
Abstract
Nanofibers of poly(vinyl alcohol) (PVA) reinforced with cellulose nanofibrils (CNFs) and/or crosslinked with maleic anhydride (MA) were produced by electrospinning technique to compare the additivation effects of the polymeric matrix. The results suggested that the PVA mass fraction equal to 14%, CNFs volumetric fraction of 3% and maleic acid at the molar ratio 20:1 are the best proportions for renewable base fibres production. In this study, the best electrospinning parameters for membranes production were obtained at the applied voltage of 24 kV, needle tip-to-collector distance of 14.5 cm, feed rate of 0.3 mL h−1 and using a plate collector. CNFs and MA additions allow to improve nanofiber thermal properties and resistance to water degradation, which result in an eco-friendlier, biocompatible and long-term biodegradable nanofiber mats with diameters of 74 ± 33 nm for water filtration purposes.
Similar content being viewed by others


1 Introduction
Recently, the production of fibres from renewable raw materials has become a research field of strong interest due to the growing environmental concerns [1, 2]. Cellulose is the most abundant natural polymer, which makes it very attractive from an economic point of view [3]. Furthermore, this polymer is an interesting raw material for the production of biocompatible and environmentally friendly products, which can be applied in different industrial fields [4, 5]. However, the use of cellulose in technical applications is still limited mostly due to its high hygroscopicity and insolubility in water as well as in numerous organic solvents. Notwithstanding, the interest in nanocellulose materials such as nanofibrils, nanocrystals or nanowhiskers has rapidly increased due to the unique physicochemical properties of such materials [6,7,8]. However, so far, few works have been done to obtain cellulose at nanoscale using environmentally friendly and sustainable methods such as the enzymatic hydrolysis [9]. Unlike conventional acid hydrolysis methods, enzymatic hydrolysis does not use solvents or chemical reagents [10]. Electrospinning is a simple and versatile technique used for the production of ultrathin and continuous nanofibers, which consists in applying a strong electric field to a polymeric solution [11, 12]. Nanofibers possess distinct properties (high specific surface area, high aspect ratio and biomimetic potential); hence, they are very useful and versatile for different areas with specific requirements, such as the development of scaffolds for tissue engineering and filtration [13,14,15,16]. However, electrospun of native cellulose is still difficult due to its high crystalline structure, which makes it insoluble in many solvents [17]. This has limited the research on cellulose derivatives soluble in water or mild solvents, namely the cellulose acetate, anionic sodium carboxymethyl cellulose, nonionic methyl cellulose, hydroxypropylmethyl cellulose, hydroxyethyl cellulose, cellulose acetate butyrate, cellulose acetate propionate and hydroxypropyl cellulose [18,19,20]. One way to overcome this drawback is the use of cellulose as reinforcing agent, avoiding therefore the use of solvents that can promote the degradation of the polymeric structure, and the introduction of additional steps to remove the solvent from the deposited fibres or to regenerate cellulose fibres [21,22,23,24]. The production of electrospun composites using renewable resources allows to obtain environmentally friendly fibres using difficult processable raw materials as cellulose [25, 26]. An improved interaction between the polymeric matrix and the reinforcing agent allows the preparation of materials with higher strength, stiffness and thermal stability, which is not the results of the sum of the individual constituent’s properties but a synergistic combination of both [27, 28]. Poly(vinyl alcohol) (PVA) is a semicrystalline polymer with excellent properties, such as the solubility in water, gas permeability, non-toxicity, biodegradability and biocompatibility [29]. PVA can be considered as an appealing polymer for composites production, because of its excellent electrospinnability, solubility in water, thermal stability, chemical resistance and good processability [30,31,32]. In spite of these advantages, when applied in water, the application of PVA can be limited by its high hydrophilic properties. In order to overcome this problem and improve the thermal and mechanical properties and water resistance of the fibres, PVA can be reinforced with cellulose nanofibrils (CNFs), since both have a great amount of hydroxyl groups, ensuring a satisfactory intermolecular interaction by hydrogen bonding [33, 34]. CNFs have been considered as a promising reinforcing agent, because they show high crystallinity, stiffness, mechanical strength, high thermal stability, low weight, renewability, biodegradability, biocompatibility and non-toxicity [35,36,37]. However, PVA nanofibers frequently need to be modified by chemical or physical crosslinking in order to additionally improve its mechanical stability and reduce water absorption [38,39,40]. The use of a chemical crosslinking agent allows the formation of three-dimensional structure which can prevent the PVA dissolution in water, due to the further decrease in the hydroxyl groups [41]. Among the various crosslinking chemical agents, maleic anhydride (MA) is one of the most effective crosslinking agents to be used for the enhancement of the electrospun PVA stability [42, 43]. The objectives of this work were: (1) to produce composite membranes from PVA and CNFs by electrospinning technique; (2) to compare the individual and combined additivation effects of the polymeric matrix with a natural filler (CNFs) and a chemical crosslinking agent (MA) in the solution parameters (viscosity and electrical conductivity); (3) to study the effect of process variables (applied voltage, feed rate, polymer concentration, tip-to-collector distance and collector type) on the morphology of the electrospun mats; (4) to investigate the reinforcing capability and crosslinking effects at level of thermal behaviour of the best mats; and (5) to characterize the presence of the additives in the matrix by chemical analysis.
2 Materials and methods
2.1 Materials
The CNFs used as reinforcing agent consist of a solution previously prepared and provided by CeNTI (V. N. de Famalicão, Portugal), with a mass concentration of 2%. The CNFs were obtained by enzymatic method. PVA (87–90% hydrolysed) with molecular weight in the range 30–70 kg mol−1, MA with molecular weight equal to 98.06 g mol−1 and sulphuric acid (H2SO4) with 95–97% concentration were provided by Sigma-Aldrich and used without further purification.
2.2 Preparation of solutions
The solutions were prepared by dissolution of PVA powder in distilled water at 80 °C under strong magnetic stirring for approximately 8 h. The PVA was added slowly, and after complete dissolution, each solution was stirred until room temperature was reached. For the preparation of the PVA/MA solutions, a proper amount of H2SO4 was added to the PVA solution to maintain the pH between 2 and 3 and catalyse the esterification reaction between the PVA and MA. After MA addition, the solution was maintained under magnetic stirring. Different CNFs volumetric fractions (yCNF) were added to PVA or PVA/MA solutions. After CNFs addition, the mixture was kept under magnetic stirring for 1 h to ensure a good homogenization. Before the electrospinning tests, solutions were submitted to an ultrasonic dispersing process during 10 min, using a Selecta ULTRASONS H–D equipment (Barcelona, Spain) for a better dispersion of CNFs. The prepared solutions were stored in a refrigerator until use.
2.3 Characterization of solutions
Viscosity and electrical conductivity were measured in a Brookfield viscometer (Fungilab Smart Series Rotational Viscometer, Barcelona, Spain) and in a conductivity meter (Thermo Scientific ORION STAR 4, Waltham, Massachusetts, EUA), respectively. The experiments were performed in three replicates and the results reported as the mean value and standard deviation. The CNFs dispersion in solutions was assessed by optical microscopy with polarized light (Optical Microscope Leica DM 2500 M, Wetzlar, Alemanha), in a dark-field mode, and transmitted light. The images were obtained by a digital image system.
2.4 Electrospinning
The electrospinning experiments were performed in a Nanon NF-103 equipment (MECC, Fukuoka, Japan) at room temperature. A 10 mL syringe and a 22 GA GP.0.16x1.0 needle were used. All the solutions were subjected to preliminary tests with variation in electrospinning parameters. The applied voltage was varied in the range 12–26 kV, feed rate was varied from 0.1 to 0.5 mL h−1, and the needle tip-to-collector distance was varied in the range 11.0–18.5 cm. An aluminium plate and a rotating cylinder were used as fibre collector at 500 rpm. In the first stage of this study, preliminary tests were performed with different concentrations of polymer and reinforcing agent in order to determine the right quantity to be used. The deposition time of the nanofibers in these experiments was 2 h. In the second stage, the equipment parameters (applied voltage, feed rate, tip-to-collector distance, collector type) were studied to develop the best formulations. These tests were performed during 8 h in order to obtain membranes with higher thickness.
2.5 Scanning electron microscopy (SEM)
Morphological analysis of nanofibers was carried out with a XL 30 ESEM Philips equipment (Amsterdam, Nederland), SEM-XL Series. Secondary electron images were taken with an acceleration voltage of 25 kV. Samples were placed in stubs previously prepared with carbon tape and then coated with gold for five cycles, each cycle lasting 60 s, in order to make them conductive. The results allowed to observe the influence of each equipment parameter in the production of fibres from the best formulations developed. Only the mats that showed the best results were selected for characterization by thermogravimetric analysis (TGA), differential scanning calorimetry (DSC) and attenuated total reflectance-Fourier-transform infrared (ATR-FTIR).
2.6 Thermogravimetric analysis (TGA)
The thermogravimetric analysis was carried on a Pyris 1 TGA (PerkinElmer, Waltham, Massachusetts, EUA) equipment. The TGA trace was obtained in the range between room temperature and 900 °C, under nitrogen atmosphere, with a feed rate of 20 mL min−1 and heating rate of 10 °C min−1. The graph was plotted as weight loss percentage versus temperature. CNFs film was obtained by drying the CNFs solution in a Petri dish for 2 days in an oven at 25 °C.
2.7 Differential scanning calorimetry (DSC) analysis
DSC analysis was carried on a PerkinElmer Diamond DSC equipment (Waltham, Massachusetts, EUA), under nitrogen atmosphere, with a feed rate of 20 mL min−1 and heating rate of 20 °C min−1. The thermogram was obtained in the range between − 50 and 200 °C and plotted as heat flow versus temperature. The upper limit was selected based on initial decomposition temperature of PVA as indicated in the product data sheet. The samples were subjected to two heat cycles and one cooling cycle, in order to eliminate the thermal history of the materials.
2.8 Attenuated total reflectance-Fourier-transform infrared spectroscopy (ATR-FTIR)
The qualitative chemical analysis of selected electrospun membranes and the pure CNFs film was performed in an ATR-FTIR spectrophotometer (PerkinElmer Spectrum 100 FTIR, Waltham, Massachusetts, EUA), equipped with a ZnSe crystal. The spectra were acquired in transmittance mode with 16 scans at a resolution of 4 cm−1 and a wavenumber range from 4000 to 600 cm−1.
3 Results and discussion
3.1 Optimization of the solution parameters
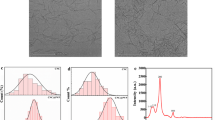
The solution parameters, mainly concentration, viscosity and conductivity, affect the size and shape of nanofibers produced by electrospinning [11]. So, the first objective of this study was the optimization of the PVA-based solutions. Solutions with different concentrations of PVA (8%, 11%, 12%, 14% and 20%) were prepared and tested. The formulation with a mass fraction of xPVA = 20% was rapidly discharged since it was impossible to electrospun due the high viscosity of the solution, which does not allow to control and maintain the PVA solution flow to the tip of the needle, and by the cohesive nature of the solution of PVA at high concentrations [44]. CNFs tested volumes (1%, 3%, 5%, 10%, 20%) showed that no dripping occurs only for mass fraction of yCNF ≤ 3%. Figure 1 allows evaluating the presence and dispersion of CNFs in the suspension samples. Under the polarized light of the optical microscope, the birefringent structures of nanocellulose appear as bright structures on a dark background [45]. In general, cellulose appears to meet an acceptable dispersion for electrospinning tests as it does not cause the solution dripping. However, it becomes necessary to submit the solution to sonication process before the production of composite nanofibers assays to achieve an acceptable dispersion. Regarding the PVA/MA formulation, better results were obtained from the solution with xPVA = 14% and molar ratio zMA = 20:1. Table 1 reports the formulations that were characterized and their electrical conductivity (σ) and viscosity (μ). Preliminary SEM analyses were performed (data not shown) for the electrospun mats to identify which were the solutions with the best spinning capacity. The increase of the xPVA up to 14% caused an increase of σ and μ, allowing more continuous and defect-free fibres. Low polymer concentrations promoted the formation of small drops, resulting in the appearance of beads in the fibre structure due to the influence of surface tension. The beads are considered defects, as they are responsible for reducing the surface area of the electrospun nanofibers [46]. The increase in viscosity above a critical value allowed the loaded jet to be properly stretched under the action of electric forces, whereby the instability of curvature was dominant, resulting in more uniform fibres. On one hand, the addition of CNFs caused a decrease in the measured parameters values. However, the decrease in viscosity was not considered significant due to low amount of CNFs in the total volume of the water suspension. On the other hand, the addition of MA to the solutions caused a significant increase in electrical conductivity. High conductivity in solution causes a higher load-carrying capacity favouring the instability of the curvature, which is the main responsible for the stretch of the fibres [47]. At the same time, the crosslinking process causes an increase in the molar mass of the polymer, resulting in an increase in the solution viscosity. Therefore, the study of the electrospinning parameters was carried out for the following formulations: solution with xPVA = 14%; solution with xPVA = 14% and yCNF = 3%; solution with xPVA = 14% and zMA = 20:1; and solution with xPVA = 14%, zMA = 20:1 and yCNF = 3%.

Optical microscopy images with magnification of × 100 obtained with polarized light from the solution samples of: a suspension of 2% CNFs; b PVA/CNFs formulation with xPVA = 14% and yCNF = 3% (mass concentration of CNFs equal to 0.06%); c PVA/MA/CNFs formulation with xPVA = 14%, zMA = 20:1 and yCNF = 3% (mass concentration of CNFs equal to 0.06%)
3.2 Study of equipment parameters
The effect of the electrospinning parameters on fibres production was evaluated by scanning electron microscopy (SEM). SEM images were obtained with two different magnifications at 1000 × and 10,000 ×. At the first resolution, it was possible to observe a larger area of deposited fibres (Tables S1–S4 in electronic supplementary material). The second resolution allowed an observation of the structure of the product in more detail (Tables 2, 3, 4, 5). The reported average diameters and standard deviations were calculated on the means of at least 30 measurements. (Bead structures were not considered.) The results clearly show the general dissimilarity of the fibres diameters not only among samples but also in the same sample. It is important to remark that the use of the rotating cylinder collector was not optimized to obtain fibre alignment, because the only aim of this study was to obtain continuous and defect-free fibres. Despite it was not possible to obtain completely free-defect membranes, the best result shows few isolate droplets in the mats structure.
Table 2 shows the effect of the equipment parameters on the morphology of the nanofibers produced from the solution with xPVA = 14%. The SEM images suggest that the decrease in the applied voltage significantly improves the structure of the obtained nanofibers (Table 2B). A similar effect was observed decreasing the tip-to-collector distance; however, there is a decrease in the fibre density on the mat surface (Table 2C). When the feed rate is increased, it can be noted a high number of brittle and broken fibres as well as an increased number of beads. This behaviour was intensified using the rotating cylinder collector (Table 2C). Overall, the best mat for xPVA = 14% was obtained at the condition reported in Table 2B (decreasing the applied voltage to 18 kV) showing uniform and almost defect-free nanofibers with diameters of 61 ± 20 nm. The initial applied electrical potential produced high values of the electrostatic and Coulombic repulsion forces. Decreasing the electrostatic force, it was possible to obtain defect-free PVA nanofibers due to the reduction in the speed and mass flow rate of the charged jet, which caused the onset for the bending instability to occur far away from the collector [48, 49].
In Table 3, it is possible to observe the fibres produced from the solution with xPVA = 14% and yCNF = 3%. The introduction of CNFs in the solution improves the formation of defect and beads formation on the mats, with no significant improvements by decreasing the applied voltage. Small improvements can be obtained reducing the tip-to-collector distance or increasing the feed rate. The fibres in these conditions show better stretching and low beads number (Table 3I, H). However, using the rotating cylinder as collector, the obtained fibres show the best results being more uniform and continuous with diameter in the range of 85 ± 39 nm (Table 3J). The successful use of a rotating target to control fibre quality and diameter and fibre alignment was previously reported [50]. The effect of rotation speed of the collector at the applied velocity (500 rpm) increases fibre uniformity and bridging but not alignment in the presence of CNFs due to the higher stretching level imposed to the fibres and to the charge dissipation process [51].
Table 4 presents the nanofibers produced from the solution with xPVA = 14% and zMA = 20:1. The presence of the crosslinker seems to decrease fibre uniformity compared to the PVA control. The decrease in the tip-to-collector distance and the introduction of a rotating cylinder collector favour the beads reduction in the mats (Table 4N, O) leading to a similar result in fibre diameters with values ranging 63 ± 21 nm and 62 ± 29 nm, respectively. The maleic acid in solution is not fully functional as crosslinker diminishing the solution viscosity. The low viscoelastic forces are insufficient to stand the stretching forces from both electrostatic and Coulombic repulsion forces leading to jet instability and beads formation for some of the fibres [52].
Table 5 displays the fibres obtained from the solution with xPVA = 14%, zMA = 20:1 and yCNF = 3%. The decrease in applied voltage caused, and the change in collector showed a significant increase in beads deposition preventing the formation of continuous fibres (Table 5Q, T). On the other hand, changing the tip-to-collector distance produced dripping of the solution dissolving parts of the mats (Table S4r in electronic supplementary material). The best results can be achieved increasing the feed rate. If the feed rate is too low, electrospinning solution cannot be properly stretched into continuous and uniform fibres as previously reported [53]. At this condition, the beads formation was significantly reduced obtaining the highest amount of continuous and almost defect-free fibres with diameters in the range of 74 ± 33 nm (Table 5S).
Preliminary tests of membrane degradation in water (data not shown) indicate that only the MA crosslinked mats (PVA/MA and PVA/CNFs/MA) maintain their structure in after 2 h in water at room temperature under a shaking orbital agitation at 100 rpm.
3.3 Thermogravimetric analysis (TGA)
TGA analysis of the PVA, PVA/CNFs, PVA/MA and PVA/MA/CNFs electrospun mats showed that the degradation takes place in three well-differentiated steps (Fig. 2). The first degradation step occurred until 100 °C for all membranes, which is associated with an initial mass loss due to moisture content [54]. The values corresponding to the mass loss of the samples are approximate values. The pure cellulose nanofibrils film (CNFs) profile showed that the mass loss (61%) occurred mostly in the second step between 337 and 385 °C with a residual mass at 900 °C of 17.3%. This is the highest residual mass obtained among all the sample and can be related to the presence of non-cellulosic components as lignin, which was not degraded during the extraction process [55]. The thermal degradation profile of PVA nanofibers showed that the greatest mass loss, about 80%, occurred within 293–343 °C, which was related to the decomposition of the side chain of the PVA. The third degradation step occurred mainly from 417 to 498 °C, which corresponded to 5.8% of the mass loss. This mass loss may be related to the degradation of the main chain of PVA [54]. The residual mass at 900 °C was of 1.8%. The TGA of the PVA/CNFs mat showed a mass loss, of 78.7%, in the range 302–351 °C with a residual mass at 900 °C of 2%. The CNFs addition seems to have contributed to the thermal stability enhancement of PVA mat. The addition of MA to PVA caused a visible change in the thermal degradation profile, whereby a significant mass loss was observed at two levels. The first level occurred between 271 and 294 °C with a mass loss of 14%. The second level was within 354–450 °C with a mass loss equal to 42%. The first degradation level is very close to the PVA degradation profile due to incomplete PVA crosslinking. However, the second level occurred at higher temperatures than the degradation of PVA mat, suggesting that the crosslinking of PVA with MA promoted an increase in the number of covalent bonds, making the membrane more stable at high temperatures [56]. The residual mass of the sample was of 3.2% at 900 °C. The PVA/MA/CNFs mat profile still showed the presence of the two degradation levels, corresponding in this case to the following: the first level of mass loss, about 15.3%, occurred within 272–300 °C; the second level corresponds to a significant mass loss, equal to 43.0%, within 353–449 °C. The addition of CNFs to crosslinked PVA caused a slight displacement of the first degradation level in direction of higher temperatures, which indicate the presence of CNFs in the matrix. However, this addition had no influence on the second degradation level, so this can only correspond to the mass loss of the crosslinked PVA. The residual mass of the sample was of 0.4% at 900 °C.

Thermal degradation profiles of the electrospun mats, given by sample mass change (m) as a function of temperature increase (T), from room temperature to 900 °C, performed at a heating rate of 10 °C min−1, in a nitrogen atmosphere
The results suggested that the effect of CNFs as reinforcing agent on the thermal stability of the mats is able to overcome the effect of the crosslinking agent in an initial stage of degradation up to about 320 °C. However, from this temperature up to 500 °C, the crosslinked membranes require higher temperatures to be degraded.
3.4 Differential scanning calorimetry (DSC) analysis
Through the DSC technique, the mats were subjected to two cycles of heating and cooling. The first heating (data not shown) was performed in order to remove the thermal history of the material. The results corresponding to the second heating cycle of the samples are shown in Fig. 3. Except the pure CNFs thermograms, which did not show any thermal or transition event, it was possible to observe the existence of a glass transition temperature (Tg) at 75 °C for PVA, 70 °C for PVA/CNFs, 73 °C for PVA/MA and 71 °C for PVA/MA/CNFs mats. It was found that the addition of materials to the PVA matrix leads to a slight decrease in the values of Tg due the loss of the regularity of the –OH groups of PVA because of crosslinking [57]. No other thermal events were detected. The PVA nanofibers produced by electrospinning have no crystalline structure, since during the PVA dissolution in water crystalline structure, destruction occurs. Furthermore, the rapid solidification of the polymer chains, by solvent evaporation, difficult the crystal growth [58]. Thus, the low degree of crystallinity and molecular orientation of PVA can contribute to the absence of crystallization exothermic peaks and melting endotherm peaks [59].

DSC curves of different samples analysed during the second heating cycle, which express the heat flow (Q) versus temperature (T). The information “Endo →” indicates the endothermic direction
3.5 Attenuated total reflectance-Fourier-transform infrared spectroscopy (ATR-FTIR)
The PVA spectrum (Fig. 4) showed dominant absorption peaks at 3323 cm−1, 2941 cm−1, 1733 cm−1, 1428 cm−1, 1090 cm−1 and 844 cm−1, which were, respectively, attributed to the ν(O–H), ν(CH2), ν(C=O), δ(CH–O–H), ν(C–O) and ν(C–C) of pure PVA [54]. The stretching vibration of the OH group corresponds to the intermolecular and intramolecular hydrogen bonds on the PVA chain. The remaining bands identified and assigned to their respective groups (CH2, C=O, CH–O–H, C–O, C–C) were associated with the corresponding groups on the PVA chain structure [58]. It was also obtained a carboxylic band at 1595 cm−1, which was attributed to acetate groups present in the PVA chain, since the polymer is not completely hydrolysed and the band at 844 cm−1 was associated with signals of the main chain of the PVA [59]. In the spectrum of pure CNFs film, it was observed mainly the band at 3332 cm−1, attributed to the stretching vibration of the OH groups, which are abundant in its structure. The PVA/CNFs spectrum showed the characteristics PVA bands because of the low amount of CNFs in the matrix. The main change was observed in the band of stretching of OH groups at 3317 cm−1, which represent the decrease in the PVA-free OH groups interacting with CNFs structure by hydrogen bonds. The addition of MA caused a decrease in the band attributed to the stretching of OH groups at 3307 cm−1. This may be related to a decrease in the OH groups due to crosslinking of the PVA by esterification reaction [59]. The band attributed to the acetate groups in PVA chain at 1595 cm−1 disappeared probably due to the overlap of the acetate groups by analogues of crosslinking agent. Moreover, the appearance of the new low intensity band at 1715 cm−1 can be associated with the C=O carbonyl bond [60]. The PVA/MA/CNFs spectrum showed the same characteristics bands of the PVA/MA spectrum. One possible explanation could be the less available OH groups in the crosslinked PVA chains to interact with CNFs [59].

FTIR spectra obtained by analysis of the membranes produced by electrospinning. The spectrum is represented by the transmittance (T) (in arbitrary units) as a function of wavenumber (\(\tilde{v}\)). The shaded area corresponds to the portion of the spectrum where the most significant changes were detected between the different samples
4 Conclusion
The best crosslinked electrospun PVA mats containing CNFs were obtained with a PVA mass fraction equal to 14%, CNFs volumetric fraction of 3% and maleic acid at the molar ratio 20:1 at the applied voltage of 24 kV, needle tip-to-collector distance of 14.5 cm, feed rate of 0.3 mL h−1 and using a plate collector. At this condition, the beads formation was limited but not totally excluded and it was possible to obtain nanofibers with a diameters of 74 ± 33 nm. CNFs and MA addition allows to improve nanofiber thermal properties of the mats. It seems that the presence of CNFs as reinforcing agent is able to compete in terms of thermal properties to the crosslinking agent effect decreasing the Tg due the loss of the regularity of the –OH groups and increasing the degradation temperature. However, despite this similar behaviour, only the MA crosslinked mat does not degrade in water due to the stabilizing effect of the newly formed carbonyl bonds as confirmed by FTIR analysis. A systematic mechanical characterization of the membranes it will be necessary to confirm if the expected mechanical improvement is due to the addition of CNFs to the matrix. These crosslinked PVA/CNFs/MA membranes with higher stability can be considered an eco-friendlier, biocompatible and biodegradable final product for filtration processes.
References
Ramesh M, Palanikumar K, Reddy KH (2017) Plant fibre based bio-composites: sustainable and renewable green materials. Renew Sustain Energy Rev 79:558–584. https://doi.org/10.1016/j.rser.2017.05.094
Ramamoorthy SK, Skrifvars M, Persson A (2015) A review of natural fibers used in biocomposites: plant, animal and regenerated cellulose fibers. Polym Rev 55(1):107–162. https://doi.org/10.1080/15583724.2014.971124
Douglass EF, Avci H, Boy R, Rojas OJ, Kotek R (2017) A review of cellulose and cellulose blends for preparation of bio-derived and conventional membranes, nanostructured thin films, and composites. Polym Rev 58(1):102–163. https://doi.org/10.1080/15583724.2016.1269124
Ummartyotin S, Manuspiya H (2015) A critical review on cellulose: from fundamental to an approach on sensor technology. Renew Sustain Energy Rev 41:402–412. https://doi.org/10.1016/j.rser.2014.08.050
Agate S, Joyce M, Lucia L, Pal L (2018) Cellulose and nanocellulose-based flexible-hybrid printed electronics and conductive composites—a review. Carbohyd Polym 198:249–260. https://doi.org/10.1016/j.carbpol.2018.06.045
Trache D, Hussin MH, Hui Chuin CT, Sabar S, Fazita MRN, Taiwo OFA, Hassan TM, Haafiz MKM (2016) Microcrystalline cellulose: isolation, characterization and bio-composites application—a review. Int J Biol Macromol 93:789–804. https://doi.org/10.1016/j.ijbiomac.2016.09.056
Oksman K, Aitomäki Y, Mathew AP, Siqueira G, Zhou Q, Butylina S, Tanpichai S, Zhou X, Hooshmand S (2016) Review of the recent developments in cellulose nanocomposite processing. Compos A Appl Sci Manuf 83:2–18. https://doi.org/10.1016/j.compositesa.2015.10.041
Nechyporchuk O, Belgacem MN, Bras J (2016) Production of cellulose nanofibrils: a review of recent advances. Ind Crops Prod 93:2–25. https://doi.org/10.1016/j.indcrop.2016.02.016
Ribeiro RSA, Pohlmann BC, Calado V, Bojorge N, Pereira N (2019) Production of nanocellulose by enzymatic hydrolysis: trends and challenges. Eng Life Sci 19(4):279–291. https://doi.org/10.1002/elsc.201800158
Karim Z, Afrin S, Husain Q, Danish R (2016) Necessity of enzymatic hydrolysis for production and functionalization of nanocelluloses. Crit Rev Biotechnol 37(3):355–370. https://doi.org/10.3109/07388551.2016.1163322
Haider A, Haider S, Kang I-K (2018) A comprehensive review summarizing the effect of electrospinning parameters and potential applications of nanofibers in biomedical and biotechnology. Arab J Chem 11(8):1165–1188. https://doi.org/10.1016/j.arabjc.2015.11.015
Ahmed FE, Lalia BS, Hashaikeh R (2015) A review on electrospinning for membrane fabrication: challenges and applications. Desalination 356:15–30. https://doi.org/10.1016/j.desal.2014.09.033
Khorshidi S, Solouk A, Mirzadeh H, Mazinani S, Lagaron JM, Sharifi S, Ramakrishna S (2016) A review of key challenges of electrospun scaffolds for tissue-engineering applications. J Tissue Eng Regen Med 10(9):715–738. https://doi.org/10.1002/term.1978
Ray SS, Chen S-S, Li C-W, Nguyen NC, Nguyen HT (2016) A comprehensive review: electrospinning technique for fabrication and surface modification of membranes for water treatment application. RSC Adv 6(88):85495–85514. https://doi.org/10.1039/c6ra14952a
Mirjalili M, Zohoori S (2016) Review for application of electrospinning and electrospun nanofibers technology in textile industry. J Nanostruct Chem 6(3):207–213. https://doi.org/10.1007/s40097-016-0189-y
Chinnappan A, Baskar C, Baskar S, Ratheesh G, Ramakrishna S (2017) An overview of electrospun nanofibers and their application in energy storage, sensors and wearable/flexible electronics. J Mater Chem C 5(48):12657–12673. https://doi.org/10.1039/c7tc03058d
Lundahl MJ, Klar V, Wang L, Ago M, Rojas OJ (2016) Spinning of cellulose nanofibrils into filaments: a review. Ind Eng Chem Res 56(1):8–19. https://doi.org/10.1021/acs.iecr.6b04010
Khoshnevisan K, Maleki H, Samadian H, Shahsavari S, Sarrafzadeh MH, Larijani B, Dorkoosh FA, Haghpanah V, Khorramizadeh MR (2018) Cellulose acetate electrospun nanofibers for drug delivery systems: applications and recent advances. Carbohyd Polym 198:131–141. https://doi.org/10.1016/j.carbpol.2018.06.072
Jedvert K, Heinze T (2017) Cellulose modification and shaping—a review. J Polym Eng 37(9):845–860. https://doi.org/10.1515/polyeng-2016-0272
Wang Y, Wang X, Xie Y, Zhang K (2018) Functional nanomaterials through esterification of cellulose: a review of chemistry and application. Cellulose 25(7):3703–3731. https://doi.org/10.1007/s10570-018-1830-3
Ohkawa K (2015) Nanofibers of cellulose and its derivatives fabricated using direct electrospinning. Molecules 20(5):9139–9154. https://doi.org/10.3390/molecules20059139
Peres BU, Vidotti HA, de Carvalho LD, Manso AP, Ko F, Carvalho RM (2019) Nanocrystalline cellulose as a reinforcing agent for electrospun polyacrylonitrile (PAN) nanofibers. J Oral Biosci 61(1):37–42. https://doi.org/10.1016/j.job.2018.09.002
Hivechi A, Bahrami SH, Siegel RA (2019) Drug release and biodegradability of electrospun cellulose nanocrystal reinforced polycaprolactone. Mater Sci Eng C 94:929–937. https://doi.org/10.1016/j.msec.2018.10.037
Enayati MS, Behzad T, Sajkiewicz PŁ, Bagheri R, Ghasemi-Mobarakeh L, Pierini F (2017) Theoretical and experimental study of the stiffness of electrospun composites of poly(vinyl alcohol), cellulose nanofibers, and nanohydroxy apatite. Cellulose 25(1):65–75. https://doi.org/10.1007/s10570-017-1601-6
Ras RHA, Tian X, Bayer IS (2017) Superhydrophobic and superoleophobic nanostructured cellulose and cellulose composites. In: Kargarzadeh H, Ahmad I, Thomas S, Dufresne A (eds) Handbook of nanocellulose and cellulose nanocomposites. Wiley, New York, pp 731–760. https://doi.org/10.1002/9783527689972.ch22
Wanasekara ND, Santos RPO, Douch C, Frollini E, Eichhorn SJ (2015) Orientation of cellulose nanocrystals in electrospun polymer fibres. J Mater Sci 51(1):218–227. https://doi.org/10.1007/s10853-015-9409-y
Zhang Z, Li Y, Chen C (2017) Synergic effects of cellulose nanocrystals and alkali on the mechanical properties of sisal fibers and their bonding properties with epoxy. Compos A Appl Sci Manuf 101:480–489. https://doi.org/10.1016/j.compositesa.2017.06.025
Yang W, Fortunati E, Dominici F, Giovanale G, Mazzaglia A, Balestra GM, Kenny JM, Puglia D (2016) Synergic effect of cellulose and lignin nanostructures in PLA based systems for food antibacterial packaging. Eur Polym J 79:1–12. https://doi.org/10.1016/j.eurpolymj.2016.04.003
Ben Halima N (2016) Poly(vinyl alcohol): review of its promising applications and insights into biodegradation. RSC Adv 6(46):39823–39832. https://doi.org/10.1039/c6ra05742j
Aslam M, Kalyar MA, Raza ZA (2018) Polyvinyl alcohol: a review of research status and use of polyvinyl alcohol based nanocomposites. Polym Eng Sci 58(12):2119–2132. https://doi.org/10.1002/pen.24855
Quinn JA, Yang Y, Buffington AN, Romero FN, Green MD (2018) Preparation and characterization of crosslinked electrospun poly(vinyl alcohol) nanofibrous membranes. Polymer 134:275–281. https://doi.org/10.1016/j.polymer.2017.11.023
Kamoun EA, Chen X, Mohy Eldin MS, Kenawy E-RS (2015) Crosslinked poly(vinyl alcohol) hydrogels for wound dressing applications: a review of remarkably blended polymers. Arab J Chem 8(1):1–14. https://doi.org/10.1016/j.arabjc.2014.07.005
Miao X, Tian F, Lin J, Li H, Li X, Bian F, Zhang X (2016) Tuning the mechanical properties of cellulose nanofibrils reinforced polyvinyl alcohol composites via altering the cellulose polymorphs. RSC Adv 6(86):83356–83365. https://doi.org/10.1039/c6ra14517e
Ghorbel N, Kallel A, Boufi S (2019) Molecular dynamics of poly(vinyl alcohol)/cellulose nanofibrils nanocomposites highlighted by dielectric relaxation spectroscopy. Compos A Appl Sci Manuf. https://doi.org/10.1016/j.compositesa.2019.05.033
Kim J-H, Shim BS, Kim HS, Lee Y-J, Min S-K, Jang D, Abas Z, Kim J (2015) Review of nanocellulose for sustainable future materials. Int J Precis Eng Manuf Green Technol 2(2):197–213. https://doi.org/10.1007/s40684-015-0024-9
Benítez AJ, Walther A (2017) Cellulose nanofibril nanopapers and bioinspired nanocomposites: a review to understand the mechanical property space. J Mater Chem A 5(31):16003–16024. https://doi.org/10.1039/c7ta02006f
Rol F, Belgacem MN, Gandini A, Bras J (2019) Recent advances in surface-modified cellulose nanofibrils. Prog Polym Sci 88:241–264. https://doi.org/10.1016/j.progpolymsci.2018.09.002
Cui Z, Zheng Z, Lin L, Si J, Wang Q, Peng X, Chen W (2018) Electrospinning and crosslinking of polyvinyl alcohol/chitosan composite nanofiber for transdermal drug delivery. Adv Polym Technol 37(6):1917–1928. https://doi.org/10.1002/adv.21850
Jain N, Singh VK, Chauhan S (2018) A review on mechanical and water absorption properties of polyvinyl alcohol based composites/films. J Mech Behav Mater. https://doi.org/10.1515/jmbm-2017-0027
Tian H, Yuan L, Wang J, Wu H, Wang H, Xiang A, Ashok B, Rajulu AV (2019) Electrospinning of polyvinyl alcohol into crosslinked nanofibers: an approach to fabricate functional adsorbent for heavy metals. J Hazard Mater. https://doi.org/10.1016/j.jhazmat.2019.120751
Niu Y, Zhang X, He X, Zhao J, Zhang W, Lu C (2015) Effective dispersion and crosslinking in PVA/cellulose fiber biocomposites via solid-state mechanochemistry. Int J Biol Macromol 72:855–861. https://doi.org/10.1016/j.ijbiomac.2014.09.042
Truong YB, Choi J, Mardel J, Gao Y, Maisch S, Musameh M, Kyratzis IL (2017) Functional cross-linked electrospun polyvinyl alcohol membranes and their potential applications. Macromol Mater Eng. https://doi.org/10.1002/mame.201700024
Samnani M, Rathod H, Raval H (2018) A novel reverse osmosis membrane modified by polyvinyl alcohol with maleic anhydride crosslinking. Mater Res Express. https://doi.org/10.1088/2053-1591/aab0e4
Thenmozhi S, Dharmaraj N, Kadirvelu K, Kim HY (2017) Electrospun nanofibers: new generation materials for advanced applications. Mater Sci Eng B 217:36–48. https://doi.org/10.1016/j.mseb.2017.01.001
Deepa B, Abraham E, Cordeiro N, Mozetic M, Mathew AP, Oksman K, Faria M, Thomas S, Pothan LA (2015) Utilization of various lignocellulosic biomass for the production of nanocellulose: a comparative study. Cellulose 22(2):1075–1090. https://doi.org/10.1007/s10570-015-0554-x
Wong D, Andriyana A, Ang BC, Chan YR, Lee JJL, Afifi AM, Verron E (2016) Surface morphology analysis and mechanical characterization of electrospun nanofibrous structure. Key Eng Mater 701:89–93. https://doi.org/10.4028/www.scientific.net/KEM.701.89
Angammana CJ, Jayaram SH (2015) Fundamentals of electrospinning and processing technologies. Part Sci Technol 34(1):72–82. https://doi.org/10.1080/02726351.2015.1043678
Supaphol P, Chuangchote S (2008) On the electrospinning of poly(vinyl alcohol) nanofiber mats: a revisit. J Appl Polym Sci 108(2):969–978. https://doi.org/10.1002/app.27664
Ding B, Kim H-Y, Lee S-C, Shao C-L, Lee D-R, Park S-J, Kwag G-B, Choi K-J (2002) Preparation and characterization of a nanoscale poly(vinyl alcohol) fiber aggregate produced by an electrospinning method. J Polym Sci B Polym Phys 40(13):1261–1268. https://doi.org/10.1002/polb.10191
Medeiros ES, Mattoso LHC, Ito EN, Gregorski KS, Robertson GH, Offeman RD, Wood DF, Orts WJ, Imam SH (2008) Electrospun nanofibers of poly(vinyl alcohol) reinforced with cellulose nanofibrils. J Biobased Mater Bioenergy 2(3):231–242. https://doi.org/10.1166/jbmb.2008.411
Park S, Park K, Yoon H, Son J, Min T, Kim G (2007) Apparatus for preparing electrospun nanofibers: designing an electrospinning process for nanofiber fabrication. Polym Int 56(11):1361–1366. https://doi.org/10.1002/pi.2345
Cai J, Chen J, Zhang Q, Lei M, He J, Xiao A, Ma C, Li S, Xiong H (2016) Well-aligned cellulose nanofiber-reinforced polyvinyl alcohol composite film: mechanical and optical properties. Carbohyd Polym 140:238–245. https://doi.org/10.1016/j.carbpol.2015.12.039
Zhang Z, Wu Y, Wang Z, Zou X, Zhao Y, Sun L (2016) Fabrication of silver nanoparticles embedded into polyvinyl alcohol (Ag/PVA) composite nanofibrous films through electrospinning for antibacterial and surface-enhanced Raman scattering (SERS) activities. Mater Sci Eng C 69:462–469. https://doi.org/10.1016/j.msec.2016.07.015
Santos C, Silva CJ, Büttel Z, Guimarães R, Pereira SB, Tamagnini P, Zille A (2014) Preparation and characterization of polysaccharides/PVA blend nanofibrous membranes by electrospinning method. Carbohyd Polym 99:584–592. https://doi.org/10.1016/j.carbpol.2013.09.008
Chirayil CJ, Joy J, Mathew L, Mozetic M, Koetz J, Thomas S (2014) Isolation and characterization of cellulose nanofibrils from Helicteres isora plant. Ind Crops Prod 59:27–34. https://doi.org/10.1016/j.indcrop.2014.04.020
Song M, Yu H, Gu J, Ye S, Zhou Y (2018) Chemical cross-linked polyvinyl alcohol/cellulose nanocrystal composite films with high structural stability by spraying Fenton reagent as initiator. Int J Biol Macromol 113:171–178. https://doi.org/10.1016/j.ijbiomac.2018.02.117
Sreedhar B, Sairam M, Chattopadhyay DK, Rathnam PAS, Rao DVM (2005) Thermal, mechanical, and surface characterization of starch-poly(vinyl alcohol) blends and borax-crosslinked films. J Appl Polym Sci 96(4):1313–1322. https://doi.org/10.1002/app.21439
Jankovic B, Pelipenko J, Skarabot M, Musevic I, Kristl J (2013) The design trend in tissue-engineering scaffolds based on nanomechanical properties of individual electrospun nanofibers. Int J Pharm 455(1–2):338–347. https://doi.org/10.1016/j.ijpharm.2013.06.083
Peresin MS, Vesterinen AH, Habibi Y, Johansson LS, Pawlak JJ, Nevzorov AA, Rojas OJ (2014) Crosslinked PVA nanofibers reinforced with cellulose nanocrystals: water interactions and thermomechanical properties. J Appl Polym Sci. https://doi.org/10.1002/app.40334
Mansur HS, Sadahira CM, Souza AN, Mansur AAP (2008) FTIR spectroscopy characterization of poly (vinyl alcohol) hydrogel with different hydrolysis degree and chemically crosslinked with glutaraldehyde. Mater Sci Eng C 28(4):539–548. https://doi.org/10.1016/j.msec.2007.10.088
Acknowledgements
The authors acknowledge the Portuguese Foundation for Science and Technology (FCT) for funding the project UID/CTM/00264/2013 and A. Zille contract IF/00071/2015.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Braga, C., Sampaio, A.R., Peixoto, J. et al. Effects of cellulose nanofibrils on the structure and properties of maleic anhydride crosslinked poly(vinyl alcohol) electrospun nanofibers. SN Appl. Sci. 1, 1640 (2019). https://doi.org/10.1007/s42452-019-1706-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-019-1706-7