
Abstract
Background
Mathematical modeling of biological regulatory networks provides valuable insights into the structural and dynamical properties of the underlying systems. While dynamic models based on differential equations provide quantitative information on the biological systems, qualitative models that rely on the logical interactions among the components provide coarse-grained descriptions useful for systems whose mechanistic underpinnings remain incompletely understood. The middle ground class of piecewise affine differential equation models was proven informative for systems with partial knowledge of kinetic parameters.
Methods
In this work we provide a comparison of the dynamic characteristics of these three approaches applied on several biological regulatory network motifs. Specifically, we compare the attractors and state transitions in asynchronous Boolean, piecewise affine and Hill-type continuous models.
Results
Our study shows that while the fixed points of asynchronous Boolean models are observed in continuous Hill-type and piecewise affine models, these models may exhibit different attractors under certain conditions.
Conclusions
Overall, qualitative models are suitable for systems with limited knowledge of quantitative information. On the other hand, when practical, using quantitative models can provide detailed information about additional real-valued attractors not present in the qualitative models.
Similar content being viewed by others


Background
Dynamic modeling approaches can be of a quantitative or qualitative nature depending on the states of the components of a system. Quantitative models, usually cast as a set of differential equations, are the most appropriate dynamic approaches for modeling real systems. However, due to unknown kinetic parameters and mechanistic details of many biological systems, these models are only feasible on smaller, well-studied systems [1–4]. Qualitative models, on the other hand, require few or no parameters, and provide coarse-grained descriptions of the underlying biological systems. At the middle ground, piecewise affine differential equation (also referred to as hybrid) models combine the qualitative description of the regulatory relationships in a network with a continuous concentration decay.
Among possible functional relationships to describe biological regulatory interactions, from simple Michaelis-Menten kinetics to non-monotonic functions corresponding to substrate inhibition [5], there is increasing evidence for the adequacy of sigmoid functions, such as Hill functions [6]. For example, Hill-type models in which the production rate of a species is modeled by a Hill function and the degradation rate is considered to be linear, have been successfully employed in modeling T cell receptor signaling [7] as well as cardiac β-adrenergic signaling networks [8]. Although the concentration of components in biological systems changes continuously over time, the input-output sigmoid curves of the regulatory interactions can be well approximated by step functions [9]. This approximation leads to hybrid models, wherein the production rates are described by logical functions and the degradation rates are considered to be linear. These models are suitable for systems for which partial knowledge of parameter values is available, and have been widely used in the literature [10–15]. In the extreme idealization case of qualitative models, called Boolean models, biological entities are characterized by binary (ON or OFF) variables and their interactions are usually expressed using the logic operators AND, OR, and NOT [16, 17]. These models are mostly suited for systems for which the values of the kinetic parameters are unknown, and have been employed in various real systems [18–24].
A comparison of these three models can provide information on the characteristics of each model, the level of detail they can provide, and on how the results of one approach can be translated into another. There have been several efforts in this direction. For example, Chaves et al. [11] proposed a method for comparison of Boolean and hybrid models by first transforming the hybrid model of a given system into a multi-level discrete model, which was translated into a Boolean model. The latter step was achieved by expanding the state space through introducing extra variables to the model, which may not be practical for modeling large-scale systems. In another study, Wittmann et al. [7] compared Boolean models with continuous Hill-type models in the context of a T cell receptor signaling network and found that the qualitative behavior of a discrete model is reproduced by its continuous homologue. In particular, they showed that Boolean stable states (also called fixed points) correspond to ‘similar’ stable steady states of normalized Hill-type models, at least for certain parameters, but continuous models may exhibit additional stable steady states [7]. Similarly, by comparing the dynamics of asynchronous Boolean models and that of the hybrid models, Jamshidi et al. [25] showed that, in general, the attractors and their reachability properties are not preserved between the two models. More recently, Chaves and Preto [26] compared the three modeling frameworks in the context of the Kai ABC oscillator, which is the core of the cyanobacterial circadian clock. Their analysis revealed that Boolean and Hill-type models of the system have similar attractors, a fixed point and a complex attractor, the latter representing stable periodic oscillations in the Hill-type formalism. These oscillations were either periodic or damped in the hybrid framework, depending on the parameters of the model [26]. Despite these efforts, there is still a need to further explore the emergent properties of these three models applied on the same system.
In this work, we provide a comparison of the dynamic characteristics of Hill-type models, hybrid models, and asynchronous Boolean models through several illustrative examples. In particular, we focus on a positive feedback loop realized by mutual inhibition, a negative feedback loop, an exclusive NOR (XNOR) gate, as well as a simplified sub-network of the T-cell large granular lymphocyte leukemia signaling network obtained from [23]. Our results show that while the fixed points of general asynchronous Boolean models are observed in continous Hill-type and hybrid models, these models may exhibit different attractors under certain conditions.
Background and Methods
Hill-type models
In general, the dynamics of a regulatory network can be described by a set of differential equations in which the rate of change of the concentration of each node at any time instant is given by a continuous function of the concentration of its regulators. This function, which can be linear or non-linear, depends on certain kinetic parameters as well. In particular, under the Hill-type formalism, the time evolution of the continuous variable \( {\overline{x}}_i \) is given by:
where λ i and γ i are synthesis and decay parameters, and \( {F}_i\left({\overline{x}}_{i_1},\dots, {\overline{x}}_{i_{m_{{}_i}}}\right)={f}_i\left({g}_{i_1}\left({\overline{x}}_{i_1}\right),\dots, {g}_{i_{m_{{}_i}}}\left({\overline{x}}_{i_{m_{{}_i}}}\right)\right) \) in which f i is defined based on the regulatory logic of node i. Specifically, an AND operator between variables \( {\overline{x}}_{i_1} \) and \( {\overline{x}}_{i_2} \) becomes \( f\left({\overline{x}}_{i_1},{\overline{x}}_{i_2}\right)=f\left({\overline{x}}_{i_1}\right)f\left({\overline{x}}_{i_2}\right) \) and an OR operator becomes \( f\left({\overline{x}}_{i_1},{\overline{x}}_{i_2}\right)=f\left({\overline{x}}_{i_1}\right)+f\left({\overline{x}}_{i_2}\right)-f\left({\overline{x}}_{i_1}\right)f\left({\overline{x}}_{i_2}\right) \). The functions \( {g}_{i_j} \) signify the Hill functions that model activating and inhibitory interactions in the network, \( {g}_{i_j}^{act}\left({\overline{x}}_{i_j}\right)=\frac{a_{i_j}{\overline{x}}_{i_j}^{n_{i_j}}}{{\overline{x}}_{i_j}^{n_{i_j}}+{k}_{i_j}^{n_{i_j}}} \) and \( {g}_{i_j}^{inh}\left({\overline{x}}_{i_j}\right)=1-\frac{a_{i_j}{\overline{x}}_{i_j}^{n_{i_j}}}{{\overline{x}}_{i_j}^{n_{i_j}}+{k}_{i_j}^{n_{i_j}}} \), respectively, where \( {n}_{i_j} \) indicates the Hill coefficient, \( {k}_{i_j} \) is the activation level of species i j at which it triggers half of the maximal activating effect on a downstream node. In order to make the comparison with Boolean models easier, the parameter \( {a}_{i_j} \) is chosen in such a way that \( {g}_{i_j}^{act}(1)=1 \) to ensure that the Hill function can attain the value of one. This normalization process, called normalized HillCubes, has been proposed and utilized previously [7, 8].
It is worth noting that one can normalize the concentration of \( {\overline{x}}_i \) to the unit interval, which reduces Eq. (1) to having the same value for the kinetic rate of synthesis and degradation [7]:
Piecewise affine differential equation (hybrid) models
The class of piecewise affine differential equation (hybrid) models, which bridges the gap between discrete and continuous models, meld the logical description of the regulatory relationships with a linear concentration decay. These models were originally proposed by Glass [13] to provide a coarse-grained description of gene regulatory networks, and since then their properties have been extensively studied in the literature [27–33]. In these models, each node is characterized by two variables: a continuous variable, \( {\overline{x}}_i \), which denotes the concentration of that component, and a discrete variable, x i , that accounts for its activity. At each time instant t, the discrete variable is defined based on the continuous variable using a threshold parameter θ i as follows:
The time evolution of the continuous variable \( {\overline{x}}_i \) is then described by the following piecewise affine differential equation:
where \( {F}_i^B \) is the Boolean function for the ith node with m i regulators, and λ i and γ i are synthesis and decay parameters. It is usually assumed that \( \max\ {\overline{x}}_i={\lambda}_i/{\gamma}_i>{\theta}_i \). An alternative is to scale the continuous variable \( {\overline{x}}_i \) by λ i /γ i in (4) and thus to consider θ i ∈ (0, 1). Similar to the Hill-type models one can normalize Eq. (4) to have only one parameter for synthesis and degradation:
We note that in a more general form, one can consider p i threshold parameters for a node if it regulates p i downstream nodes [29]; in this paper, however, we consider the case of one threshold per node.
In this framework, the state space of the system is partitioned into different domains bounded by threshold hyperplanes. The domains where no variable takes a threshold value are called regular domains or boxes, and the ones where at least one variable has a threshold value (i.e., the threshold hyperplanes and their intersections) are referred to as switching domains [27]. On the regular domains, differential Eq. (5) has a unique solution, which can be obtained analytically. However, this equation is not defined on the switching domains. The Filippov approach [34] was adapted to define the solutions on the switching domains by extending the piecewise affine differential equation into a differential inclusion [33].
Boolean models
In Boolean models, the state of each node is described by a binary variable that takes the values 1 (ON) and 0 (OFF). Time is considered to be discrete and is implemented using synchronous or asynchronous updating algorithms. In general, the future state of a node, \( {x}_i^{*} \), is given by a Boolean function, \( {F}_i^B \), of the current states of its regulators:
where \( {F}_i^B \) is usually expressed using AND, OR, and NOT operators. The collection of the total number of states for a system, along with the set of possible transitions among these states, forms the state transition graph of the system. The terminal strongly connected components of this graph are attractors (asymptotic behaviors) of the system, which are classified as fixed points (stable steady states) or complex attractors (also called limit cycles in the context of synchronous Boolean models). The detailed properties of Boolean models have been summarized in several recent review articles [35–39]. These models are increasingly employed in modeling systems wherein the values of the kinetic parameters are missing. In order to overcome the complexity associated with the exponential size of the state transition graphs of these models, a number of network reduction methods have been proposed [40–44].
Comparison of the three models
In order to compare the dynamic properties of the above three models, we first construct a discretized state transition graph from the continuous state space of the hybrid and Hill-type models. To this end, different domains of the state space (defined based upon the threshold values θ i or k i in the respective hybrid or Hill-type models, which can correspond to the intrinsic thresholds in Boolean models above which a node is ON) are considered as the discrete states in a state transition graph [13]. For example, the 01 state in the discretized state transition graph of a two-node network corresponds to the domain defined by 0 ≤ x 1 ≤ θ 1 and θ 2 ≤ x 2 ≤ 1 in the respective hybrid model’s state space. The discrete domains in the Hill-type model are defined similarly by replacing θ i by k i . Whenever there is a trajectory traversing from one domain to another, an edge from the corresponding discrete state to the other can be considered. In order to form such discretized state space, here we focused on all possible transitions among the regular domains, assuming that the switching domains of the hybrid models are transparent, that is, adjacent regular domains have vector fields which can be naturally continued from one domain to the next.
We used numerical simulations to obtain the discretized state transition graphs corresponding to the hybrid and Hill-type models. We obtained trajectories of a system by using the fourth-order Runge-Kutta algorithm starting from the initial states that are vertices of the unit cube. The integration was done from t = 0 to t = 50 with Δt = 0.01 time steps. For each state, a total of 10,000 threshold parameters were sampled uniformly randomly from the (0,1) interval. An edge in the discretized state transition graph was drawn from one state to another if parameter values were found for which the transition was possible. We note, however, that transitions occurring on a parameter set of measure zero in the space of all possible trajectories are negligible and can be removed from the state transition graph [28]. An example is the case when the transition happens only if the variables simultaneously reach their thresholds. In addition, we discarded the transitions that happened for less than 5 % of the threshold parameters. For simplicity, the α i parameters in Eqs. (2) and (5) were considered to equal one, and all the Hill coefficients were fixed at n i = 4. This choice was made based on the distribution of reported Hill coefficients, ranging from close to one to ten or more, with a mean of 3.2 [6]. Additionally, in a model of the Kai ABC oscillator in the cyanobacterial circadian clock, a Hill coefficient of 4 reproduced known experimental results the best [26]. The numerical simulations in this study were performed in Python. The phase plane analysis was done using the R package PhaseR.
Results
Transcriptional regulatory networks contain a small set of recurring regulation patterns referred to as network motifs, which are observed much more often than would be expected in random networks [45, 46]. These network motifs were detected in diverse organisms from Escherichia coli and yeast to plants and humans [46–49]. Here, we considered three network motifs observed in real or synthetic biological networks, namely a mutual inhibitory loop that leads to overall positive feedback, a negative feedback loop, and an XNOR gate. We also considered a simplified sub-network of the T cell large granular lymphocyte leukemia (T-LGL) signaling network obtained from [23].
Mutual inhibitory loop
Feedback loops are ubiquitous network motifs in biological systems whose sign is determined based on the parity of the number of negative interactions in the cycle. A positive/negative feedback loop has an even/odd number of negative interactions. Positive feedback loops can exhibit ultrasensitivity [50, 51], are associated with cellular differentiation and memory [52] and signal amplification [53], and are a necessary condition for multi-stability [54–58]. Bistability, wherein the system toggles between two stable steady states, is important in understanding many biological phenomena such as decision-making processes in cell cycle progression, cellular differentiation, and apoptosis [59, 60]. A genetic toggle switch in Escherichia coli has been synthesized using two repressible promoters arranged in a mutually inhibitory network [61].
Here we considered a mutual inhibitory positive feedback loop as given in Fig. 1a. We have compared the dynamics of this regulatory network motif under the three dynamic formalisms, Boolean, hybrid, as well as Hill-type models by constructing their corresponding discrete state transition graphs (see Background and Methods for details). Our results indicate that all three models exhibit two stable states (fixed points), 01 and 10 (Fig. 1b). A phase plane representation of the Hill-type model for the case of k i = 0.5 is shown in Additional file 1: Figure S1A. Thus this exemplifies the case when there is a one-to-one correspondence between the attractors of the three models.

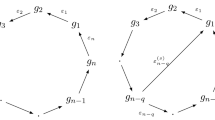
Dynamic analysis of a mutual inhibitory positive feedback loop. a A mutual inhibitory positive feedback loop and the corresponding equations and b state transition graphs for Boolean, hybrid, and Hill-type models. The left or right binary digit of the node identifier indicates the state of A or B, respectively. Each node in the discretized hybrid or Hill-type state transition graph corresponds to a regular domain in the respective state space
Negative feedback loop
Negative feedback loops play a determinant role in the dynamics of a network. For example, it has been conjectured that negative feedback loops are necessary for sustained oscillations [9, 58]. In particular, in the context of Boolean networks, it has been shown that as the number of independent negative feedback loops increases, the number of limit cycles tends to decrease while their length tends to increase [62].
Here we considered a two-element negative feedback loop as depicted in Fig. 2a. In this case, the asynchronous Boolean model converges to a limit cycle, but the Hill-type model exhibits a unique fixed point for each threshold (independent of the choice of the initial states), wherein at least one of the variables is close to its threshold value. A phase plane representation of the Hill-type model for the case of k i = 0.5 is shown in Additional file 1: Figure S1B. The hybrid model has small amplitude oscillations around threshold intersections. Based on a theorem by Glass and Pasternack [63], stating that if a Boolean model has a cyclic attractor, for the associated hybrid model all the trajectories in the regions of the state space corresponding to this cyclic attractor either approach a unique stable limit cycle or asymptotically approach a threshold intersection, we conclude that these oscillations asymptotically converge to a steady state corresponding to a threshold intersection. As such, this example shows that, for the parameters considered here, oscillations present in the asynchronous Boolean models may be absent from the hybrid or continuous Hill-type models.

Dynamic analysis of a negative feedback loop. a A negative feedback loop and the corresponding equations and b state transition graphs for Boolean, hybrid, and Hill-type models. The left or right binary digit of the node identifier indicates the state of A or B, respectively. The black-filled node in the hybrid and Hill-type state transition graphs represent threshold-dependent, real-valued fixed points
Exclusive NOR (XNOR) gate
The exclusive NOR (or for short, XNOR) gate is a digital logic gate with two or more inputs and one output, which evaluates as TRUE when all of its inputs are TRUE or when all of its inputs are FALSE. XNOR gates play a role in circuits that perform arithmetic operations such as full adders [64] and have been engineered as a synthetic genetic logic to realize simpler, independent control of biological processes [65, 66]. For example, Bonnet et al. [65] engineered an Escherichia coli transcriptional circuit that realizes this gate as well as other two-variable logic gates.
Here we considered a two-node network with both nodes regulated by an XNOR gate (Fig. 3a). Our simulations indicate that the Boolean model exhibits a fixed point, wherein both nodes stabilize in an ON state, and a complex attractor. The Hill-type model has two stable fixed points, including the fixed point of the Boolean model and an additional real-valued fixed point for each threshold, which is not necessarily a threshold intersection. A phase plane representation of the Hill-type model for the case of k i = 0.5 is shown in Additional file 1: Figure S1C. In the hybrid model, in addition to the fixed point of the Boolean model, there are small-amplitude oscillations that asymptotically approach a threshold intersection. This example shows that while the fixed point and the oscillatory attractor co-exist in the Boolean framework, the corresponding hybrid and Hill-type models, for the parameters considered here, exhibit only steady states.

Dynamic analysis of an XNOR gate. a An XNOR gate and the corresponding equations and b state transition graphs for Boolean, hybrid, and Hill-type models. Both nodes A and B have the same Boolean equations, and similar corresponding hybrid and Hill equations; so only one is shown. The left or right binary digit of the node identifier indicates the state of A or B, respectively. The black-filled node in the Hybrid and Hill-type state spaces represents a threshold-dependent, real-valued fixed point
Simplified T-cell large granular lymphocyte (T-LGL) leukemia network
Cytotoxic T cells, which induce the self-destruction of infected cells, are a central part of the immune system's response to infection. Cytotoxic T cells normally undergo activation-induced cell death (apoptosis) after fighting infection, but in the disease T-cell large granular lymphocyte (T-LGL) leukemia, these cells avoid cell death and remain long-term competent [67]. A Boolean network model of cytotoxic T cell signaling that reproduces the known experimental results was previously constructed by Zhang et al. through an extensive curation of the available experimental literature [68]. This network consists of 60 nodes including proteins, mRNAs, and small molecules, such as lipids. There are also a few abstract nodes in the network such as “Stimuli,” which represents antigen stimulation, and “Apoptosis,” which denotes programmed cell death. Dynamic analysis of this network model [23, 68] revealed that in the sustained presence of the external signals IL15 and Stimuli the system has two attractors: one that recapitulates the survival phenotype seen in T-LGL leukemia, and a second one that corresponds to apoptosis. In a previous work [23] we showed that a sub-network of six nodes can recapitulate the original system’s dynamic repertoire.
Here we considered a further simplified sub-network of the T-LGL leukemia signaling network obtained from [23] by removing three additional nodes (Fas, Flip, and Apoptosis). This 3-node sub-network and the Boolean rules governing the nodes’ states are given in Fig. 4a. Hybrid and Hill equations can be easily obtained as described in Background and Methods. As we can see in Fig. 4b, all three models result in two fixed points. Fixed point 100 is consistent with the T-LGL attractor, while the 001 fixed point corresponds to the apoptosis attractor. The state transition graph of the Boolean and hybrid models are the same while the Hill-type state space has more transitions compared to the other two models. Two transitions, one from the 001 domain to 000 and one from 100 to 000, which happen for 24 % and 11 % of the parameters, respectively, are not expected as 001 and 100 are fixed points. This might be partly due to the fact that trajectories get close to the switching domains before they converge to the fixed point.

Dynamic analysis of a simplified T-LGL signaling network. a The T-LGL network and the corresponding Boolean rules (the hybrid and Hill-type equations are not shown for simplicity) and b state transition graphs for Boolean, hybrid, and Hill-type models. The digits of the node identifiers from left to right indicate the state of S1P, Ceramide, and DISC, respectively
Discussion
Here we compared the qualitative dynamics and emerging properties of three methods, Boolean, hybrid and Hill-type models applied on three different network motifs as well as a simplified sub-network in the context of leukemia. We found that the fixed points of Boolean models are conserved in the hybrid model as well as the normalized Hill-type model. However, both hybrid and Hill models can have additional real-valued fixed points not observed in the respective Boolean models. These additional fixed points seem to correspond to the oscillations present in the respective Boolean model, essentially replacing sustained oscillations with damped oscillations that asymptotically approach a threshold crossing. Further rigorous analysis is needed to understand this relationship.
We note that here we only focused on the dynamic transitions among the regular domains of the hybrid/Hill-type models; however, investigating the behavior of a system along the switching domains can provide additional differences between qualitative and quantitative models. For networks without a self-loop, all the switching domains in the state space of the hybrid model are transparent, that is, adjacent regular domains have vector fields that can be naturally continued [13], and thus no further analysis on these domains is needed. This was the case for the first three examples that we considered here, however, such analysis might provide further information on the T-LGL leukemia signaling network.
Given that in this work we only considered the initial states that are vertices of the unit cube, further analysis is needed to fully characterize the behavior of a system under hybrid and Hill-type formalisms. Moreover, one can add another level of complexity to the hybrid models by considering ternary states (low, medium and high) in the discrete component of these models. For example, a recent study proposed a hybrid model with ternary states for the efficiency of promoters and ribosome binding sites to capture the dynamics of synthetic genetic circuits [15]. Finally, variations in the synthesis and decay parameters of hybrid and Hill-type models may provide additional dynamic properties that cannot be captured using qualitative models alone.
In modeling the dynamics of a biological regulatory network, one usually starts with a qualitative description of the system and makes it more quantitative as quantitative and kinetic experimental data become available. If the value of the majority of the kinetic parameters is known, comparison of the modeling results with available experimental observations can help to estimate the value of the remaining parameters or constrain the ranges of parameter values [14]. As a detailed, quantitative model with many parameters has a high degree of uncertainty when many of the parameter values are not known, taking the opposite route of transitioning to more abstract models that have fewer parameters can also be beneficial [8, 69]. Qualitative and quantitative models share the potential shortcoming that their trajectories reflect only a few of the many trajectories possible for a real system (e.g., considering cell-to-cell variability captured by recent single-cell analysis [70, 71]), and in the absence of a detailed experimental investigation of the system it is not known which trajectories are the most relevant.
Conclusions
In this work, we provided a comparison of the dynamic characteristics of qualitative and quantitative models through several illustrative examples. Our study showed that the qualitative models could successfully recapitulate the stable steady states of the respective quantitative models, which are the dominant attractors of biological systems, representing cellular states and cell types. This makes qualitative models, such as Boolean models, more attractive for systems with limited knowledge of quantitative information. On the other hand, when practical, using quantitative models, such as Hill-type models, can provide detailed information about additional real-valued attractors not present in the discrete models, as well as on the trajectories of the system. When partial quantitative information is available, hybrid models can successfully recapitulate the dynamic characteristics of the respective Hill-type model. It is important to note that the model choice also depends on the questions to be addressed and on the type of predictions that are aimed to make. For example, Boolean models are suitable to identify targets whose genetic knockout or pharmacological inhibition drives the system to a desired attractor. A recent study [72] has shown that such predictions made by an asynchronous Boolean model were validated in the corresponding Hill-type model as well. If more quantitative predictions are desired, hybrid and Hill-type models are more appropriate.
References
Barkai N, Leibler S. Robustness in simple biochemical networks. Nature. 1997;387(6636):913–7.
Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298(5596):1241–5.
Tyson JJ. Modeling the cell division cycle: cdc2 and cyclin interactions. Proc Natl Acad Sci U S A. 1991;88(16):7328–32.
von Dassow G, Meir E, Munro EM, Odell GM. The segment polarity network is a robust developmental module. Nature. 2000;406(6792):188–92.
Copeland RA. Enzymes: a practical introduction to structure, mechanism, and data analysis. 2nd ed: Wiley; 2000.
Zhang Q, Bhattacharya S, Andersen ME. Ultrasensitive response motifs: basic amplifiers in molecular signalling networks. Open biology. 2013;3(4):130031.
Wittmann DM, Krumsiek J, Saez-Rodriguez J, Lauffenburger DA, Klamt S, Theis FJ. Transforming Boolean models to continuous models: methodology and application to T-cell receptor signaling. BMC Syst Biol. 2009;3:98.
Kraeutler MJ, Soltis AR, Saucerman JJ. Modeling cardiac beta-adrenergic signaling with normalized-Hill differential equations: comparison with a biochemical model. BMC Syst Biol. 2010;4:157.
Thomas R, D'Ari R. Biological feedback. Boca Raton: CRC Press; 1990.
Chaves M, Sontag ED, Albert R. Methods of robustness analysis for Boolean models of gene control networks. Syst Biol. 2006;153(4):154–67.
Chaves M, Tournier L, Gouze JL. Comparing Boolean and piecewise affine differential models for genetic networks. Acta Biotheor. 2010;58(2-3):217–32.
de Jong H, Geiselmann J, Batt G, Hernandez C, Page M. Qualitative simulation of the initiation of sporulation in Bacillus subtilis. Bull Math Biol. 2004;66(2):261–99.
Glass L. Combinatorial and topological methods in nonlinear chemical-kinetics. J Chem Phys. 1975;63(4):1325–35.
Thakar J, Saadatpour-Moghaddam A, Harvill ET, Albert R. Constraint-based network model of pathogen-immune system interactions. J R Soc Interface. 2009;6(36):599–612.
Zomorrodi AR, Maranas CD. Coarse-grained optimization-driven design and piecewise linear modeling of synthetic genetic circuits. Eur J Oper Res. 2014;237(2):665–76.
Kauffman SA. Metabolic stability and epigenesis in randomly constructed genetic nets. J Theor Biol. 1969;22(3):437–67.
Thomas R. Boolean formalization of genetic control circuits. J Theor Biol. 1973;42(3):563–85.
Albert R, Othmer HG. The topology of the regulatory interactions predicts the expression pattern of the segment polarity genes in Drosophila melanogaster. J Theor Biol. 2003;223(1):1–18.
Christensen TS, Oliveira AP, Nielsen J. Reconstruction and logical modeling of glucose repression signaling pathways in Saccharomyces cerevisiae. BMC Syst Biol. 2009;3:7.
Giacomantonio CE, Goodhill GJ. A Boolean model of the gene regulatory network underlying Mammalian cortical area development. PLoS Comput Biol. 2010;9(6):e1000936.
Helikar T, Konvalina J, Heidel J, Rogers JA. Emergent decision-making in biological signal transduction networks. Proc Natl Acad Sci U S A. 2008;105(6):1913–8.
Mendoza L, Thieffry D, Alvarez-Buylla ER. Genetic control of flower morphogenesis in Arabidopsis thaliana: a logical analysis. Bioinformatics. 1999;15(7-8):593–606.
Saadatpour A, Wang RS, Liao A, Liu X, Loughran TP, Albert I, et al. Dynamical and structural analysis of a T cell survival network identifies novel candidate therapeutic targets for large granular lymphocyte leukemia. PLoS Comput Biol. 2011;7(11):e1002267.
Saez-Rodriguez J, Simeoni L, Lindquist JA, Hemenway R, Bommhardt U, Arndt B, et al. A logical model provides insights into T cell receptor signaling. PLoS Comput Biol. 2007;3(8):e163.
Jamshidi S, Siebert H, Bockmayr A. Comparing discrete and piecewise affine differential equation models of gene regulatory networks, IPCAT. Cambridge: Springer; 2012.
Chaves M, Preto M. Hierarchy of models: from qualitative to quantitative analysis of circadian rhythms in cyanobacteria. Chaos. 2013;23(2):025113.
Casey R, de Jong H, Gouze JL. Piecewise-linear models of genetic regulatory networks: Equilibria and their stability. J Math Biol. 2006;52(1):27–56.
Chaves M, Farcot E, Gouze JL, editors. Transition probabilities for piecewise affine models of genetic networks. Proc Int Symp Mathematical Theory of Networks and Systems 2010; Budapest, Hungary.
de Jong H, Gouze JL, Hernandez C, Page M, Sari T, Geiselmann J. Qualitative simulation of genetic regulatory networks using piecewise-linear models. Bull Math Biol. 2004;66(2):301–40.
de Jong H, Page M. Search for steady states of piecewise-linear differential equation models of genetic regulatory networks. IEEE/ACM Trans Comput Biol Bioinform. 2008;5(2):208–22.
Farcot E. Limit cycles in piecewise-affine gene network models with multiple interaction loops. Int J Syst Sci. 2010;41(1):119–30.
de Jong H, Geiselmann J, Hernandez C, and Page M. Genetic network analyzer: Qualitative simulation of genetic regulatory networks. Bioinformatics. 2003;19(3):336–344.
Gouze JL, Sari T. A class of piecewise linear differential equations arising in biological models. Dynam Sys Intern J. 2002;17(4):299–316.
Filippov AF. Differential equations with discontinuous righthand sides. Dordrecht: Kluwer Academic Publisher; 1988.
Morris MK, Saez-Rodriguez J, Sorger PK, Lauffenburger DA. Logic-based models for the analysis of cell signaling networks. Biochemistry. 2010;49(15):3216–24.
Saadatpour A, Albert R. Boolean modeling of biological regulatory networks: a methodology tutorial. Methods. 2013;62(1):3–12.
Wang RS, Saadatpour A, Albert R. Boolean modeling in systems biology: an overview of methodology and applications. Phys Biol. 2012;9(5):055001.
Helikar T, Kochi N, Konvalina J, Rogers JA. Boolean modeling of biochemical networks. Open Bioinformatics J. 2011;5:16–25.
Bornholdt S. Boolean network models of cellular regulation: prospects and limitations. J R Soc Interface. 2008;5 Suppl 1:S85–94.
Naldi A, Remy E, Thieffry D, Chaouiya C. Dynamically consistent reduction of logical regulatory graphs. Theoret Comput Sci. 2011;412(21):2207–18.
Richardson KA. Simplifying Boolean networks. Adv Complex Systems. 2005;8:365–81.
Saadatpour A, Albert R, Reluga TC. A reduction method for Boolean network models proven to conserve attractors. SIAM J on Applied Dynamical Systems. 2013;12(4):1997–2011.
Veliz-Cuba A. Reduction of Boolean network models. J Theor Biol. 2011;289:167–72.
Zanudo JGT, Albert R. An effective network reduction approach to find the dynamical repertoire of discrete dynamic networks. Chaos. 2013;23(2):025111.
Alon U. Network motifs: theory and experimental approaches. Nat Rev Genet. 2007;8(6):450–61.
Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U. Network motifs: simple building blocks of complex networks. Science. 2002;298(5594):824–7.
Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298(5594):799–804.
Saddic LA, Huvermann B, Bezhani S, Su Y, Winter CM, Kwon CS, et al. The LEAFY target LMI1 is a meristem identity regulator and acts together with LEAFY to regulate expression of CAULIFLOWER. Development. 2006;133(9):1673–82.
Saint-Andre V, Federation AJ, Lin CY, Abraham BJ, Reddy J, Lee TI, et al. Models of human core transcriptional regulatory circuitries. Genome Res. 2016;26(3):385–96.
Ferrell Jr JE, Machleder EM. The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science. 1998;280(5365):895–8.
Sneppen K, Micheelsen MA, Dodd IB. Ultrasensitive gene regulation by positive feedback loops in nucleosome modification. Mol Syst Biol. 2008;4:182.
Thomas R, Kaufman M. Multistationarity, the basis of cell differentiation and memory. I. Structural conditions of multistationarity and other nontrivial behavior. Chaos. 2001;11(1):170–9.
Freeman M. Feedback control of intercellular signalling in development. Nature. 2000;408(6810):313–9.
Cinquin O, Demongeot J. Positive and negative feedback: striking a balance between necessary antagonists. J Theor Biol. 2002;216(2):229–41.
Reichard A, Comet J-P. Necessary conditions for multistationarity in discrete dynamical systems. Discrete Applied Mathematics. 2007;155:2403–13.
Remy E, Ruet P, Thieffry D. Graphic requirements for multistability and attractive cycles in a Boolean dynamical framework. Adv Appl Math. 2008;41:335–50.
Soule C. Graphic requirements for multistationarity. ComPlexUs. 2003;1:123–33.
Thomas R. On the relation between the logical structure of systems and their ability to generate multiple steady states and sustained oscillations. In: Numerical Methods in the Study of Critical Phenomena. 9. Berlin: Springer Verlag; 1981. p. 180-93.
Eissing T, Conzelmann H, Gilles ED, Allgower F, Bullinger E, Scheurich P. Bistability analyses of a caspase activation model for receptor-induced apoptosis. J Biol Chem. 2004;279(35):36892–7.
Wilhelm T. The smallest chemical reaction system with bistability. BMC Syst Biol. 2009;3:90.
Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000;403(6767):339–42.
Sontag E, Veliz-Cuba A, Laubenbacher R, Jarrah AS. The effect of negative feedback loops on the dynamics of boolean networks. Biophys J. 2008;95(2):518–26.
Glass L, Pasternack JS. Stable oscillations in mathematical models of biological control systems. J Math Biol. 1978;6(3):207–23.
Bui HT, Wang Y, Jiang Y. Design and analysis of low-power 10-transistor full adders using novel XOR-XNOR gates. IEEE Trans On Circuits Systems II. 2002;49(1):25–30.
Bonnet J, Yin P, Ortiz ME, Subsoontorn P, Endy D. Amplifying genetic logic gates. Science. 2013;340(6132):599–603.
Siuti P, Yazbek J, Lu TK. Synthetic circuits integrating logic and memory in living cells. Nat Biotechnol. 2013;31(5):448–52.
Sokol L, Loughran TP Jr. Large granular lymphocyte leukemia. Oncologist. 2006;11(3):263–73.
Zhang R, Shah MV, Yang J, Nyland SB, Liu X, Yun JK, et al. Network model of survival signaling in large granular lymphocyte leukemia. Proc Natl Acad Sci U S A. 2008;105(42):16308–13.
Davidich M, Bornholdt S. The transition from differential equations to Boolean networks: a case study in simplifying a regulatory network model. J Theor Biol. 2008;255(3):269–77.
Saadatpour A, Lai S, Guo G, Yuan GC. Single-cell analysis in cancer genomics. Trends Genet. 2015;31(10):576–86.
Sandberg R. Entering the era of single-cell transcriptomics in biology and medicine. Nat Methods. 2014;11(1):22–4.
Zanudo JG, Albert R. Cell fate reprogramming by control of intracellular network dynamics. PLoS Comput Biol. 2015;11(4):e1004193.
Acknowledgements
This work was supported by NSF grants IIS-1161007 and PHY-1205840 to RA. The authors would like to thank Dr. Madalena Chaves for useful discussions.
Authors’ contributions
RA and AS conceived and designed the study. AS performed the simulations. AS and RA analyzed the data and drafted the manuscript. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1: Figure S1.
Phase plane representations of the Hill-type models presented in Figs. 1, 2, and 3. (A-C) Phase planes, nullclines (red lines, whose intersections determine the equilibria), stable (filled red circles) and unstable (open red circles) equilibrium points and representative trajectories (black lines) in the case of k i = 0.5 for the (A) mutual inhibitory positive feedback loop given in Fig. 1, (B) negative feedback loop given in Fig. 2, and (C) XNOR gate given in Fig. 3. (PDF 504 kb)
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Saadatpour, A., Albert, R. A comparative study of qualitative and quantitative dynamic models of biological regulatory networks. EPJ Nonlinear Biomed Phys 4, 5 (2016). https://doi.org/10.1140/epjnbp/s40366-016-0031-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1140/epjnbp/s40366-016-0031-y