
Abstract
Citrus fruits and their by-products such as concentrated juices and essential oils are important intermediate by-products in the food industry that can selectively accumulate plant protection agrochemicals employed in their production. They are very difficult matrices for pesticide residue analysis due their high number and concentration of phytochemicals that could hamper the determinations. The fruit processing leads to the concentration/dilution or elimination of some of these natural products that change totally the nature of the matrix where the pesticides partition unevenly. Looking at the industrial process of the fruit, a unified vision for the pesticide residues analysis throughout the lemon fruit chain production, useful for the routine analysis of the above mentioned three matrices is presented. The driven concept is the minimization of matrix effects through sample dilution of the concentrated by-products, either after sample treatment or not. This approach will contribute to the maintenance of the whole instrumental system. QuEChERS AOAC 2007.01 was selected as the most suitable protocol for routine determination of, residues of 16 the pesticides most commonly used in the fruits during the citrus production through liquid chromatography coupled to tandem mass spectrometry (LC–MS/MS). The same protocol was applied for pesticide residue analysis in concentrated juice after diluting 4 times the sample to minimize the matrix effects. For the analysis of lemon essential oils, the dilution and shoot procedure proved to be useful for LC–MS/MS and gas chromatography–mass spectrometry determination. The three methodologies were validated following SANTE guidelines, with quantitation limits below the established European Union and Codex Alimentarius maximum residue limits. The developed methodologies are useful tools for the routine control analysis of pesticide residues in lemon matrices, allowing high sample throughput and enhancing labs productivity.
Similar content being viewed by others
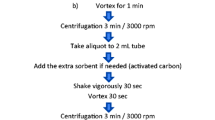

1 Introduction
Citrus fruits and their by-products chain of value are consumed throughout the entire world. The global citrus production involves around 124,246.0 thousand tons [1]. Nowadays in Uruguay, citrus production represents 2% of the gross value of agricultural production products [2] in two large producing areas that differ in terms of surface, production specificity and scale of production. Particularly, southern Uruguay is specialized in the cultivation of lemon as the main species that is consumed either as fruit or processed for juices, juice concentrates, oils, pectin base and pellets for animal feed [2].
In order to preserve the fruit quality and avoid pests attack, citrus cultivation commonly involves the use of specific technological packages but many pesticide residues remain on the different citrus species even though there are many biological, physicochemical and mechanical factors that can cause their dissipation [3, 4]. These residues can threaten human health but also hamper international trade, forcing the monitoring of the remaining residues in both, fruits and products derived from the citrus chain production. The omnipresence of pesticide residues in citrus products was pointed out as over a total of 8920 analyzed citrus fruits in South Africa, 84% contained traces of imidazole-benzimidazole fungicides and 14% of organophosphorus insecticides [5]. In China, imazalil and thiabendazole residues were determined in 19 orange and 21 mandarin samples using UHPLC-QTOF-MS and automated MS/MS library searching [6]. Imazalil was found in all the samples from different countries of a common, internationally sold carbonated orange juice except the sample of US where no natural juice is used in this production [7] Furthermore, the European Pharmacopoeia considers that pesticide residues analysis should be carried out in cold pressed essential oils,such as the citric ones, due to the potential harm they pose when included in pharmaceutical formulations [8].
Several analytical methodologies have been developed for the determination of pesticide residues in mandarins, oranges and other citrus fruits [9,10,11,12], but only a few studied systematically lemon as a matrix. Most of these reports cover the analysis of pesticide residues of imazalil, thiabendazole, carbendazim and 2-phenylphenol in lemon fruit [13,14,15,16]. In every case, the complexity of citrus fruits as an analytical matrix for pesticide residue analysis had been pointed out, either using LC–MS or GC–MS determinations [17, 18]. Citrus fruits have been analyzed for pesticide residues through a multitude of different approaches. Nowadays, the most commonly applied are those based in QuEChERS templates, which is a miniaturized sample preparation scheme, particularly useful to be coupled to mass spectrometry detectors. The idea is that, after extraction with a suitable solvent, being ACN the most popular (Ethyl Acetate and Acetone can also be used), a dispersive step of the organic solvent is performed using salts and absorbents that try to minimize the co-extractives load directed to the instrumental system. This point stems for the most common problem faced when studying pesticide residues analysis in citrus fruits, the matrix effects. Roughly, a QuEChERS citrus extract for pesticide residue analysis contains up to 3000 co-extracted compounds, some of them at high concentration levels that coelute with the analytes along the chromatographic run and affects or interfere with the residue determinations. Several studies on matrix effects during LC–MS/MS either QqQ or ToF [17, 19, 20] have been reported. They all show the strong incidence of co-extractives in pesticide residues determination.
But also the original QuEChERS approach proved to be unsatisfactory for citrus fruit pesticide residues analysis as some widely used pesticides in citrus production could not be determined properly. That is the case of pre-harvest fungicides (captan and folpet) or post-harvest ones, such as imazalil or prochloraz. The high acidity of the matrix either protonates the azole hampering their quantitative extraction by the organic solvent or facilitates their degradation in the low pH of comminuted citrus fruits. Two main variations based on pH adjustment of the extracting solution were developed that are now the official methods in USA (AOAC2007.01) and EU (CEN 15662) respectively. The main difference between both methods is the final pH for extraction that is reached using different buffering systems generated in situ. In AOAC 2007.01, the acetate buffer reaches a pH of 4.5–5 whereas in the CEN method the citrate buffer yields a pH of 5–5.55 getting an extraction of pH dependent or sensitive pesticides. Several adsorbents have been used for PRA in citrus fruits, such as ZrO2, Zsep, carbon nanotubes, alumina and florisil, combined or not with the traditional QuEChERS absorbents PSA; RPC18 and GCB, with different success [18]. None of them proved to be better enough to replace them in pesticide residue studies in citrus fruits.
Within the citrus chain production, fresh juice, concentrated juice, essential oils, and pellets are elaborated, among other goods. The basis for concentrated juice production is to increase the dry matter content and decrease the water content of juices, in order to extend shelf life and improve transportation and storage properties [21]. On an industrial scale, essential oils are obtained as a by-product of fruit juice production [22]. The presence, distribution and persistence of pesticide residues in these by-products are due to the production process and the physicochemical properties of those compounds [23].
In the case of citrus by-products, different methodologies have been developed for the determination of pesticide residues in citrus juices [16, 24, 25], while few works report methodologies for pesticide residues determination in concentrated lemon juice, a very acid matrix [26, 27]. On the other hand, essential oils (EOs) are extracted from citrus peel and the concentration of lipophilic pesticides is much higher than that found in the fruit [28]. Pesticide residues in EOs are widely studied by different researchers who have found organochlorine [29] and organophosphorus residues [30] among others. A review of the presence and evolution of pesticides and plasticizers in Italian essential oils according the type of citrus EO was published by Saitta and his group in 2012 [31]. The developed sample preparations for EOs are diverse; from solid/liquid extraction either using cartridge based SPE or Matrix Solid Dispersion (MSPD) [28, 32] o a sample dilution step after oil evaporation as applied by Fillatre et al. [33].
The aim of this work was to develop and validate suitable analytical methodologies to be applied routinely throughout the lemon fruit and by-products productive chain, like the concentrated juice and the essential oil.
2 Materials and methods
2.1 Reagents and apparatus
HPLC grade ethyl acetate (EtOAc) and acetonitrile (MeCN), free of pesticide residues, were purchased from Pharmco Products Inc. (Brookfield, CT. USA) and Sigma-Aldrich (Steinheim, Germany), respectively. These solvents were employed both in liquid chromatography and during the extraction step. Sodium citrate dihydrate and anhydrous magnesium sulphate from J.T.Mallinckrodt Baker Inc. (Phillipsburg, NJ, USA) were used. Sodium chloride and sodium acetate were obtained from Carlo Erba Reagents S.A.S (Val de Reuil Cedex, Italy). Di-sodium hydrogen citrate 1.5 hydrate was purchased from Scharlau (Barcelona, Spain).
Deionized water was obtained using a Thermo Scientific (Marietta, OH, USA) EASY pure RoDi Ultrapure water purification system. Formic acid p.a. 88% was obtained from Macron Chemicals (The Netherlands). Glacial acetic acid (HAc) from Dorwill (Buenos Aires, Argentina), RP-C18, amino sorbent (PSA, 40–60 μm) and graphitized carbon black (GCB, 120–400 mesh) were from Scharlau (Barcelona, Spain).
Reference standards (purity > 98%) were purchased from Dr. Ehrenstofer (Augsburg, Germany) and were stored at − 40 °C. Stock solutions (1000–2000 µg mL−1) of each pesticide were prepared by dissolving the standards in acetonitrile or ethyl acetate and stored in glass vials at − 40 °C. A working solution at 10 µg mL−1, was prepared by diluting stock solutions.
A SL16 centrifuge from Thermo IEC HN-SII (Langenselbold, Germany), a vortex mixer Wisd VM 10, a Turbovap® Biotage LV evaporator (Charlotte, NC, United States) and an ultrasonic bath Wisd WUC-A03H from Daihan Scientific Co. Ltda. (Gangwon-do, Korea) were used. Analytical balances were SHIMADZU AUX220, readability, 0.1 mg (Kyoto, Japan) and a SIMADZU TXB622L, readability 0.01 g (Cebu, Philippines).
2.2 Instrumentation
2.2.1 LC–MS/MS operating conditions
The HPLC–MS/MS analysis was performed with a HPLC Agilent 1200, equipped with a quaternary pump, degasser and a thermostatted autosampler coupled to a triple quadrupole API 4000 (4000 Q-TRAP ABSCIEX). Electrospray ionization (ESI) was used in positive and negative mode and the triple quadrupole operated in MRM (Multiple Reaction Monitoring). The column used was a Zorbax Eclipse XDB-C18, 150 mm long, 4.6 mm i.d. and 5 µm particle size, which was kept at 40 °C during the analysis.
Methods 1 and 2 used gradients of water with 0.1% HAc (A) and acetonitrile (B) and operated in positive and negative electrospray ionization mode respectively. All methods had a 0.6 mL min−1 mobile phase flow and 5 µL of each sample were injected.
The elution process for each chromatographic method was different with method 1 starting with 70% of mobile phase A, which was dropped to 0% in 12 min (3.50 min hold) then it rose back to 70% in 1.50 min (4 min hold) for a total run time of 21 min, while method 2 started with 70% of mobile phase A (1 min hold), dropped to 0% in 3.50 min then rose back to 70% in 2.50 min (3 min hold) for a total run time of 10 min. The source temperature was set to 500 °C, ionization voltage was 5000 V, a nitrogen curtain at 20 psi and air as nebulization gas at 50 psi were used. A retention time window of 90 s was set for each pesticide and data analysis was performed with Biosystems Analyst 1.5 software. The mass spectrometer conditions are presented in Table 1.
2.2.2 GC–MS operating conditions
GC–MS pesticide analysis was performed on a Shimadzu GC–MS QP2010 operating in electron impact ionization (EI) mode with ionization energy of 70 eV. The injection volume was 1 µL of each sample in split mode at 280 °C to an Rtx-5MS fused silica capillary column (30 m × 0.25 mm diameter and film thickness of 0.25 µm) with ultrapure helium as gas carrier. The interface and ion source were set at 280 and 230 °C respectively. The oven temperature gradient started at 120 °C for 5 min, increased to 190 °C with a 10 °C min−1 rate (1 min hold), then to 250 °C at a 5 °C min−1 rate (5 min hold) and finally increased to 300 at a 5 °C min−1 rate (5 min hold) for a total run time of 45 min. Detection was performed in SIM (Single Ion Monitoring) acquisition mode. GC data was processed with GC–MS Solution software 4.11 SU2 version. Pesticides detection parameters for both analytical systems are shown in Table 1.
2.3 Sample preparation
2.3.1 Lemon fruit and concentrated juice
2.3.1.1 Preliminary tests
Ethyl acetate sample preparation protocol (EtOAc method) [3] and acetate QuEChERS AOAC [34] using in both cases 10 g of previously homogenized sample were evaluated. The sample preparation of QuEChERS AOAC is described in concentrated juice session.
For the EtOAc method, the sample preparation consisted in weighing 10 g of sample into 50 mL PP conic tubes where 10 mL of EtOAc was added and skaked by hand for 1 min. The salting-out was performed with 8 g of anhydrous MgSO4 and 1.5 g of NaCl. It was then shaken for 5 min manually, placed in an ultrasonic bath for 15 min and centrifuged for 5 min at 2264×g. In this case, no sample clean up step was applied.
For the concentrated juice, 3 extraction methodologies based on the different variations of the QuEChERS method were evaluated: the original one [35], the buffered versions [34, 36]. Conditions were then optimized for validation of the chosen method.
Using the original version of QuEChERS [35], 5 g of concentrated juice were weighed in a 50 mL tube and then 5 g of distilled water was added. After that, 10 mL of MeCN were added and shaken manually for 1 min. Then, 4 g of anhydrous MgSO4 and 1 g of NaCl were added. It was then shaken manually for 5 min and placed on an ultrasonic bath for 15 min. Finally, the tubes were centrifuged for 10 min at 4620×g. Afterwards, part of the supernatant was transferred to 15 mL PP disposable tubes with previously weighed sorbents: 25 mg of PSA and 150 mg of anhydrous MgSO4 per milliliter of supernatant.
For the citrate QuEChERS version [36], a similar procedure was performed, differing in the formation of a buffer in situ. The main difference is the usage of 1 g of NaCl, 4 g of anhydrous MgSO4, 1 g trisodium citrate dehydrate and 0.5 g disodium hydrogencitrate sesquihydrate as extraction salts. For acetate QuEChERS (AOAC) [34] the procedure is similar to the first method, changing the extraction solvent to MeCN acidified with 1% of HAc. Another difference is the use of 1 g of sodium acetate and 4 g of anhydrous MgSO4 as phase partition salts.
For both modified QuEChERS, during the clean-up step, 150 mg of anhydrous MgSO4 and 50 mg of PSA were used per milliliter of supernatant.
In all cases, for the LC–MS/MS analysis, 1 mL of the supernatant was taken and filtered through a 22 µm syringe filtered and 500 µL were placed in an autosampler vial for further analysis.
2.3.1.2 Final sample preparation methods
In order to obtain an homogenized samples of lemon fruit, each randomly selected fruit was cut in four pieces, two opposite quarters were selected for analysis and the other two were discarded as reported in previous work [3]. The laboratory sample was then composed and homogenized.
For both fruit and concentrated juice the acetate QuEChERS (AOAC) [34] method was used. For lemon, the procedure started with weighing 10 g of the previously homogenized sample into a 50 mL tube, after that, 10 mL of MeCN acidified with 1% HAc were added and shaken manually for 1 min. Then, 4 g of anhydrous MgSO4 and 1 g of sodium acetate were added. They were then shaken manually for 5 min and placed on an ultrasonic bath for 15 min. Finally, the tubes were centrifuged for 10 min at 4620×g. Afterwards, part of the supernatant was transferred to 15 mL PP disposable tubes with previously weighed sorbents: 150 mg of anhydrous MgSO4 and 50 mg of PSA were used per milliliter of supernatant.
For concentrated juice, the sample preparation step was modified from the original AOAC method, instead of 10 g of sample, 2.5 g of concentrated juice were weighted and 7.5 g of water were added. Apart from that, the method remained the same.
In both cases, for the LC–MS/MS analysis, 1 mL of the supernatant was taken and filtered through a 22 µm syringe filter and 500 µL were placed in an autosampler vial for further analysis.
2.3.2 Essential oils
For the pesticide residues determination on essential oils, a dilution x5 of the sample and subsequent direct injection protocol was used. First, the sample was vortexed and filtered through a 0.45 µm syringe filter, an aliquot of 200 µL was taken and put into an autosampler vial then 800 µL of the appropriated solvent: MeCN or EtOAc for LC and GC analysis respectively were added.
For precision studies, lemon essential oils that had been spiked with the selected pesticides at concentrations ranging from 5 to 200 µg L−1 were diluted with 800 µL of the appropriate solvent. Due to the complexity of essential oils and that the composition could be different according to the extraction method used, the citrus type [22] and the geographical areas from where the fruit was grown [37] plus the difficulties of finding a blank sample, the standard addition calibration technique was used for quantification purposes. To accomplish this, instead of using 800 µL of solvent, the appropriate amount of pesticide mix was used together with the needed solvent to reach 800 µL.
The trueness of the methods was evaluated as recovery percentages of all pesticides at 100 μg kg−1 concentration level. The spike process was performed with a mix of pesticides prepared from standards solutions of each pesticide.
2.4 Validation procedure
A thorough and complete method validation for assaying 17 pesticides in lemon matrices was done following the SANTE guidelines for the Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed [38]. In order to validate the methods, parameters as selectively, trueness, repeatibility, linearity, matrix effect, and quantification limit were studied. The selectivity of the method was determined by measuring the level of interfering components in a lemon blank (a fruit free of pesticides). Trueness was estimated as the average of five replicates percentage of recovery for each studied pesticide. The quantification was performed using matrix matched calibration curves and matrix effect (ME) percentage was studied by comparison of the slopes of the calibration curves in solvent and in matrix and calculated as:
In order to minimize matrix effects, the linearity was studied by matrix-matched standard calibrations. The quantification limit (LOQ) was established as the minimum value where the trueness and precision are in accordance with the SANTE guidelines criteria for a given compound (recoveries percentage from 70 to 120% and relative standard deviation lower than 20%) [38].
2.5 Real samples
A total of 6 samples of lemon fruit, 5 of concentrated juice and 101 of essential oils were analyzed. Fruits were purchased from different local markets in Paysandú (Uruguay) and as well as essential oils. The concentrated lemon juices were provided by a local industry. All samples were stored in their original packaging under the recommended conditions until use.
3 Results and discussion
For the evaluation of the best performance of the analytical methods, a representative group of 17 pesticides belonging to different chemical families were selected. The choosing of the pesticides to be analyzed was based, not only on their applicability in pre and post-harvest citrus production in Uruguay (reported in SATA guide [39]), but also on their probability of being found in said matrices according to previous findings in bibliographic data. Pesticides such as neonicotinoids, organophosphates, carboxamides, imidazoles, strobilurins, and benzimidazoles were included. Table 1 shows the selected pesticides.
3.1 Chromatographic and mass spectrometry conditions
Two chromatographic methods were used due to the physicochemical characteristics of the selected pesticides. For the LC configuration the optimization process was made using direct infusion with electrospray (ESI) and negative and positive acquisition modes were employed. Pesticides LC and GC detection parameters are shown in Table 1.
3.2 Preliminary tests and method validation
3.2.1 Lemon fruit
For the analysis of pesticide residues in lemon fruit, two reported methodologies were evaluated: EtOAc [3] and QuEChERS AOAC. The EtOAc method showed recoveries for 3 of the 16 studied pesticides in this matrix, that did not meet the SANTE document requirements criteria (pesticide recoveries between 70 and 120% and relative standard deviations (RSD) < 20%) [38]. Imazalil, carbendazim, and thiabendazole (imidazole and benzimidazole chemistry family), whose pKa is 6.53, 4.2, and 4.73 [40] respectively, were lost during the extraction step due to the low pH of the matrix. As stated above, the QuEChERS AOAC method stabilizes the matrix pH through an in situ generated buffer that allows the quantitative extraction of low-pH susceptible pesticides and it was chosen for its validation.
Method selectivity was studied, and no interfering ions with those of pesticides were found in the blank sample. Percentages of recovery and RSD at two concentration levels (10 and 50 µg kg−1) together with matrix effect obtained in LC–MS/MS for each pesticide under study are shown in Table 2. Based on the results from matrix matched and solvents calibration curves, it can be concluded that a linear behavior is verified. The limit of quantitation of all the studied analytes was 10 µg kg−1. As it was expected when the electrospray interface is used, the matrix effects cause suppression of the analytical signal for 83% of the studied compounds. Nevertheless, the observed matrix effect of imidacloprid is enhancement of the signal with a value of 96%. These results comply with a recent work reported by our group, where using LC-AHRMS-TOF, a positive value of 84% was found for this compound in lemon fruit [17].
3.2.2 Concentrated juice
Concentrated juices are defined by the Uruguayan National Bromatological Regulation (UNBR) as “… products obtained by concentrating vegetable juices having a minimum volume ratio with the original juice of 50% (1–2), except for citrus fruits, which shall be 33% (1–3) and that comply with the requirements of this regulation …” [41]. Lemon juice contains flavonoids, carotenoids, fatty acids, vitamins among others metabolites [42], in the case of concentrated juices these compounds are at higher concentration, and as a consequence, higher matrix effects are expected. The selective removal of compounds at higher concentration levels in comparison with the studied pesticides could be a difficult task. In addition, during the concentration process, some artifacts, conjugates and decomposition compounds are formed, turning the matrix even more complex. The three procedures described above based on QuEChERS methodology were evaluated in terms of their ability to provide good analyte extraction and maximizing the removal of co-extractives that could interfere. Trueness and precision were evaluated at 100 µg kg−1 concentration level. Also, matrix effects were studied and a strong suppression of the signal was observed. From the three selected methods, all pesticides gave recoveries ≥ 100% whereas the AOAC version, shown lower percentage of pesticides with matrix effect ≥ 50%. A strategy of “reconstructing” the original juice in terms of total solid content was followed aiming to minimize this effect. Sample dilution has proven to minimize matrix effects during ESI and, therefore, ion suppression phenomena could be overcome and higher sensitivity achieved.
Therefore, when validating the QuEChERS AOAC method, the sample preparation step was modified and 2.5 g of concentrated juice were weighted and 7.5 g of water were added. (See Fig. 1). The trueness of the method was studied at 3 concentration levels: 10, 100 and 300 µg kg−1. Linear ranges were from 10 to 300 µg kg−1 for all analytes with the exception of imidacloprid (from 100 to 300 µg kg−1) with residuals lower than 20%. All figure of merit are presented in Table 3.

Percentage of pesticides with matrix effect (%) lower and higher than 20, using 5 g (a) and 2.5 g (b) of concentrated juice during QuEChERS acetate method
The chemical composition of the concentrated juice is less complex than that of the whole fruit but the juice has a higher solid content and potentially interfering compounds. Matrix effects were minimized with the exception of carbendazim and thiabendazole that showed − 39 and − 48% matrix effects, all the studied pesticides suffered no matrix effects. Sample dilution eliminated the high matrix effect suffered by imidacloprid in the whole fruit, as well as most of the other matrix effects observed during its analysis.
3.2.3 Essential oils
For the analysis of lemon essential oils 16 LC amenable pesticides and one GC amenable pesticide: 2-phenylphenol (OPP) were selected. This fungicide is commonly applied in citrus post- harvest step and OPP residues in lemon, orange and mandarin essential oils were reported by Ito et al. [43]. The proposed method is a five times simple dilution of the sample and injection in the LC-MSMS. In this work, after the sample dilution and injection in the instrument, the repeatability of the method was studied at 5 concentration levels: 10, 25, 50, 100 and 200 µg L−1 (n = 3). The results showed good repeatability for all the studied analytes (0.6–9.8% range). The LOQ of the method were 10 µg L−1 for 73%, 25 µg L−1 for 13% and 50 µg L−1 for 6% of the analytes while for OPP the LOQ was 100 µg L−1. Although the overall dropping down of matrix effects to negligible ones, boscalid and iprodione suffered of a great signal suppression due to matrix effect that prevented their identification at concentrations below 100 µg L−1, and could not be analyzed. The linear ranges were from LOQ to 200 µg L−1 for LC amenable analytes and from LOQ to 500 µg L−1 for OPP that was analyzed by GC–MS, with residuals yielding deviations lower than 20% in all cases (See Table 4).
3.3 Real sample analysis
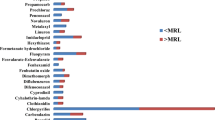
Real samples of lemon fruit (6 in total) were analyzed. Iprodione (0.04 mg kg−1), boscalid (0.24 mg kg−1) and pyraclostrobin (0.11 mg kg−1) residues were detected. On the other hand, 101 EOs samples were analyzed. The post-harvest pesticides most commonly found were imazalil (0.15–2.20 mg L−1), prochloraz (0.01–35.9 mg L−1), 2-phenylphenol (0.10–52.1 mg L−1) and propiconazole (0.01–15.0 mg L−1). Also residues of pyraclostrobin and difenoconazole, that are usually used during pre-harvest were found. Considering the amount of EO is 0.1% v/p of the fruit, and that the oil is separated by centrifugation, citrus EOs concentrate lipophilic and not ionizable pesticides. Therefore, low traces of pesticides in the fruit can reach very high levels in the EOs. In order to compare the results with the established MRLs it is necessary to take into account the essential oil density to convert mg L−1 into mg kg−1 (consider it when comparing the results with the MRLs). In this work, we consider a value of 0.85 kg L−1 based in the Clará research [44].
Five concentrated lemon juices were analyzed. Most countries rely on regulations for processed foods and should comply with the maximum limits for pesticides. However, the Codex Alimentarius [45] and European Legislation [46] do not provide MRLs data for concentrated juices but state that the appropriate MRL applied to concentrated juice corresponds to the MRL value for each pesticide in a raw agricultural commodity.
In this work the MRL of each pesticide reported by the Codex Alimentarius for lemon was used and the MRL value for concentrated juice is: MRL in lemon juice (µg kg−1) = MRL in lemon fruit (µg kg−1). Propiconazole, imazalil, pyrimethanil and imidacloprid residues were found in commercial samples. In all cases, the found concentrations were below the MRLs. Figure 2 shows the chromatographic signal of pyrimethanil standard at the LOQ level (µg kg−1) and a pyrimethanil signal present in a concentrated lemon juice at 46.6 µg kg−1 concentration level.

Pyrimethanil residue identification in concentrated lemon juice by LC–MS/MS. Chromatographic signal in a matrix matched calibration (10 µg kg−1) and in a real sample at concentration level of 46.6 µg kg−1
4 Conclusions
Specific analytical methodologies were optimized and validated for lemon fruits and the main by-products obtained in its production chain. They are suitable protocols for the routine analysis of these commodities. They entail the use of common reactives and simple procedures. The sample dilution prior the extraction step and analysis were successfully applied for the analysis of lemon by-products, concentrated juices and essential oils. The dilution step minimizes the matrix effects, and generally a better performance is observed, with the exception of boscalid and iprodione in EO. When analyzing the concentrated juice, the dilution x 4 in the extraction step allowed a smooth application of the acetate buffered QuEChERS protocol. Matrix effects were minimal after dilution. On the other hand, the analysis of essential oils involved the simple dilution of the sample before determination and it can represent great benefit for laboratories as it allows increasing samples throughput. The buffered acetate QuEChERS was the most effective protocol to analyze pH-dependent pesticides in lemon fruit. The analytical scope of the method is the same, representing the most commonly applied pesticides in citrus production chain. They are currently applied to the routine determination of pesticide residues in lemon commodities for export. Pesticide residues (imazalil, pyraclostrobin, pyrimethanil, prochloraz, difenoconazole, 2-phenylphenol) were found on the three evaluated matrices. In lemon fruits and concentrated juices the residues found were at concentrations below the MRLs established by the Codex Alimentarius and European Union (EU). In the case of EOs, prochloraz, 2-phenylphenol and propiconazole exceed the lemon EU MRLs.
References
FAO (2017) Citrus fruit-fresh and processed. Statistical Bulletin. Rome (2016)
INIA (2017) Programas Nacionales de Investigación. Producción Citrícola. Antecedentes. www.inia.org.uy. Accessed 17 Jan 2017
Besil N, Pérez-Parada A, Cesio V, Varela P, Rivas F, Heinzen H (2016) Degradation of imazalil, orthophenylphenol and pyrimethanil in Clementine mandarins under conventional postharvest industrial conditions at 4 °C. Food Chem 194:1132–1137. https://doi.org/10.1016/j.foodchem.2015.08.111
Sun D, Zhu Y, Pang J, Zhou Z, Jiao B (2016) Residue level, persistence and safety of spirodiclofen-pyridaben mixture in citrus fruits. Food Chem 194:805–810. https://doi.org/10.1016/j.foodchem.2015.08.044
Mutengwe MT, Chidamba L, Korsten L (2016) Pesticide residue monitoring on South African fresh produce exported over a 6-year period. J Food Prot 79(10):1759–1766. https://doi.org/10.4315/0362-028X.JFP-16-022
Wang Z, Cao Y, Ge N, Liu X, Chang Q, Fan C, Pang GF (2016) Wide-scope screening of pesticides in fruits and vegetables using information-dependent acquisition employing UHPLC-QTOF-MS and automated MS/MS library searching. Anal Bioanal Chem 408(27):7795–7810. https://doi.org/10.1007/s00216-016-9883-3
Garcia-Reyes JF, Gilbert López B, Molina-Díaz A, Fernández-Alba A (2009) Determination of pesticide residues in fruit-based soft drinks, vol 80. https://doi.org/10.1021/ac8012708
Klier B, Knodler M, Peschke J, Riegert U, Steinhoff B (2015) Pesticide residues in essential oils: evaluation of a database. Pharmeuropa Bio Sci Notes 2015:131–149
Uclés A, Herrera López S, Dolores Hernando M, Rosal R, Ferrer C, Fernández-Alba AR (2015) Application of zirconium dioxide nanoparticle sorbent for the clean-up step in post-harvest pesticide residue analysis. Talanta 144:51–61. https://doi.org/10.1016/j.talanta.2015.05.055
Soler C, Mañes J, Picó Y (2005) Routine application using single quadrupole liquid chromatography–mass spectrometry to pesticides analysis in citrus fruits. J Chromatogr A 1088(1–2):224–233. https://doi.org/10.1016/j.chroma.2005.03.106
Sun D, Pang J, Jiao B, Zhao Q, Zhou Z (2015) Multiresidue method for the simultaneous determination of 16 acaricides by modified quick, easy, cheap, effective, rugged, and safe extraction and ultra-high performance liquid chromatography coupled with tandem mass spectrometry in citrus. J Sep Sci 38(22):3845–3851. https://doi.org/10.1002/jssc.201500837
Valenzuela AI, Lorenzini R, Redondo MJ, Font G (1999) Matrix solid-phase dispersion microextraction and determination by high-performance liquid chromatography with UV detection of pesticide residues in citrus fruit. J Chromatogr A 839(1–2):101–107. https://doi.org/10.1016/S0021-9673(99)00055-2
Prousalis KP, Polygenis DA, Syrokou A, Lamari FN, Tsegenidis T (2004) Determination of carbendazim, thiabendazole, and o-phenylphenol residues in lemons by HPLC following sample clean-up by ion-pairing. Anal Bioanal Chem 379(3):458–463. https://doi.org/10.1007/s00216-004-2614-1
Navickiene S, Ribeiro ML (2002) A simple and rapid extraction for gas chromatographic determination of thiabendazole and imazalil residues in lemons. J Braz Chem Soc 13(5):592–596
Yamazaki Y, Ninomiya T (1996) Determination of imazalil residues in lemons by gas chromatography with nitrogen-phosphorus detection. J AOAC Int 79(3):787–790
Boeris V, Arancibia JA, Olivieri AC (2014) Determination of five pesticides in juice, fruit and vegetable samples by means of liquid chromatography combined with multivariate curve resolution. Anal Chim Acta 814:23–30. https://doi.org/10.1016/j.aca.2014.01.034
Besil N, Cesio V, Heinzen H, Fernandez-Alba AR (2017) Matrix effects and interferences of different citrus fruit coextractives in pesticide residue analysis using ultrahigh-performance liquid chromatography-high-resolution mass spectrometry. J Agric Food Chem 65(23):4819–4829. https://doi.org/10.1021/acs.jafc.7b00243
Niell S, Besil N, Colazzo M, Cesio V, Heinzen H (2017) QuEChERS and other MRM sample preparation methods SPE, SPME, DLLME, SBSE, ASE, MAE, SFE. In: Heinzen H, Nollet L, Fernandez-Alba AR (eds) Multiresidue methods for the analysis of pesticide residues in food, vol 1. CRC Press, Boca Raton, p 452
Ferrer C, Lozano A, Agüera A, Girón AJ, Fernández-Alba AR (2011) Overcoming matrix effects using the dilution approach in multiresidue methods for fruits and vegetables. J Chromatogr A 1218(42):7634–7639. https://doi.org/10.1016/j.chroma.2011.07.033
Stahnke H, Kittlaus S, Kempe G, Alder L (2012) Reduction of matrix effects in liquid chromatography-electrospray ionization-mass spectrometry by dilution of the sample extracts: how much dilution is needed? Anal Chem 84(3):1474–1482. https://doi.org/10.1021/ac202661j
Horváth-Kerkai E, Stéger-Máté M (2012) Manufacturing fruit beverages and concentrates. In: Sinha N, Sidhu J, Barta J, Wu J, Cano M (eds) Handbook of fruits and fruit processing, 2nd edn. Wiley-Blackwell, Oxford, p 223. https://doi.org/10.1002/9781118352533.ch13
Arce A, Soto A (2008) Citrus Essential oils: extraction and deterpenation, vol 2. Citrus 1. Tree and forestry science and biotechnology. vol Special Issue 1. Global Sciences Book, London
Montti M, Visciglio S, Raviol F, Subovich G, Munitz M (2013) Incidencia de la carga inicial de pesticidas en fruta sobre los niveles residuales en aceites esenciales cítricos. Ciencia, Docencia y Tecnología 47:187–218
Dreassi E, Zanfini A, Zizzari AT, La Rosa C, Botta M, Corbini G (2010) Lc/Esi/Ms/Ms determination of postharvest fungicide residues in citrus juices. LWT Food Sci Technol 43(9):1301–1306. https://doi.org/10.1016/j.lwt.2010.04.010
Rizzetti TM, Kemmerich M, Martins ML, Prestes OD, Adaime MB, Zanella R (2016) Optimization of a QuEChERS based method by means of central composite design for pesticide multiresidue determination in orange juice by UHPLC-MS/MS. Food Chem 196:25–33. https://doi.org/10.1016/j.foodchem.2015.09.010
Sannino A, Bolzoni L, Bandini M (2004) Application of liquid chromatography with electrospray tandem mass spectrometry to the determination of a new generation of pesticides in processed fruits and vegetables. J Chromatogr A 1036(2):161–169. https://doi.org/10.1016/j.chroma.2004.02.078
Sannino A (2007) Determination of three natural pesticides in processed fruit and vegetables using high-performance liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom 21(13):2079–2086. https://doi.org/10.1002/rcm.3051
Barrek S, Paisse O, Grenier-Loustalot MF (2003) Analysis of pesticide residues in essential oils of citrus fruit by GC-MS and HPLC-MS after solid-phase extraction. Anal Bioanal Chem 376(2):157–161. https://doi.org/10.1007/s00216-003-1899-9
Saitta M, Di Bella G, Salvo F, Lo Curto S, Dugo G (2000) Organochlorine pesticide residues in Italian citrus essential oils, 1991-1996. J Agric Food Chem 48(3):797–801. https://doi.org/10.1021/jf990331z
Alves AAR, Rezende MJC, Hovell AMC, Bizzo HR, Oliveira ACL, Rodrigues SV, Rezende CM (2012) Comparison between GC-MS-SIM and GC-ECD for the determination of residues of organochlorine and organophosphorus pesticides in brazilian citrus essential oils. J Braz Chem Soc 23(2):306–314. https://doi.org/10.1590/S0103-50532012000200017
Saitta M, Di Bella G, Dugo G (2012) Pesticides and plasticizers in Citrus essential oils: an ordinary history of research. J Essent Oil Res 24(2):171–180. https://doi.org/10.1080/10412905.2012.659522
Pérez-Parada A, Colazzo M, Besil N, Heinzen H, Dellacassa E, Cesio V, Fernández A (2011) Pesticide residues in natural products with pharmaceutical use: occurrence, analytical advances and perspectives. In: Stoytcheva M (ed) Pesticides in the modern world—trends in pesticides analysis, vol 1. Intech, Rijeka, pp 357–390
Fillatre Y, Rondeau D, Daguin A, Communal PY (2016) A workflow for multiclass determination of 256 pesticides in essential oils by liquid chromatography tandem mass spectrometry using evaporation and dilution approaches: application to lavandin, lemon and cypress essential oils. Talanta 149:178–186. https://doi.org/10.1016/j.talanta.2015.11.052
Lehotay SJ, O’Neil M, Tully J, García AV, Contreras M, Mol H, Heinke V, Anspach T, Lach G, Fussell R, Mastovska K, Poulsen ME, Brown A, Hammack W, Cook JM, Alder L, Lindtner K, Vila MG, Hopper M, De Kok A, Hiemstra M, Schenck F, Williams A, Parker A (2007) Determination of pesticide residues in foods by acetonitrile extraction and partitioning with magnesium sulfate: collaborative study. J AOAC Int 90(2):485–520
Anastassiades M, Lehotay SJ, Štajnbaher D, Schenck FJ (2003) Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J AOAC Int 86(2):412–431
Anastassiades M, Scherbaum E, Tasdelen B, Stajnbaher D (2007) Recent developments in QuEChERS methodology for pesticide multiresidue analysis. In: Ohkawa HMH, Lee PW (eds) Pesticide chemistry: crop protection, public health, environmental safety. Wiley-VCH, Weinheim, pp 439–458
Dellacassa E, Lorenzo D, Moyna P, Verzera A, Mondello L, Dugo P (1997) Uruguayan essential oils. Part VI. Composition of lemon oil. Flavour Fragr J 12(4):247–255. https://doi.org/10.1002/(sici)1099-1026(199707)12:4<247:aid-ffj639>3.0.co;2-v
DG-SANTE (2015) Guidance document on analytical quality control and method validation procedures for pesticides residues analysis in food and feed. Document SANTE/11945/2015. European Commission
Guía_SATA (2016-2017) Guía Uruguaya para la protección y nutrición vegetal. http://www.laguiasata.com. Accessed Oct 2018
BCPC (2012) A world compendium the pesticide manual, 17th edn. BCPC, United Kingdom
MSP (1994) Reglamento Bromatológico Nacional. Decreto 315/994. 3er Edition edn. IMPO, Montevideo, Uruguay
Uçan F, Ağçam E, Akyildiz A (2016) Bioactive compounds and quality parameters of natural cloudy lemon juices. J Food Sci Technol 53(3):1465–1474. https://doi.org/10.1007/s13197-015-2155-y
Ito Y, Toyoda M, Suzuki H, Iwaida M (1977) A systematic analysis of diphenyl, o-phenylphenol and thiabendazole in citrus oil. Food Hyg Saf Sci 18(5):450–454. https://doi.org/10.3358/shokueishi.18.450
Clará RA, Gómez Marigliano AC, Sólimo HN (2011) Physicochemical properties and vapor-liquid equilibrium data for steam-distilled lemon essential oil. Latin Am Appl Res 40(1):61–66
Codex_Alimentarius (2003) Maximum residue limits for processed or ready to eat foods or feeds. vol CX/PR 03/17. The Netherlands
European_Commission (2005) Regulation (EC) No 396/2005 of the European Parliament and of the Council of 23 February 2005 on maximum residue levels of pesticides in or on food and feed of plant and animal origin and amending Council Directive 91/414/EEC. Official Journal of the European Union
Acknowledgements
N. Besil be grateful to Agencia Nacional de Investigación e Innovación for her scholarship POS_NAC_2012_1_9348, Programa de Apoyo a las Ciencias Básicas (PEDECIBA). The authors are grateful to Instituto Nacional de Investigación Agropecuaria for lemon samples. S. Rezende be thankful to Departamento de Química del Litoral for her ungraduate thesis supervision.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical approval
This research does not have any studies with human subjects or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Besil, N., Rezende, S., Alonzo, N. et al. Analytical methods for the routinely evaluation of pesticide residues in lemon fruits and by products. SN Appl. Sci. 1, 618 (2019). https://doi.org/10.1007/s42452-019-0626-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-019-0626-x