulator-like LIF has a dose–response role, with a high concentration inducing bone resorption and a lower dose promoting bone formation [135, 136]. However, although these studies have demonstrated the pathophysiological effects of these cytokines on bone cells, their exact roles (mechanistic pathways) in the development of osteoporosis have not been elucidated. Furthermore, a growing number of clinical studies have demonstrated not only the concomitance of regional osteoporosis with regional inflammation, but also the association between systemic osteoporosis and events of systemic inflammation, which exposes osteoporosis to the effects of general in vivo AGEs [116].
The detrimental effects of AGEs on osteoblast function also include increased apoptosis signalling by the activation of RAGE and other receptors such as growth factor receptors [55]. Figure 4 shows that this involves the regulation of various autocrines and paracrines, such as insulin-like growth factor I (IGF-I), IL-6 and transforming growth factor-β (TGF-β) [119, 123, 130, 137, 138]. For example, it has been reported that the physiological level of CML-cross-linked collagen can stimulate apoptosis in various osteoblastic cell cultures and that the signalling is mediated through RAGE by stimulating both p38 MAPK and JNK [114, 126]). The enhanced osteoblast apoptosis by AGEs contributes to the mechanisms of the development of osteoporosis [139].

Effect of AGEs on apoptosis signalling. AP-1, activator protein 1; ERK, extracellular signal-regulated protein kinases; IGF-I, insulin-like growth factor I; IL-6, interleukin-6; JAK, Janus kinase; JNK, c-Jun N-terminal kinases; MEK, mitogen-activated protein kinase; NF-κB, nuclear factor kappa B; p38 MAPK, p38 mitogen-activated protein kinase; RAGE, receptor for AGEs; STAT3, signal transducers and activators of transcription 3; TGF-β, transforming growth factor-β
AGEs and joint health
Osteoarthritis (OA) is one of the most frequent chronic diseases of the elderly. It is characterised by the softening, ulceration and disintegration of articular cartilage, as well as by the formation of outgrowths of bone and cartilage at the joint margins [96]. It is a major source of pain, disability and socioeconomic cost worldwide [140]. The exact pathophysiological mechanisms of OA are not completely clear; they can be complex and multifactorial, including genetic, biological and biomechanical components. However, age has been recognised as one of the main risk factors for the development of OA [141]. In fact, the majority of people older than 60 years have OA in at least one joint [142] and the development of OA is strongly correlated with chronological age [143]. Therefore, age-related changes in articular cartilage are likely to play a role in the aetiology of OA.
One of the major age-related changes in articular cartilage is increased levels of cross-linked AGEs. From age 20 onward, AGEs accumulate in collagen and proteoglycans in articular cartilage. The prevalence of OA increases with age and coincides with the accumulation of AGEs in articular cartilage [144, 145]. Recent studies have found that the increased AGEs levels can negatively modify articular cartilage by increasing its stiffness, elevating chondrocyte-mediated proteoglycan degradation, decreasing proteoglycan synthesis and inducing the degradation of the extracellular matrix (ECM) of cartilage [141, 146, 147]).
The ECM of cartilage is degraded by matrix metalloproteinases (MMPs), which can be induced by cytokines. The accumulation of AGEs in articular cartilage may increase oxidative stress and stimulate the expression of cytokines, which can affect the turnover of the ECM [148]. A study in which mice were fed a high AGEs diet showed the up-regulation of syndecan-4 and MMP-3; this was proposed for the activation of aggrecanase II (a disintegrin and metalloproteinase with thrombospondin motifs 5, ADAMTS-5), which is a critical phenomenon in the development of OA [149, 150]. In human C28/I2 chondrocytes, the blocking of RAGE prevented the increase in ADAMTS-5, syndecan-4, MMP-1 and MMP-3 in vitro after treatment with AGE-modified BSA [150]. As articular cartilage is one of the tissues containing the greatest amounts of AGEs in the body, AGE-related negative effects on the synthesis and degradation of proteoglycan in articular cartilage and ECM turnover are likely to be important contributors to the development of OA [141, 151, 152]. However, the causative link between AGEs and OA, the pathophysiological mechanisms and the pathways by which dietary AGEs affect joint tissue such as cartilage are not yet completely clear.
AGEs and skeletal muscle
Sarcopenia, as loss of muscle mass, strength and endurance, is an important factor causing mobility difficulties such as low strength, decreased lower extremity performance, slow walking speed and physical disability [113, 153]. It is estimated that there could be an average of 5% loss of muscle mass every 10 years after 40 years of age, with a decrease in muscle cross-sectional area, loss of muscle fibre and fibre atrophy, and that this may get worse after age 65 [154, 155]. In fact, sarcopenia occurs to some degree as a consequence of aging in all individuals, but can be accelerated by a variety of factors including inactivity, poor nutrition, increased oxidative stress and chronic disease conditions [155].
Recently, it has been hypothesised that AGEs could play a role in the pathogenesis of sarcopenia through cross-linking tissues in skeletal muscle, AGE–RAGE-mediated inflammation and endothelial dysfunction in the microcirculation of skeletal muscle [156, 157]. Both animal studies and human studies have reported increased levels of AGEs in skeletal muscle with aging [117, 157,158,159]. The accumulated AGEs may contribute to increased stiffness in muscle tissue, similar to that in articular cartilage, and may also reduce the viscoelastic properties of muscle and hence impair muscle function. The AGEs cross-linked with connective tissue surrounding muscle fibres may contribute to the decline in muscle function with aging [157]. Furthermore, when AGEs accumulate in endothelial cells, they up-regulate inflammation through RAGE and contribute to endothelial dysfunction, which leads to loss of muscle mass and strength [156]. In fact, some studies have reported that the elevated serum AGEs levels are associated with sarcopenia-related outcomes, such as poor grip strength, slow walking speed and increased muscle weakness [153, 160].
To summarise, AGEs accumulate in bones, joints and skeletal muscles. They may play important roles in the development of osteoporosis, osteoarthritis and sarcopenia with aging. More prospective studies are needed to determine whether elevated serum AGEs levels predict a decline in mobility measures. In addition, intervention studies are required to investigate the beneficial effects of low endogenous AGEs levels and the effects of the accumulation of AGEs on mobility outcomes.
Current therapeutic and nutritional anti-AGEs approaches
Therapeutic approaches
As AGEs contribute to the development of chronic diseases such as diabetes, cardiovascular disease and renal impairment by their impacts on oxidative stress and inflammation, they are considered to be promising drug targets for a potential therapeutic approach, which aims to prevent diabetic or other pathogenic complications. In fact, it has been reported that such pharmaceutical interventions effectively prevent and treat diabetic and other pathogenic complications [161,162,163]. A large number of compounds have been reported to play a potential role against AGEs [164], e.g. aminoguanidine [163, 165, 166], vitamins (e.g. thiamine, pyridoxamine, citric acid) [167,168,169], anti-inflammatory drugs with anti-glycation properties (e.g. aspirin, tenilsetam) [170, 171], antidiabetic drug (thiazolidinediones) [172], anti-hypertensive drugs with AGEs inhibition activity (e.g. angiotensin converting enzyme inhibitors) [173], HMA-CoA reductase inhibitors (Statins) [174], antioxidant agents (carnosine, flavonoids, curcumin) [175, 176], and chelators with AGE inhibition properties (carnosine, pyridoxamine) [177]. Results obtained from preclinical evaluation studies demonstrated anti-AGEs activity of these compounds but a clear mechanism of action has not been elucidated [79, 164]. The effects of many of these substances require further validation by clinical studies. We will discuss the most relevant compounds in this section.
Both synthetic and natural compounds have been evaluated as inhibitors against the formation of AGEs and their cross-linking to proteins. There are different inhibitory mechanisms, including inhibiting AGEs formation, accelerating the catabolism of existing AGEs or AGEs cross-links and blocking the biological response of AGEs [167]. Aminoguanidine, a synthetic compound, exhibited AGE inhibitory effect in rats [163]. The possible mechanism is that a nucleophile hydrazine group in aminoguanidine binds to carbonyl groups, leading to the decrease of accumulation of AGE-mediated collagen cross-linking [163]. Following a series of preclinical and small clinical studies [161, 178,179,180,181,182,183,184,185,186,187,188], two large clinical trials the ACTION trial and the ACTION-II trial [165, 166] were conducted to target at the anti-AGEs effects of aminoguanidine in patients with diabetes. An expected significant beneficial effect of aminoguanidine in preventing the progression of disease, however, failed to be observed. In ACTION-II study, patients treated with aminoguanidine frequently reported side effects, which included flu-like symptoms, liver abnormalities, gastrointestinal disorders and anemia [166]. Considering the toxic and potential side effects of synthetic molecules, natural products are preferable.
With respect to the in vivo mechanisms of AGEs formation, antioxidants can protect against the glycation of proteins that is caused by the exposure of protein to glucose under oxidative conditions. However, studies confirm the inhibitory function of antioxidants only on the formation of glycoxidation products but not for the glycation of proteins [176]. Conflicting results have been obtained when antioxidants, such as α-tocopherol, retinol and ascorbic acid, have been used for treating diabetic complications such as cataracts [189,190,191]. Only flavonoids have shown promising evidence that demonstrates their inhibitory effect on AGEs formation and the prevention of some complications of diabetes [192,193,194,195].
Some metal chelators may also indirectly inhibit AGEs formation by blocking ROS and free transition metal ions, which have been recognised as key players in advanced glycation [167]. They are applied mainly as drugs that are commonly used for the treatment of diabetic complications. Some natural compounds, e.g. citric acid, can be considered to be an AGE inhibitor because of its non-specific metal-chelating activity. In addition, pyridoxamine (a natural derivative of vitamin B6), as a dietary supplement, proved to be a potent inhibitor of AGEs formation with a stronger effect than aminoguandine [164]. Pyridoxamine can chelate metal ions that catalyse Amadori reactions, reduce the generation of dicarbonyl intermediates, and hence can inhibit the formation of AGEs [196]. It was revealed in preclinical studies as an effective agent in prevention of kidney and cardiovascular disease, reducing cross-linking of collagen, decreasing CML and CEL levels, and regulating oxidative stress [197,198,199]. Clinical studies on pyridoxamine, however, failed to demonstrate the anti-AGEs effects [200, 201].
Another vitamin derivative, bentotiamine, is a fat-soluble derivative of thiamine, found to be able to prevent diabetes-induced CML in rats [164, 202]. The plausible mechanisms appear to be the activation of transketolase, which targets the precursors of AGEs towards the activation of pentose-5-phosphate pathway, blocking other pathways involved in production of AGEs [203, 204]. However, conflicting evidences have been obtained from human studies about anti-AGEs effect of benfotiamine. An RCT study on patients with T2DM and nephropathy treated with benfotiamine found no significant impact in levels of CML, CEL, 5-hydro-5-methylimidazolone and other inflammatory markers [205]. Similar results were also observed in another RCT, in which inflammatory markers and AGEs levels were found no difference between treated patients and controls [206]. In contrast, a study revealed that benfotiamine was able to significantly reduce serum markers of endothelial dysfunction and AGEs levels [207].
Some AGEs inhibitors directly scavenge the reactive carbonyls. These molecules have one or more nucleophilic centres that display the capacity to trap different carbonyls [167]. In addition to pharmaceutical agents such as aminoguaidine, natural compounds such as thiamine (vitamin B1), benfotiamine, pyridoxamine and some natural polyphenols, have demonstrated effective inhibitory effects on the glycation of proteins by scavenging carbonyl species both in vitro and in vivo [203, 208,209,210]. For example, theaflavins from black tea and epicatechins from green tea effectively trapped MG in vitro, and have been suggested as potential AGEs inhibitors for in vivo studies [211].
Besides exogenous AGEs inhibitors, other inhibitors that are involved in the catabolism of AGEs mainly serve as physiological reducing agents, antioxidant enzymes and agents in the detoxification system. As discussed in Section “Endogenous Formation of AGEs”., they work as a defense system, balancing the AGEs in vivo pool.
Furthermore, AGEs breakers, which aim to break the AGEs cross-links, have been proposed. However, although the mechanism is very promising, as the recovery of oxidised proteins would be expected, the real effects of the proposed AGEs breakers are unlikely to be the result of the cleavage or the reversal of existing protein–AGE cross-linking; instead, they will have more direct effects on the formation of AGEs, such as their antioxidant and chelating effects and their reaction mechanism with dicarbonyl intermediates in the Maillard reaction [167].
In addition, there are AGEs inhibitor pharmaceuticals that target the RAGE–AGE axis. They work as either antagonists of RAGE or antagonists of circulating AGEs [212,213,214,215]. Interestingly, some antibacterial proteins, in particular lactoferrin and lysozyme, bind to AGEs with high affinity, before cellular uptake or their cross-linking to proteins [44, 45, 216]. Lysozyme also accelerates the renal clearance of AGEs and suppresses intracellular AGE-mediated signalling [49]. As lactoferrin is a milk protein and lysozyme has already been applied in infant formula, these two proteins may be considered to be functional ingredients that can be added to food as AGEs inhibitors [217, 218]. In fact, a registered patent (US 5891341 A; Li et al., 1999) uses the similar molecular domains of lactoferrin and lysozyme to remove AGEs.
Dietary approaches
Cooking strategies such as brief heating time, low temperatures, high moisture, and/or exposure to an acidic solution are effective in suppressing generation of new AGEs in food [1, 4]. Cooking methods involving high temperature such as frying, grilling, roasting and broiling propagate the production of dietary AGEs compared to low temperature cooking methods such as boiling, steaming, poaching and stewing. For example, AGEs detected in roasted or broiled chicken increase about four times than the same piece meat cooked by poaching or steaming [1, 4]. Preexposure to acidified environment (marinades such as lemon juice and vinegar) can be encouraged to inhibit the new formation of dietary AGEs. For example, unmarinated beef contains more than half the amount of AGEs than marinated beef [1].
The dietary AGEs database indicated that cuisines involving high consumption of fish, legumes, whole grains, low-fat milk products, fruits and vegetables, such as Mediterranean and Asian cuisines, are more favourable in reducing dietary AGEs as compared to diets featured with solid fats, fatty meats, full-fat dairy products and highly processed foods. These recommendations are in good agreement with dietary guidelines set by organizations such as the American Heart Association [219], the American Institute for Cancer Research [220], and the American Diabetes Association [221].
Dietary AGEs interventions can minimise the absorption of AGEs via the gastrointestinal tract and reduce the levels of circulating AGEs. Therefore, long term intervention with a low AGEs diet may also reduce the pool of AGEs in vivo. Table 2 lists human intervention studies in which a low dietary AGEs intake was compared with a high AGEs load. The dietary AGEs levels correlated with serum concentrations of AGEs in both healthy individuals and people with different disease conditions. Reduced serum AGEs levels were associated with improvements in levels of inflammatory markers and mediators, such as vascular cell adhesion molecule 1 (VCAM-1), NF-κB and TNF-α. These low AGEs diets may provide an important adjunct to interventions directly towards the inhibition of endogenous AGEs [40].
Conclusion
Further considerations
As discussed above, intervention studies with low dietary AGEs intakes have been conducted in healthy individuals and patients with different diseases, measuring serum AGEs levels, inflammatory status and other AGEs markers in PBMCs, such as mRNA RAGE, mRNA AGER1, TNF-α and NF-κB. Furthermore, the effects of AGEs inhibitors, as pharmaceutical applications, have also been investigated in human studies that targeted diabetic complications. However, only a limited number of studies have focused on measuring the effects of low AGEs levels or AGEs inhibitors on mobility, although many observational human studies and in vitro studies have reported the correlation of AGEs with and the contribution of AGEs to mobility, particular in diseases such as osteoporosis, cartilage degradation, osteoarthritis and sarcopenia [56, 96, 113, 114, 124, 125, 130, 146, 147, 150, 153, 157, 222].
It should be noted that there is insufficient information from previous animal and human studies for use as a reference to determine the intervention period. Although serum AGEs levels can be easily affected by a lower AGEs diet or AGEs inhibitors, it may take longer to see the changes in certain organs or tissues, as a result of a reduction in AGEs accumulation. For example, the intervention period for the use of anti-AGE drugs in rats varied from 10 to 32 weeks, when AGEs levels in the kidney were measured [107, 223, 224]. Therefore, a long intervention period in which to observe changed AGEs levels in skin, bones, joints and muscles can be expected.
More prospective studies are needed to determine whether changed serum AGEs and/or skin autofluorescence predict different mobility measures. In addition, human intervention studies are required to investigate the beneficial effects of exogenous AGEs inhibitors on mobility outcomes.
Abbreviations
- 3-DG:
-
3-deoxyglucosone
- AcLDL:
-
Acetylated LDL
- AFGP:
-
Alkyl formyl glycosyl pyrrole
- AGER1:
-
Advanced glycation end product receptor-1
- AGER2:
-
Advanced glycation end product receptor-2
- AGER3:
-
Advanced glycation end product receptor-3
- AGEs:
-
Advanced glycation end products
- ALI:
-
Arginine-lysine imidazole
- AP-1:
-
Activator protein 1
- Cdc42-Rac:
-
Cell division control protein 42 homolog-Rac
- CEL:
-
Nɛ-Carboxyethyllysine
- CML:
-
Nɛ-carboxymethyllysine
- CVD:
-
Cardiovascular diseases
- ELISA:
-
Enzyme-linked immunosorbent assays
- ERK 1/2:
-
Extracellular signal-regulated protein kinases 1 and 2
- FEEL-1 and FEEL-2:
-
Link domain-containing scavenger receptor-1 and -2
- FOXO:
-
Forkhead box protein O subclass
- GC:
-
Gas chromatography
- GO:
-
Glyoxal
- GOLD:
-
Glyoxal-lysine-dimer
- HMW:
-
High molecular weight
- HPLC:
-
High-performance liquid chromatography
- IGF-I:
-
Insulin-like growth factor I
- IL-6:
-
Interleukin-6
- IR:
-
Insulin resistance
- ISRE:
-
Interferon-stimulated response elements
- JAK/STAT:
-
Janus kinase/signal transducers and activators of transcription
- JNK:
-
c-Jun N-terminal kinase
- LDL:
-
Low density lipoprotein;LIF: leukaemia inhibitory factor
- LMW:
-
Low molecular weight
- MG:
-
Methylglyoxal
- MMPs:
-
Metalloproteinases
- MOLD:
-
Methylglyoxal-lysine dimer
- MS:
-
Mass spectrometry
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NF-κB:
-
Nuclear factor kappa B
- OA:
-
Osteoarthritis
- OS:
-
Oxidative stress
- OxLDL:
-
Oxidised LDL
- p38 MAPK:
-
p38 mitogen-activated protein kinase
- PBMCs:
-
Peripheral blood mononuclear cells
- PKC:
-
Protein kinase C
- RAGE:
-
Receptor of advanced glycation end products
- ROS:
-
Reactive oxygen species
- SAF:
-
Skin autofluorecence
- SIRT1:
-
Sirtuin-1
- SR-A:
-
Scavenger receptor class A
- SR-B:
-
Scavenger receptor class B
- SR-BI:
-
Scavenger receptor class B Type I
- SR-E:
-
Scavenger receptor class E
- STAT3:
-
Signal transducers and activators of transcription 3
- TGF-β:
-
Transforming growth factor-β
- TIRAP-MyD88:
-
Toll-interleukin 1 receptor domain containing adaptor protein and myeloid differentiation primary response protein 88
- TNF-α:
-
Tumour necrosis factor alpha
- VCAM-1:
-
Vascular cell adhesion molecule 1
References
Uribarri J, Woodruff S, Goodman S, Cai W, Chen X, Pyzik R, Yong A, Striker GE, Vlassara H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J Am Diet Assoc. 2010;110:911–6 e912.
Vistoli G, De Maddis D, Cipak A, Zarkovic N, Carini M, Aldini G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): an overview of their mechanisms of formation. Free Radic Res. 2013;47(Suppl 1):3–27.
Erbersdobler HF, Somoza V. Forty years of furosine - forty years of using Maillard reaction products as indicators of the nutritional quality of foods. Mol Nutr Food Res. 2007;51:423–30.
Goldberg T, Cai W, Peppa M, Dardaine V, Baliga BS, Uribarri J, Vlassara H. Advanced glycoxidation end products in commonly consumed foods. J Am Diet Assoc. 2004;104:1287–91.
Chao PC, Huang CN, Hsu CC, Yin MC, Guo YR. Association of dietary AGEs with circulating AGEs, glycated LDL, IL-1alpha and MCP-1 levels in type 2 diabetic patients. Eur J Nutr. 2010;49:429–34.
Singh VP, Bali A, Singh N, Jaggi AS. Advanced glycation end products and diabetic complications. Korean J Physiol Pharmacol. 2014;18(1):14.
Simm A. Protein glycation during aging and in cardiovascular disease. J Proteome. 2013;92:248–59.
Takeuchi M, Takino J, Yamagishi S. Involvement of the toxic AGEs (TAGE)-RAGE system in the pathogenesis of diabetic vascular complications: a novel therapeutic strategy. Curr Drug Targets. 2010;11:1468–82.
Wang Z, Jiang Y, Liu N, Ren L, Zhu Y, An Y, Chen D. Advanced glycation end-product Nepsilon-carboxymethyl-lysine accelerates progression of atherosclerotic calcification in diabetes. Atherosclerosis. 2012;221:387–96.
Liu SH, Sheu WH, Lee MR, Lee WJ, Yi YC, Yang TJ, Jen JF, Pan HC, Shen CC, Chen WB, et al. Advanced glycation end product Nepsilon-carboxymethyllysine induces endothelial cell injury: the involvement of SHP-1-regulated VEGFR-2 dephosphorylation. J Pathol. 2013;230:215–27.
van Boekel MA. Formation of flavour compounds in the Maillard reaction. Biotechnol Adv. 2006;24:230–3.
Vlassara H, Uribarri J. Advanced glycation end products (AGE) and diabetes: cause, effect, or both? Curr Diabetes Rep. 2014;14:453 013–0453-0451.
Cai W, Gao QD, Zhu L, Peppa M, He C, Vlassara H. Oxidative stress-inducing carbonyl compounds from common foods: novel mediators of cellular dysfunction. Mol Med. 2002;8:337–46.
O'Brien J, Morrissey PA. Nutritional and toxicological aspects of the Maillard browning reaction in foods. Crit Rev Food Sci Nutr. 1989;28:211–48.
Poulsen MW, Hedegaard RV, Andersen JM, de Courten B, Bugel S, Nielsen J, Skibsted LH, Dragsted LO. Advanced glycation endproducts in food and their effects on health. Food Chem Toxicol. 2013;60:10–37.
Faist V, Erbersdobler HF. Metabolic transit and in vivo effects of melanoidins and precursor compounds deriving from the Maillard reaction. Ann Nutr Metab. 2001;45:1–12.
Koschinsky T, He CJ, Mitsuhashi T, Bucala R, Liu C, Buenting C, Heitmann K, Vlassara H. Orally absorbed reactive glycation products (glycotoxins): an environmental risk factor in diabetic nephropathy. Proc Natl Acad Sci U S A. 1997;94:6474–9.
Forster A, Kuhne Y, Henle T. Studies on absorption and elimination of dietary maillard reaction products. Ann N Y Acad Sci. 2005;1043:474–81.
Foerster A, Henle T. Glycation in food and metabolic transit of dietary AGEs (advanced glycation end-products): studies on the urinary excretion of pyrraline. Biochem Soc Trans. 2003;31:1383–5.
He C, Sabol J, Mitsuhashi T, Vlassara H. Dietary glycotoxins: inhibition of reactive products by aminoguanidine facilitates renal clearance and reduces tissue sequestration. Diabetes. 1999;48:1308–15.
Cai W, He JC, Zhu L, Chen X, Zheng F, Striker GE, Vlassara H. Oral glycotoxins determine the effects of calorie restriction on oxidant stress, age-related diseases, and lifespan. Am J Pathol. 2008;173:327–36.
Vlassara H, Cai W, Crandall J, Goldberg T, Oberstein R, Dardaine V, Peppa M, Rayfield EJ. Inflammatory mediators are induced by dietary glycotoxins, a major risk factor for diabetic angiopathy. Proc Natl Acad Sci U S A. 2002;99:15596–601.
Uribarri J, Peppa M, Cai W, Goldberg T, Lu M, He C, Vlassara H. Restriction of dietary glycotoxins reduces excessive advanced glycation end products in renal failure patients. J Am Soc Nephrol. 2003;14:728–31.
Uribarri J, Peppa M, Cai W, Goldberg T, Lu M, Baliga S, Vassalotti JA, Vlassara H. Dietary glycotoxins correlate with circulating advanced glycation end product levels in renal failure patients. Am J Kidney Dis. 2003;42:532–8.
Uribarri J, Cai W, Peppa M, Goodman S, Ferrucci L, Striker G, Vlassara H. Circulating glycotoxins and dietary advanced glycation endproducts: two links to inflammatory response, oxidative stress, and aging. J Gerontol A Biol Sci Med Sci. 2007;62:427–33.
Prasad K, Dhar I, Caspar-Bell G. Role of advanced glycation end products and its receptors in the pathogenesis of cigarette smoke-induced cardiovascular disease. Int J Angiol. 2015;24:75–80.
Cerami C, Founds H, Nicholl I, Mitsuhashi T, Giordano D, Vanpatten S, Lee A, Al-Abed Y, Vlassara H, Bucala R, et al. Tobacco smoke is a source of toxic reactive glycation products. Proc Natl Acad Sci U S A. 1997;94:13915–20.
Nicholl ID, Bucala R. Advanced glycation endproducts and cigarette smoking. Cell Mol Biol. 1998;44:1025–33.
Wautier MP, Tessier FJ, Wautier JL. Advanced glycation end products: a risk factor for human health. Ann Pharm Fr. 2014;72:400–8.
Arsov S, Graaff R, van Oeveren W, Stegmayr B, Sikole A, Rakhorst G, Smit AJ. Advanced glycation end-products and skin autofluorescence in end-stage renal disease: a review. Clin Chem Lab Med. 2014;52:11–20.
Bakker SF, Tushuizen ME, Gozutok E, Ciftci A, Gelderman KA, Mulder CJ, Simsek S. Advanced glycation end products (AGEs) and the soluble receptor for AGE (sRAGE) in patients with type 1 diabetes and coeliac disease. Nutr Metab Cardiovasc Dis. 2015;25:230–5.
Del Turco S, Basta G. An update on advanced glycation endproducts and atherosclerosis. Biofactors. 2012;38:266–74.
Kellow NJ, Savige GS. Dietary advanced glycation end-product restriction for the attenuation of insulin resistance, oxidative stress and endothelial dysfunction: a systematic review. Eur J Clin Nutr. 2013;67:239–48.
Nagai R, Shirakawa J, Ohno R, Moroishi N, Nagai M. Inhibition of AGEs formation by natural products. Amino Acids. 2014;46:261–6.
Rahbar S, Blumenfeld O, Ranney HM. Studies of an unusual hemoglobin in patients with diabetes mellitus. Biochem Biophys Res Commun. 1969;36:838–43.
Ahmed N, Argirov OK, Minhas HS, Cordeiro CA, Thornalley PJ. Assay of advanced glycation endproducts (AGEs): surveying AGEs by chromatographic assay with derivatization by 6-aminoquinolyl-N-hydroxysuccinimidyl-carbamate and application to Nepsilon-carboxymethyl-lysine- and Nepsilon-(1-carboxyethyl)lysine-modified albumin. Biochem J. 2002;364:1–14.
Ward MS, Fortheringham AK, Cooper ME, Forbes JM. Targeting advanced glycation endproducts and mitochondrial dysfunction in cardiovascular disease. Curr Opin Pharmacol. 2013;13:654–61.
Thornalley PJ, Langborg A, Minhas HS. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J. 1999;344:109–16.
Fu MX, Requena JR, Jenkins AJ, Lyons TJ, Baynes JW, Thorpe SR. The advanced glycation end product, Nepsilon-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J Biol Chem. 1996;271:9982–6.
Forbes JM, Soldatos G, Thomas MC. Below the radar: advanced glycation end products that detour “around the side”: is HbA(1c) not an accurate enough predictor of long term progression and glycaemic control in diabetes? Clin Biochem Rev. 2005;26:123–34.
Bunn HF. Non-enzymatic glycosylation of protein: a form of molecular aging. Schweiz Med Wochenschr. 1981;111:1503–7.
Nowotny K, Jung T, Höhn A, Weber D, Grune T. Advanced Glycation End Products and Oxidative Stress in Type 2 Diabetes Mellitus. Biomolecules. 2015;5(1):194–222.
Rabbani N, Thornalley PJ. Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids. 2012;42:1133–42.
Liu H, Zheng F, Cao Q, Ren B, Zhu L, Striker G, Vlassara H. Amelioration of oxidant stress by the defensin lysozyme. Am J Physiol Endocrinol Metab. 2006;290:E824–32.
Vlassara H, Uribarri J, Cai W, Striker G. Advanced glycation end product homeostasis: exogenous oxidants and innate defenses. Ann N Y Acad Sci. 2008;1126:46–52.
Uribarri J, Cai W, Ramdas M, Goodman S, Pyzik R, Chen X, Zhu L, Striker GE, Vlassara H. Restriction of advanced glycation end products improves insulin resistance in human type 2 diabetes: potential role of AGER1 and SIRT1. Diabetes Care. 2011;34:1610–6.
Stirban A, Gawlowski T, Roden M. Vascular effects of advanced glycation endproducts: clinical effects and molecular mechanisms. Mol Metab. 2013;3:94–108.
Bohlender JM, Franke S, Stein G, Wolf G. Advanced glycation end products and the kidney. Am J Physiol Renal Physiol. 2005;289:F645–59.
Zheng F, Cai W, Mitsuhashi T, Vlassara H. Lysozyme enhances renal excretion of advanced glycation endproducts in vivo and suppresses adverse age-mediated cellular effects in vitro: a potential AGE sequestration therapy for diabetic nephropathy? Mol Med. 2001;7:737–47.
Tanaka S, Avigad G, Eikenberry EF, Brodsky B. Isolation and partial characterization of collagen chains dimerized by sugar-derived cross-links. J Biol Chem. 1988;263:17650–7.
Charonis AS, Reger LA, Dege JE, Kouzi-Koliakos K, Furcht LT, Wohlhueter RM, Tsilibary EC. Laminin alterations after in vitro nonenzymatic glycosylation. Diabetes. 1990;39:807–14.
Avery NC, Bailey AJ. The effects of the Maillard reaction on the physical properties and cell interactions of collagen. Pathologie-biologie. 2006;54:387–95.
Wang X, Shen X, Li X, Agrawal CM. Age-related changes in the collagen network and toughness of bone. Bone. 2002;31:1–7.
Saito M, Fujii K, Mori Y, Marumo K. Role of collagen enzymatic and glycation induced cross-links as a determinant of bone quality in spontaneously diabetic WBN/Kob rats. Osteoporos Int. 2006;17:1514–23.
Alikhani M, Alikhani Z, Boyd C, MacLellan CM, Raptis M, Liu R, Pischon N, Trackman PC, Gerstenfeld L, Graves DT. Advanced glycation end products stimulate osteoblast apoptosis via the MAP kinase and cytosolic apoptotic pathways. Bone. 2007;40:345–53.
Sanguineti R, Storace D, Monacelli F, Federici A, Odetti P. Pentosidine effects on human osteoblasts in vitro. Ann N Y Acad Sci. 2008;1126:166–72.
Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014;2:411–29.
Ramasamy R, Yan SF, Schmidt AM. Advanced glycation endproducts: from precursors to RAGE: round and round we go. Amino Acids. 2012;42:1151–61.
Barbezier N, Tessier FJ, Chango A. Receptor of advanced glycation endproducts RAGE/AGER: an integrative view for clinical applications. Ann Biol Clin (Paris). 2014;72:669–80.
Grimm S, Ott C, Horlacher M, Weber D, Hohn A, Grune T. Advanced-glycation-end-product-induced formation of immunoproteasomes: involvement of RAGE and Jak2/STAT1. Biochem J. 2012;448:127–39.
Tamura Y, Adachi H, Osuga J, Ohashi K, Yahagi N, Sekiya M, Okazaki H, Tomita S, Iizuka Y, Shimano H, et al. FEEL-1 and FEEL-2 are endocytic receptors for advanced glycation end products. J Biol Chem. 2003;278:12613–7.
Ohgami N, Nagai R, Ikemoto M, Arai H, Kuniyasu A, Horiuchi S, Nakayama H. CD36, a member of class B scavenger receptor family, is a receptor for advanced glycation end products. Ann N Y Acad Sci. 2001;947:350–5.
Ohgami N, Nagai R, Miyazaki A, Ikemoto M, Arai H, Horiuchi S, Nakayama H. Scavenger receptor class B type I-mediated reverse cholesterol transport is inhibited by advanced glycation end products. J Biol Chem. 2001;276:13348–55.
Jono T, Miyazaki A, Nagai R, Sawamura T, Kitamura T, Horiuchi S. Lectin-like oxidized low density lipoprotein receptor-1 (LOX-1) serves as an endothelial receptor for advanced glycation end products (AGE). FEBS Lett. 2002;511:170–4.
Lu C, He JC, Cai W, Liu H, Zhu L, Vlassara H. Advanced glycation endproduct (AGE) receptor 1 is a negative regulator of the inflammatory response to AGE in mesangial cells. Proc Natl Acad Sci U S A. 2004;101:11767–72.
Vlassara H. The AGE-receptor in the pathogenesis of diabetic complications. Diabetes Metab Res Rev. 2001;17:436–43.
Schmidt AM, Yan SD, Brett J, Mora R, Nowygrod R, Stern D. Regulation of human mononuclear phagocyte migration by cell surface-binding proteins for advanced glycation end products. J Clin Invest. 1993;91:2155–68.
Tanaka N, Yonekura H, Yamagishi S, Fujimori H, Yamamoto Y, Yamamoto H. The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-alpha through nuclear factor-kappa B, and by 17beta-estradiol through Sp-1 in human vascular endothelial cells. J Biol Chem. 2000;275:25781–90.
Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, Nagashima M, Morser J, Arnold B, Preissner KT, et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med. 2003;198:1507–15.
Ottum MS, Mistry AM. Advanced glycation end-products: modifiable environmental factors profoundly mediate insulin resistance. J Clin Biochem Nutr. 2015;57:1–12.
Caron AZ, He X, Mottawea W, Seifert EL, Jardine K, Dewar-Darch D, Cron GO, Harper ME, Stintzi A, McBurney MW. The SIRT1 deacetylase protects mice against the symptoms of metabolic syndrome. FASEB J. 2014;28:1306–16.
Giorgi C, Agnoletto C, Baldini C, Bononi A, Bonora M, Marchi S, Missiroli S, Patergnani S, Poletti F, Rimessi A, et al. Redox control of protein kinase C: cell- and disease-specific aspects. Antioxid Redox Signal. 2010;13:1051–85.
Gopalakrishna R, Jaken S. Protein kinase C signaling and oxidative stress. Free Radic Biol Med. 2000;28:1349–61.
Ou X, Lee MR, Huang X, Messina-Graham S, Broxmeyer HE. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells. 2014;32:1183–94.
Ragheb R, Shanab GM, Medhat AM, Seoudi DM, Adeli K, Fantus IG. Free fatty acid-induced muscle insulin resistance and glucose uptake dysfunction: evidence for PKC activation and oxidative stress-activated signaling pathways. Biochem Biophys Res Commun. 2009;389:211–6.
Rimessi A, Rizzuto R, Pinton P. Differential recruitment of PKC isoforms in HeLa cells during redox stress. Cell Stress Chaperones. 2007;12:291–8.
Salminen A, Kaarniranta K, Kauppinen A. Crosstalk between oxidative stress and SIRT1: impact on the aging process. Int J Mol Sci. 2013;14:3834–59.
Meerwaldt R, Graaff R, Oomen PHN, Links TP, Jager JJ, Alderson NL, Thorpe SR, Baynes JW, Gans ROB, Smit AJ. Simple non-invasive assessment of advanced glycation endproduct accumulation. Diabetologia. 2004;47:1324–30.
Stirban A, Gawlowski T, Roden M. Vascular effects of advanced glycation endproducts: clinical effects and molecular mechanisms. Mol Metab. 2014;3:94–108.
Thornalley PJ. Measurement of protein glycation, glycated peptides, and glycation free adducts. Perit Dial Int. 2005;25:522–33.
Genuth S, Sun W, Cleary P, Sell DR, Dahms W, Malone J, Sivitz W, Monnier VM, Group DSCAS. Glycation and carboxymethyllysine levels in skin collagen predict the risk of future 10-year progression of diabetic retinopathy and nephropathy in the diabetes control and complications trial and epidemiology of diabetes interventions and complications participants with type 1 diabetes. Diabetes. 2005;54:3103–11.
Da Moura Semedo C, Webb M, Waller H, Khunti K, Davies M. Skin autofluorescence, a non-invasive marker of advanced glycation end products: clinical relevance and limitations. Postgrad Med J. 2017;93:289–94.
den Hollander NC, Mulder DJ, Graaff R, Thorpe SR, Baynes JW, Smit GP, Smit AJ. Advanced glycation end products and the absence of premature atherosclerosis in glycogen storage disease Ia. J Inherit Metab Dis. 2007;30:916–23.
Meerwaldt R, Hartog JW, Graaff R, Huisman RJ, Links TP, den Hollander NC, Thorpe SR, Baynes JW, Navis G, Gans RO, et al. Skin autofluorescence, a measure of cumulative metabolic stress and advanced glycation end products, predicts mortality in hemodialysis patients. J Am Soc Nephrol. 2005;16:3687–93.
Koetsier M, Lutgers HL, de Jonge C, Links TP, Smit AJ, Graaff R. Reference values of skin autofluorescence. Diabetes Technol Ther. 2010;12:399–403.
Gerrits EG, Lutgers HL, Kleefstra N, Graaff R, Groenier KH, Smit AJ, Gans RO, Bilo HJ. Skin autofluorescence: a tool to identify type 2 diabetic patients at risk for developing microvascular complications. Diabetes Care. 2008;31:517–21.
Meerwaldt R, Lutgers HL, Links TP, Graaff R, Baynes JW, Gans RO, Smit AJ. Skin autofluorescence is a strong predictor of cardiac mortality in diabetes. Diabetes Care. 2007;30:107–12.
Smit AJ, Smit JM, Botterblom GJ, Mulder DJ. Skin autofluorescence based decision tree in detection of impaired glucose tolerance and diabetes. PLoS One. 2013;8:e65592.
Fokkens BT, Smit AJ. Skin fluorescence as a clinical tool for non-invasive assessment of advanced glycation and long-term complications of diabetes. Glycoconj J. 2016;33:527–35.
Lutgers HL, Graaff R, Links TP, Ubink-Veltmaat LJ, Bilo HJ, Gans RO, Smit AJ. Skin autofluorescence as a noninvasive marker of vascular damage in patients with type 2 diabetes. Diabetes Care. 2006;29:2654–9.
Vlassara H, Cai W, Goodman S, Pyzik R, Yong A, Chen X, Zhu L, Neade T, Beeri M, Silverman JM, et al. Protection against loss of innate defenses in adulthood by low advanced glycation end products (AGE) intake: role of the antiinflammatory AGE receptor-1. J Clin Endocrinol Metab. 2009;94:4483–91.
Yaffe K, Lindquist K, Schwartz AV, Vitartas C, Vittinghoff E, Satterfield S, Simonsick EM, Launer L, Rosano C, Cauley JA, et al. Advanced glycation end product level, diabetes, and accelerated cognitive aging. Neurology. 2011;77:1351–6.
Heier M, Margeirsdottir HD, Torjesen PA, Seljeflot I, Stensaeth KH, Gaarder M, Brunborg C, Hanssen KF, Dahl-Jorgensen K. The advanced glycation end product methylglyoxal-derived hydroimidazolone-1 and early signs of atherosclerosis in childhood diabetes. Diab Vasc Dis Res. 2015;12:139–45.
Kuhla A, Ludwig SC, Kuhla B, Munch G, Vollmar B. Advanced glycation end products are mitogenic signals and trigger cell cycle reentry of neurons in Alzheimer's disease brain. Neurobiol Aging. 2015;36:753–61.
Gaens KH, Stehouwer CD, Schalkwijk CG. Advanced glycation endproducts and its receptor for advanced glycation endproducts in obesity. Curr Opin Lipidol. 2013;24:4–11.
Saudek DM, Kay J. Advanced glycation endproducts and osteoarthritis. Curr Rheumatol Rep. 2003;5:33–40.
de Groot L, Hinkema H, Westra J, Smit AJ, Kallenberg CG, Bijl M, Posthumus MD. Advanced glycation endproducts are increased in rheumatoid arthritis patients with controlled disease. Arthritis Res Ther. 2011;13:R205.
Rajaobelina K, Cougnard-Gregoire A, Delcourt C, Gin H, Barberger-Gateau P, Rigalleau V. Autofluorescence of skin advanced glycation end products: marker of metabolic memory in elderly population. J Gerontol A Biol Sci Med Sci. 2015;70:841–6.
Birlouez-Aragon I, Saavedra G, Tessier FJ, Galinier A, Ait-Ameur L, Lacoste F, Niamba CN, Alt N, Somoza V, Lecerf JM. A diet based on high-heat-treated foods promotes risk factors for diabetes mellitus and cardiovascular diseases. Am J Clin Nutr. 2010;91:1220–6.
Jerums G, Panagiotopoulos S, Forbes J, Osicka T, Cooper M. Evolving concepts in advanced glycation, diabetic nephropathy, and diabetic vascular disease. Arch Biochem Biophys. 2003;419:55–62.
Peppa M, Uribarri J, Cai W, Lu M, Vlassara H. Glycoxidation and inflammation in renal failure patients. Am J Kidney Dis. 2004;43:690–5.
Baumann M. Role of advanced glycation end products in hypertension and cardiovascular risk: human studies. J Am Soc Hypertens. 2012;6:427–35.
Yamagishi S, Matsui T, Fukami K. Role of receptor for advanced glycation end products (RAGE) and its ligands in Cancer risk. Rejuvenation Res. 2015;18:48–56.
Gugliucci A, Menini T. The axis AGE-RAGE-soluble RAGE and oxidative stress in chronic kidney disease. Adv Exp Med Biol. 2014;824:191–208.
Henle T, Miyata T. Advanced glycation end products in uremia. Adv Ren Replace Ther. 2003;10:321–31.
Cai W, He JC, Zhu L, Chen X, Wallenstein S, Striker GE, Vlassara H. Reduced oxidant stress and extended lifespan in mice exposed to a low glycotoxin diet: association with increased AGER1 expression. Am J Pathol. 2007;170:1893–902.
Forbes JM, Cooper ME, Thallas V, Burns WC, Thomas MC, Brammar GC, Lee F, Grant SL, Burrell LM, Jerums G, et al. Reduction of the accumulation of advanced glycation end products by ACE inhibition in experimental diabetic nephropathy. Diabetes. 2002;51:3274–82.
Schiekofer S, Franke S, Andrassy M, Chen J, Rudofsky G, Schneider JG, von Eynatten M, Wendt T, Morcos M, Kientsch-Engel R, et al. Postprandial mononuclear NF-kappaB activation is independent of the AGE-content of a single meal. Exp Clin Endocrinol Diabetes. 2006;114:160–7.
Peppa M, He C, Hattori M, McEvoy R, Zheng F, Vlassara H. Fetal or neonatal low-glycotoxin environment prevents autoimmune diabetes in NOD mice. Diabetes. 2003;52:1441–8.
Sandu O, Song K, Cai W, Zheng F, Uribarri J, Vlassara H. Insulin resistance and type 2 diabetes in high-fat-fed mice are linked to high glycotoxin intake. Diabetes. 2005;54:2314–9.
Vlassara H, Uribarri J, Ferrucci L, Cai W, Torreggiani M, Post JB, Zheng F, Striker GE. Identifying advanced glycation end products as a major source of oxidants in aging: implications for the management and/or prevention of reduced renal function in elderly persons. Semin Nephrol. 2009;29:594–603.
Milaneschi Y, Tanaka T, Ferrucci L. Nutritional determinants of mobility. Curr Opin Clin Nutr Metab Care. 2010;13:625–9.
Lauretani F, Russo CR, Bandinelli S, Bartali B, Cavazzini C, Di Iorio A, Corsi AM, Rantanen T, Guralnik JM, Ferrucci L. Age-associated changes in skeletal muscles and their effect on mobility: an operational diagnosis of sarcopenia. J Appl Physiol (1985). 2003;95:1851–60.
Hein G, Wiegand R, Lehmann G, Stein G, Franke S. Advanced glycation end-products pentosidine and N epsilon-carboxymethyllysine are elevated in serum of patients with osteoporosis. Rheumatology. 2003;42:1242–6.
Hein GE. Glycation endproducts in osteoporosis--is there a pathophysiologic importance? Clin Chim Acta. 2006;371:32–6.
Ginaldi L, Di Benedetto MC, De Martinis M. Osteoporosis, inflammation and ageing. Immun Ageing. 2005;2:14.
Snow LM, Fugere NA, Thompson LV. Advanced glycation end-product accumulation and associated protein modification in type II skeletal muscle with aging. J Gerontol A Biol Sci Med Sci. 2007;62:1204–10.
PVaT N. Osteoporosis. Neuroradiology. 2011;53:S185–9.
Sanguineti R, Puddu A, Mach F, Montecucco F, Viviani GL. Advanced glycation end products play adverse proinflammatory activities in osteoporosis. Mediat Inflamm. 2014;2014:975872.
Yang X, Mostafa AJ, Appleford M, Sun LW, Wang X. Bone formation is affected by matrix advanced glycation end products (AGEs) in vivo. Calcif Tissue Int. 2016;99:373–83.
Odetti P, Rossi S, Monacelli F, Poggi A, Cirnigliaro M, Federici M, Federici A. Advanced glycation end products and bone loss during aging. Ann N Y Acad Sci. 2005;1043:710–7.
Katayama Y, Akatsu T, Yamamoto M, Kugai N, Nagata N. Role of nonenzymatic glycosylation of type I collagen in diabetic osteopenia. J Bone Miner Res. 1996;11:931–7.
Katayama Y, Celic S, Nagata N, Martin TJ, Findlay DM. Nonenzymatic glycation of type I collagen modifies interaction with UMR 201-10B preosteoblastic cells. Bone. 1997;21:237–42.
McCarthy AD, Etcheverry SB, Bruzzone L, Lettieri G, Barrio DA, Cortizo AM. Non-enzymatic glycosylation of a type I collagen matrix: effects on osteoblastic development and oxidative stress. BMC Cell Biol. 2001;2:16.
McCarthy AD, Uemura T, Etcheverry SB, Cortizo AM. Advanced glycation endproducts interefere with integrin-mediated osteoblastic attachment to a type-I collagen matrix. Int J Biochem Cell Biol. 2004;36:840–8.
Hein G, Weiss C, Lehmann G, Niwa T, Stein G, Franke S. Advanced glycation end product modification of bone proteins and bone remodelling: hypothesis and preliminary immunohistochemical findings. Ann Rheum Dis. 2006;65:101–4.
Yamamoto T, Ozono K, Miyauchi A, Kasayama S, Kojima Y, Shima M, Okada S. Role of advanced glycation end products in adynamic bone disease in patients with diabetic nephropathy. Am J Kidney Dis. 2001;38:S161–4.
Cortizo AM, Lettieri MG, Barrio DA, Mercer N, Etcheverry SB, McCarthy AD. Advanced glycation end-products (AGEs) induce concerted changes in the osteoblastic expression of their receptor RAGE and in the activation of extracellular signal-regulated kinases (ERK). Mol Cell Biochem. 2003;250:1–10.
McCarthy AD, Etcheverry SB, Cortizo AM. Advanced glycation endproduct-specific receptors in rat and mouse osteoblast-like cells: regulation with stages of differentiation. Acta Diabetol. 1999;36:45–52.
McCarthy AD, Etcheverry SB, Cortizo AM. Effect of advanced glycation endproducts on the secretion of insulin-like growth factor-I and its binding proteins: role in osteoblast development. Acta Diabetol. 2001;38:113–22.
Valcourt U, Merle B, Gineyts E, Viguet-Carrin S, Delmas PD, Garnero P. Non-enzymatic glycation of bone collagen modifies osteoclastic activity and differentiation. J Biol Chem. 2007;282:5691–703.
De Martinis M, Di Benedetto MC, Mengoli LP, Ginaldi L. Senile osteoporosis: is it an immune-mediated disease? Inflamm Res. 2006;55:399–404.
Lencel P, Magne D. Inflammaging: the driving force in osteoporosis? Med Hypotheses. 2011;76:317–21.
Mellis DJ, Itzstein C, Helfrich MH, Crockett JC. The skeleton: a multi-functional complex organ: the role of key signalling pathways in osteoclast differentiation and in bone resorption. J Endocrinol. 2011;211:131–43.
Sims NA, Johnson RW. Leukemia inhibitory factor: a paracrine mediator of bone metabolism. Growth factors (Chur, Switzerland). 2012;30:76–87.
Kozawa O, Otsuka T, Uematsu T. Leukemia inhibitory factor enhances bFGF-induced IL-6 synthesis in osteoblasts: involvement of JAK2/STAT3. Cell Signal. 2002;14:311–5.
Adams GR. Invited review: autocrine/paracrine IGF-I and skeletal muscle adaptation. J Appl Physiol. 2002;93:1159–67.
Grivennikov S, Karin M. Autocrine IL-6 signaling: a key event in tumorigenesis? Cancer Cell. 2008;13:7–9.
Hock JM, Krishnan V, Onyia JE, Bidwell JP, Milas J, Stanislaus D. Osteoblast apoptosis and bone turnover. J Bone Miner Res. 2001;16:975–84.
Glyn-Jones S, Palmer AJ, Agricola R, Price AJ, Vincent TL, Weinans H, Carr AJ. Osteoarthritis. Lancet. 2015;386(9991):376–87.
DeGroot J, Verzijl N, Jacobs KM, Budde M, Bank RA, Bijlsma JW, TeKoppele JM, Lafeber FP. Accumulation of advanced glycation endproducts reduces chondrocyte-mediated extracellular matrix turnover in human articular cartilage. Osteoarthr Cartil. 2001;9:720–6.
Felson DT, Zhang Y. An update on the epidemiology of knee and hip osteoarthritis with a view to prevention. Arthritis Rheum. 1998;41:1343–55.
Felson DT, Zhang Y, Hannan MT, Naimark A, Weissman BN, Aliabadi P, Levy D. The incidence and natural history of knee osteoarthritis in the elderly. The Framingham osteoarthritis study. Arthritis Rheum. 1995;38:1500–5.
Pokharna HK, Pottenger LA. Nonenzymatic glycation of cartilage proteoglycans: an in vivo and in vitro study. Glycoconj J. 1997;14:917–23.
Verzijl N, DeGroot J, Oldehinkel E, Bank RA, Thorpe SR, Baynes JW, Bayliss MT, Bijlsma JW, Lafeber FP, Tekoppele JM. Age-related accumulation of Maillard reaction products in human articular cartilage collagen. Biochem J. 2000;350(Pt 2):381–7.
Steenvoorden MM, Huizinga TW, Verzijl N, Bank RA, Ronday HK, Luning HA, Lafeber FP, Toes RE, DeGroot J. Activation of receptor for advanced glycation end products in osteoarthritis leads to increased stimulation of chondrocytes and synoviocytes. Arthritis Rheum. 2006;54:253–63.
DeGroot J, Verzijl N, Bank RA, Lafeber FP, Bijlsma JW, TeKoppele JM. Age-related decrease in proteoglycan synthesis of human articular chondrocytes: the role of nonenzymatic glycation. Arthritis Rheum. 1999;42:1003–9.
Xie J, Mendez JD, Mendez-Valenzuela V, Aguilar-Hernandez MM. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal. 2013;25:2185–97.
Echtermeyer F, Bertrand J, Dreier R, Meinecke I, Neugebauer K, Fuerst M, Lee YJ, Song YW, Herzog C, Theilmeier G, et al. Syndecan-4 regulates ADAMTS-5 activation and cartilage breakdown in osteoarthritis. Nat Med. 2009;15:1072–6.
Ruike TL, D J; Grosjean, F; Esposito, V; Majeska, R J; Iatridis, J C; Vlassara, H; Striker, G E; Sun, H B: Accumulation of Advanced Glycation Endproducts Induces Cartilage Degradation via Activation of Syndecan-4-ADAMTS-5 Pathway. In: ORS 2012 Annual Meeting. 2012. Orthopeadic Research Society 2012 Annual Meeting. Poster No. 0701.
Verzijl N, DeGroot J, Thorpe SR, Banks RA, Shaw JN, Lyons TJ, Bijlsma JW, Lafeber FP, Baynes JW, TeKoppele JM. Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem. 2000;275:39027–31.
Sell DR, Monnier VM. Structure elucidation of a senescence cross-link from human extracellular matrix. Implication of pentoses in the aging process. J Biol Chem. 1989;264:21597–602.
Dalal M, Ferrucci L, Sun K, Beck J, Fried LP, Semba RD. Elevated serum advanced glycation end products and poor grip strength in older community-dwelling women. J Gerontol A Biol Sci Med Sci. 2009;64:132–7.
Tzankoff SP, Norris AH. Effect of muscle mass decrease on age-related BMR changes. J Appl Physiol Respir Environ Exerc Physiol. 1977;43:1001–6.
Greenlund LJ, Nair KS. Sarcopenia--consequences, mechanisms, and potential therapies. Mech Ageing Dev. 2003;124:287–99.
Payne GW. Effect of inflammation on the aging microcirculation: impact on skeletal muscle blood flow control. Microcirculation. 2006;13:343–52.
Haus JM, Carrithers JA, Trappe SW, Trappe TA. Collagen, cross-linking, and advanced glycation end products in aging human skeletal muscle. J Appl Physiol. 2007;103:2068–76.
Reddy GK. Cross-linking in collagen by nonenzymatic glycation increases the matrix stiffness in rabbit achilles tendon. Exp Diabesity Res. 2004;5:143–53.
de la Maza MP, Uribarri J, Olivares D, Hirsch S, Leiva L, Barrera G, Bunout D. Weight increase is associated with skeletal muscle immunostaining for advanced glycation end products, receptor for advanced glycation end products, and oxidation injury. Rejuvenation Res. 2008;11:1041–8.
Semba RD, Bandinelli S, Sun K, Guralnik JM, Ferrucci L. Relationship of an advanced glycation end product, plasma carboxymethyl-lysine, with slow walking speed in older adults: the InCHIANTI study. Eur J Appl Physiol. 2010;108:191–5.
Brownlee M, Vlassara H, Kooney A, Ulrich P, Cerami A. Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science. 1986;232:1629–32.
Vasan S, Zhang X, Zhang X, Kapurniotu A, Bernhagen J, Teichberg S, Basgen J, Wagle D, Shih D, Terlecky I, et al. An agent cleaving glucose-derived protein crosslinks in vitro and in vivo. Nature. 1996;382:275–8.
Li YM, Steffes M, Donnelly T, Liu C, Fuh H, Basgen J, Bucala R, Vlassara H. Prevention of cardiovascular and renal pathology of aging by the advanced glycation inhibitor aminoguanidine. Proc Natl Acad Sci U S A. 1996;93:3902–7.
Nenna A, Spadaccio C, Lusini M, Ulianich L, Chello M, Nappi F. Basic and clinical research against advanced glycation end products (AGEs): new compounds to tackle cardiovascular disease and diabetic complications. Recent Adv Cardiovasc Drug Discov. 2015;10:10–33.
Bolton WK, Cattran DC, Williams ME, Adler SG, Appel GB, Cartwright K, Foiles PG, Freedman BI, Raskin P, Ratner RE, et al. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am J Nephrol. 2004;24:32–40.
Freedman BI, Wuerth JP, Cartwright K, Bain RP, Dippe S, Hershon K, Mooradian AD, Spinowitz BS. Design and baseline characteristics for the aminoguanidine clinical trial in overt type 2 diabetic nephropathy (ACTION II). Control Clin Trials. 1999;20:493–510.
Aldini G, Vistoli G, Stefek M, Chondrogianni N, Grune T, Sereikaite J, Sadowska-Bartosz I, Bartosz G. Molecular strategies to prevent, inhibit, and degrade advanced glycoxidation and advanced lipoxidation end products. Free Radic Res. 2013;47:93–137.
Booth AA, Khalifah RG, Hudson BG. Thiamine pyrophosphate and pyridoxamine inhibit the formation of antigenic advanced glycation end-products: comparison with aminoguanidine. Biochem Biophys Res Commun. 1996;220:113–9.
Thornalley PJ. The potential role of thiamine (vitamin B1) in diabetic complications. Curr Diabetes Rev. 2005;1:287–98.
Contreras I, Reiser KM, Martinez N, Giansante E, Lopez T, Suarez N, Postalian S, Molina M, Gonzalez F, Sanchez MR, et al. Effects of aspirin or basic amino acids on collagen cross-links and complications in NIDDM. Diabetes Care. 1997;20:832–5.
Munch G, Taneli Y, Schraven E, Schindler U, Schinzel R, Palm D, Riederer P. The cognition-enhancing drug tenilsetam is an inhibitor of protein crosslinking by advanced glycosylation. J Neural Transm Park Dis Dement Sect. 1994;8:193–208.
Tan KCB, Chow WS, Tso AWK, Xu A, Tse HF, Hoo RLC, Betteridge DJ, Lam KSL. Thiazolidinedione increases serum soluble receptor for advanced glycation end-products in type 2 diabetes. Diabetologia. 2007;50:1819–25.
Sebekova K, Gazdikova K, Syrova D, Blazicek P, Schinzel R, Heidland A, Spustova V, Dzurik R. Effects of ramipril in nondiabetic nephropathy: improved parameters of oxidatives stress and potential modulation of advanced glycation end products. J Hum Hypertens. 2003;17:265–70.
Tam HL, Shiu SW, Wong Y, Chow WS, Betteridge DJ, Tan KC. Effects of atorvastatin on serum soluble receptors for advanced glycation end-products in type 2 diabetes. Atherosclerosis. 2010;209:173–7.
Lin J, Tang Y, Kang Q, Chen A. Curcumin eliminates the inhibitory effect of advanced glycation end-products (AGEs) on gene expression of AGE receptor-1 in hepatic stellate cells in vitro. Lab Investig. 2012;92:827–41.
Augustyniak A, Bartosz G, Cipak A, Duburs G, Horakova L, Luczaj W, Majekova M, Odysseos AD, Rackova L, Skrzydlewska E, et al. Natural and synthetic antioxidants: an updated overview. Free Radic Res. 2010;44:1216–62.
Reddy VP, Garrett MR, Perry G, Smith MA. Carnosine: a versatile antioxidant and antiglycating agent. Sci Aging Knowl Environ. 2005;2005:pe12.
Birrell AM, Heffernan SJ, Kirwan P, McLennan S, Gillin AG, Yue DK. The effects of aminoguanidine on renal changes in a baboon model of type 1 diabetes. J Diabetes Complicat. 2002;16:301–9.
Kern TS, Engerman RL. Pharmacological inhibition of diabetic retinopathy: aminoguanidine and aspirin. Diabetes. 2001;50:1636–42.
Degenhardt TP, Fu MX, Voss E, Reiff K, Neidlein R, Strein K, Thorpe SR, Baynes JW, Reiter R. Aminoguanidine inhibits albuminuria, but not the formation of advanced glycation end-products in skin collagen of diabetic rats. Diabetes Res Clin Pract. 1999;43:81–9.
Yamauchi A, Takei I, Makita Z, Nakamoto S, Ohashi N, Kiguchi H, Ishii T, Koike T, Saruta T. Effects of aminoguanidine on serum advanced glycation endproducts, urinary albilmin excretion, mesangial expansion, and glomerular basement membrane thickening in Otsuka long-Evans Tokushima fatty rats. Diabetes Res Clin Pract. 1997;34:127–33.
Schmidt RE, Dorsey DA, Beaudet LN, Reiser KM, Williamson JR, Tilton RG. Effect of aminoguanidine on the frequency of neuroaxonal dystrophy in the superior mesenteric sympathetic autonomic ganglia of rats with streptozocin-induced diabetes. Diabetes. 1996;45:284–90.
Miyauchi Y, Shikama H, Takasu T, Okamiya H, Umeda M, Hirasaki E, Ohhata I, Nakayama H, Nakagawa S. Slowing of peripheral motor nerve conduction was ameliorated by aminoguanidine in streptozocin-induced diabetic rats. Eur J Endocrinol. 1996;134:467–73.
Fu MX, Wells-Knecht KJ, Blackledge JA, Lyons TJ, Thorpe SR, Baynes JW. Glycation, glycoxidation, and cross-linking of collagen by glucose. Kinetics, mechanisms, and inhibition of late stages of the Maillard reaction. Diabetes. 1994;43:676–83.
Huijberts MS, Wolffenbuttel BH, Boudier HA, Crijns FR, Kruseman AC, Poitevin P, Levy BI. Aminoguanidine treatment increases elasticity and decreases fluid filtration of large arteries from diabetic rats. J Clin Invest. 1993;92:1407–11.
Edelstein D, Brownlee M. Aminoguanidine ameliorates albuminuria in diabetic hypertensive rats. Diabetologia. 1992;35:96–7.
Soulis-Liparota T, Cooper M, Papazoglou D, Clarke B, Jerums G. Retardation by aminoguanidine of development of albuminuria, mesangial expansion, and tissue fluorescence in streptozocin-induced diabetic rat. Diabetes. 1991;40:1328–34.
Hammes HP, Martin S, Federlin K, Geisen K, Brownlee M. Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc Natl Acad Sci U S A. 1991;88:11555–8.
Valero MP, Fletcher AE, De Stavola BL, Vioque J, Alepuz VC. Vitamin C is associated with reduced risk of cataract in a Mediterranean population. J Nutr. 2002;132:1299–306.
Ravindran RD, Vashist P, Gupta SK, Young IS, Maraini G, Camparini M, Jayanthi R, John N, Fitzpatrick KE, Chakravarthy U, et al. Inverse association of vitamin C with cataract in older people in India. Ophthalmology. 2011;118:1958–65 e1952.
Nourmohammadi I, Modarress M, Khanaki K, Shaabani M. Association of serum alpha-tocopherol, retinol and ascorbic acid with the risk of cataract development. Ann Nutr Metab. 2008;52:296–8.
Anusiri P, Choodej S, Chumriang P, Adisakwattana S, Pudhom K. Inhibitory effects of flavonoids from stem bark of Derris indica on the formation of advanced glycation end products. J Ethnopharmacol. 2014;158:437–41.
Urios P, Grigorova-Borsos AM, Sternberg M. Flavonoids inhibit the formation of the cross-linking AGE pentosidine in collagen incubated with glucose, according to their structure. Eur J Nutr. 2007;46:139–46.
Urios P, Kassab I, Grigorova-Borsos AM, Guillot R, Jacolot P, Tessier F, Peyroux J, Sternberg M. A flavonoid fraction purified from Rutaceae aurantiae (Daflon(R)) inhibiting AGE formation, reduces urinary albumin clearance and corrects hypoalbuminemia in normotensive and hypertensive diabetic rats. Diabetes Res Clin Pract. 2014;105:373–81.
Odetti PR, Borgoglio A, De Pascale A, Rolandi R, Adezati L. Prevention of diabetes-increased aging effect on rat collagen-linked fluorescence by aminoguanidine and rutin. Diabetes. 1990;39:796–801.
Metz TO, Alderson NL, Thorpe SR, Baynes JW. Pyridoxamine, an inhibitor of advanced glycation and lipoxidation reactions: a novel therapy for treatment of diabetic complications. Arch Biochem Biophys. 2003;419:41–9.
Alderson NL, Chachich ME, Youssef NN, Beattie RJ, Nachtigal M, Thorpe SR, Baynes JW. The AGE inhibitor pyridoxamine inhibits lipemia and development of renal and vascular disease in Zucker obese rats. Kidney Int. 2003;63:2123–33.
Chang KC, Liang JT, Tsai PS, Wu MS, Hsu KL. Prevention of arterial stiffening by pyridoxamine in diabetes is associated with inhibition of the pathogenic glycation on aortic collagen. Br J Pharmacol. 2009;157:1419–26.
Degenhardt TP, Alderson NL, Arrington DD, Beattie RJ, Basgen JM, Steffes MW, Thorpe SR, Baynes JW. Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat. Kidney Int. 2002;61:939–50.
Lewis EJ, Greene T, Spitalewiz S, Blumenthal S, Berl T, Hunsicker LG, Pohl MA, Rohde RD, Raz I, Yerushalmy Y, et al. Pyridorin in type 2 diabetic nephropathy. J Am Soc Nephrol. 2012;23:131–6.
Williams ME, Bolton WK, Khalifah RG, Degenhardt TP, Schotzinger RJ, McGill JB. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am J Nephrol. 2007;27:605–14.
Stracke H, Hammes HP, Werkmann D, Mavrakis K, Bitsch I, Netzel M, Geyer J, Kopcke W, Sauerland C, Bretzel RG, et al. Efficacy of benfotiamine versus thiamine on function and glycation products of peripheral nerves in diabetic rats. Exp Clin Endocrinol Diabetes. 2001;109:330–6.
Babaei-Jadidi R, Karachalias N, Ahmed N, Battah S, Thornalley PJ. Prevention of incipient diabetic nephropathy by high-dose thiamine and benfotiamine. Diabetes. 2003;52:2110–20.
Hammes HP, Du X, Edelstein D, Taguchi T, Matsumura T, Ju Q, Lin J, Bierhaus A, Nawroth P, Hannak D, et al. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med. 2003;9:294–9.
Alkhalaf A, Kleefstra N, Groenier KH, Bilo HJ, Gans RO, Heeringa P, Scheijen JL, Schalkwijk CG, Navis GJ, Bakker SJ. Effect of benfotiamine on advanced glycation endproducts and markers of endothelial dysfunction and inflammation in diabetic nephropathy. PLoS One. 2012;7:e40427.
Fraser DA, Diep LM, Hovden IA, Nilsen KB, Sveen KA, Seljeflot I, Hanssen KF. The effects of long-term oral benfotiamine supplementation on peripheral nerve function and inflammatory markers in patients with type 1 diabetes: a 24-month, double-blind, randomized, placebo-controlled trial. Diabetes Care. 2012;35:1095–7.
Stirban A, Negrean M, Stratmann B, Gawlowski T, Horstmann T, Gotting C, Kleesiek K, Mueller-Roesel M, Koschinsky T, Uribarri J, et al. Benfotiamine prevents macro- and microvascular endothelial dysfunction and oxidative stress following a meal rich in advanced glycation end products in individuals with type 2 diabetes. Diabetes Care. 2006;29:2064–71.
Tarwadi KV, Agte VV. Effect of micronutrients on methylglyoxal-mediated in vitro glycation of albumin. Biol Trace Elem Res. 2011;143:717–25.
Voziyan PA, Hudson BG. Pyridoxamine as a multifunctional pharmaceutical: targeting pathogenic glycation and oxidative damage. Cell Mol Life Sci. 2005;62:1671–81.
Wu CH, Yen GC. Inhibitory effect of naturally occurring flavonoids on the formation of advanced glycation endproducts. J Agric Food Chem. 2005;53:3167–73.
Lo CY, Li S, Tan D, Pan MH, Sang S, Ho CT. Trapping reactions of reactive carbonyl species with tea polyphenols in simulated physiological conditions. Mol Nutr Food Res. 2006;50:1118–28.
Sabbagh MN, Agro A, Bell J, Aisen PS, Schweizer E, Galasko D. PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer disease. Alzheimer Dis Assoc Disord. 2011;25:206–12.
Rao NV, Argyle B, Xu X, Reynolds PR, Walenga JM, Prechel M, Prestwich GD, MacArthur RB, Walters BB, Hoidal JR, et al. Low anticoagulant heparin targets multiple sites of inflammation, suppresses heparin-induced thrombocytopenia, and inhibits interaction of RAGE with its ligands. Am J Physiol Cell Physiol. 2010;299:C97–110.
Kumano-Kuramochi M, Ohnishi-Kameyama M, Xie Q, Niimi S, Kubota F, Komba S, Machida S. Minimum stable structure of the receptor for advanced glycation end product possesses multi ligand binding ability. Biochem Biophys Res Commun. 2009;386:130–4.
Basta G. Receptor for advanced glycation endproducts and atherosclerosis: from basic mechanisms to clinical implications. Atherosclerosis. 2008;196:9–21.
Li YM, Tan AX, Vlassara H. Antibacterial activity of lysozyme and lactoferrin is inhibited by binding of advanced glycation-modified proteins to a conserved motif. Nat Med. 1995;1:1057–61.
Eschenburg G, Heine W, Peters E. Fecal sIgS and lysozyme excretion in breast feeding and formula feeding. Kinderarztliche Praxis. 1990;58:255–60.
Huang J, Wu L, Yalda D, Adkins Y, Kelleher SL, Crane M, Lonnerdal B, Rodriguez RL, Huang N. Expression of functional recombinant human lysozyme in transgenic rice cell culture. Transgenic Res. 2002;11:229–39.
American Heart Association Nutrition Committee A, Lichtenstein AH, Appel LJ, Brands M, Carnethon M, Daniels S, Franch HA, Franklin B, Kris-Etherton P, Harris WS, et al. Diet and lifestyle recommendations revision 2006: a scientific statement from the American Heart Association nutrition committee. Circulation. 2006;114:82–96.
Marmot M, Atinmo T, Byers T, Chen J, Hirohata T, Jackson A, James W, Kolonel L, Kumanyika S, Leitzmann C, et al. Food, Nutrition, Physical Activity, and the Prevention of Cancer: a Global Perspective. (WCRF/AICR Expert Report). In: World Cancer Research Fund / American Institute for Cancer Research. Washington: American Institute for Cancer Research (AICR); 2007.
American Diabetes Association A, Bantle JP, Wylie-Rosett J, Albright AL, Apovian CM, Clark NG, Franz MJ, Hoogwerf BJ, Lichtenstein AH, Mayer-Davis E, et al. Nutrition recommendations and interventions for diabetes: a position statement of the American Diabetes Association. Diabetes Care. 2008;31 Suppl 1:S61–78.
Liu FC, Hung LF, Wu WL, Chang DM, Huang CY, Lai JH, Ho LJ. Chondroprotective effects and mechanisms of resveratrol in advanced glycation end products-stimulated chondrocytes. Arthritis Res Ther. 2010;12:R167.
Menini S, Iacobini C, Ricci C, Blasetti Fantauzzi C, Pugliese G. Protection from diabetes-induced atherosclerosis and renal disease by D-carnosine-octylester: effects of early vs late inhibition of advanced glycation end-products in Apoe-null mice. Diabetologia. 2015;58:845–53.
Rahbar S, Figarola JL. Novel inhibitors of advanced glycation endproducts. Arch Biochem Biophys. 2003;419:63–79.
Harcourt BE, Sourris KC, Coughlan MT, Walker KZ, Dougherty SL, Andrikopoulos S, Morley AL, Thallas-Bonke V, Chand V, Penfold SA, et al. Targeted reduction of advanced glycation improves renal function in obesity. Kidney Int. 2011;80:190–8.
Cai W, He JC, Zhu L, Peppa M, Lu C, Uribarri J, Vlassara H. High levels of dietary advanced glycation end products transform low-density lipoprotein into a potent redox-sensitive mitogen-activated protein kinase stimulant in diabetic patients. Circulation. 2004;110:285–91.
Negrean M, Stirban A, Stratmann B, Gawlowski T, Horstmann T, Gotting C, Kleesiek K, Mueller-Roesel M, Koschinsky T, Uribarri J, et al. Effects of low- and high-advanced glycation endproduct meals on macro- and microvascular endothelial function and oxidative stress in patients with type 2 diabetes mellitus. Am J Clin Nutr. 2007;85:1236–43.
Stirban A, Negrean M, Stratmann B, Gotting C, Salomon J, Kleesiek K, Tschoepe D. Adiponectin decreases postprandially following a heat-processed meal in individuals with type 2 diabetes: an effect prevented by benfotiamine and cooking method. Diabetes Care. 2007;30:2514–6.
Stirban A, Negrean M, Gotting C, Uribarri J, Gawlowski T, Stratmann B, Kleesiek K, Koschinsky T, Vlassara H, Tschoepe D. Dietary advanced glycation endproducts and oxidative stress: in vivo effects on endothelial function and adipokines. Ann N Y Acad Sci. 2008;1126:276–9.
Stirban A, Negrean M, Gotting C, Stratmann B, Gawlowski T, Mueller-Roesel M, Kleesiek K, Koschinsky T, Tschoepe D. Leptin decreases postprandially in people with type 2 diabetes, an effect reduced by the cooking method. Horm Metab Res. 2008;40:896–900.
Acknowledgements
The funding support of Nutrition Scientific Research Foundation of BY-HEALTH is gratefully acknowledged.
Funding
Nutrition Scientific Research Foundation of BY-HEALTH.
Author information
Authors and Affiliations
Contributions
JC and XZ participated in writing of the review and collecting some relevant literature. XL and CB critiqued and provided valuable input in addition to some corrections. JC and XZ wrote and prepared the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
All authors have approved the manuscript for publication.
Competing interests
Professor Xu Lin, one of the authors, is co-editors-in-chief of Nutrition & Metabolism.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chen, JH., Lin, X., Bu, C. et al. Role of advanced glycation end products in mobility and considerations in possible dietary and nutritional intervention strategies. Nutr Metab (Lond) 15, 72 (2018). https://doi.org/10.1186/s12986-018-0306-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12986-018-0306-7