
Abstract
Purpose of review
In this review, we aim to discuss the role of the bone marrow microenvironment in supporting hematopoiesis, with particular focus on the contribution of the endothelial niche in dictating hematopoietic stem cell (HSC) fate.
Recent findings
Evidence gathered in the past two decades revealed that specific cell types within the bone marrow niche influence the hematopoietic system. Endothelial cells have emerged as a key component of the HSC niche, directly affecting stem cell quiescence, self-renewal, and lineage differentiation. Physiological alterations of the bone marrow niche occurring in aging have been described to be sufficient to promote functional aging of young HSCs. Furthermore, a growing body of evidence suggests that aberrant activation of endothelial-derived signaling pathways can aid or trigger neoplastic transformation.
Summary
Several groups have contributed to the characterization of the different cell types that comprise the complex bone marrow environment, whose function was long perceived as an undiscernible sum of many parts. Further studies will need to uncover niche cell-type-specific pathways, in order to provide new targets and therapeutic options that aim at withdrawing the microenvironmental support to malignant cells while sparing normal HSCs.
Similar content being viewed by others

Introduction
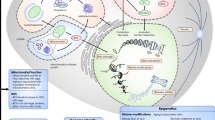
For many years, the bone marrow (BM) niche was considered an inert scaffold for hematopoietic stem cells (HSCs), but in the past two decades, it has been recognized as a complex and dynamic tissue providing the right “soil” to ensure the fulfillment of the HSC potential. Many molecular and cellular components contribute to this plastic environment. In this review, we discuss the recent advances that had shed light on the complexity of the BM niche; in particular, we will focus on the role of the endothelial niche, which sustain HSCs throughout their lifespan, since the emergence of definitive HSCs during embryonic development. Furthermore, we aim at reviewing how the process of aging re-shapes the BM endothelial niche and consequently affects hematopoiesis, impairing tissue homeostasis. Advancing age is irrefutably accompanied by an increased incidence of cancer, thus making critical the discussion on how alterations of the BM microenvironment may instruct and/or support the emergence of aberrant disease-initiating leukemia stem cells (LSC). Here, we will examine several lines of evidence that associate the genetic changes in BM non-hematopoietic cells and the leukemia-induced BM niche remodeling with the emergence of leukemia cells endowed with stem cell-like properties.
The Hematopoietic Stem Cell Niche
In the mouse embryo, definitive HSCs emerge in the aorta-gonad-mesonephros (AGM) region around embryonic day (E) 11 [1], as a result of endothelial-to-hematopoietic transitions [2,3,4]. These HSCs then migrate to the fetal liver, where they undergo a considerable expansion [5]. Eventually, HSCs colonize the BM concurrently with marrow vascularization (E16.5) [6]. The BM niche thereafter provides a critical multicellular microenvironment throughout adult life, regulating HSC quiescence, proliferation, mobilization, and lineage differentiation. In addition to endothelial cells, other cell types are determinant for the life-long regeneration of the blood system, including perivascular mesenchymal stem cells [7,8,9], adipocytes [10], mature osteoblasts [11•, 12•], non-myelinating Schwann cells [13], sympathetic nerves [14, 15], and hematopoietic-derived differentiated cells such as megakaryocytes [16, 17], granulocytes [18], and regulatory T cells [19, 20].
It was initially presumed that the HSC niche comprised mainly osteolineage cells [21, 22], whereas the BM vasculature served as a critical cellular hub for the regeneration of the hematopoietic system following myelosuppressive damage [23]. However, technical advances in imaging and the refinement of cell surface markers enriching for HSCs demonstrated that quiescent HSCs reside in close proximity to vascular niches [24, 25], including endothelial cells and perivascular cells [26]. Furthermore, the conditional deletion of key factors, such as stem cell-active cytokine (C-X-C motif) ligand 12 (CXCL12, also known as SDF1α) from mineralized osteoblasts or Osterix-expressing osteoprogenitor cells [11•], and of stem cell factor (SCF, known as KITL) from Col2.3-expressing osteoblasts [12•] has been reported to not significantly affect HSC frequency and function. Notably, these findings showed that the expression of CXCL12 from cells in the perivascular region, including endothelial cells and mesenchymal progenitors [11•], and of SCF from leptin receptor (Lepr)-expressing perivascular stromal cells and endothelial cells [12•] supported HSCs. The hypothesized contribution of mature osteolineage cells to the maintenance of HSC was additionally lessened by more recent studies combining whole-mount confocal immunofluorescence imaging with 3D computational modeling. These studies found that HSCs preferentially localized in endosteal zones, in close contact with microvessels [27, 28••], consistent with an ancillary role for mature osteoblasts in HSC control. Rather, it emerged that immature osteoprogenitor cells are HSC regulators. These osteolineage-committed progenitors are comprised in mesenchymal stem cells and are often localized in perivascular regions [29].
The Bone Marrow Endothelial Niche
Endothelial cells are specialized cells that form the inner lining of all blood vessels and support tissue growth and repair [30]. Within the BM microenvironment, endothelial cells are organized in a hierarchical structure of central, longitudinal arteries that give rise to smaller arterioles, which transition to the venous circulation in proximity of the endosteum, and then into sinusoid vessels that extend back in the BM cavity [27]. Recent data has postulated that the endothelial niche can be divided into two distinct pro-HSC niches, the arteriolar niche that is identified by the VEcadherin+ (CDH5) CD31+ Endomucin+/− SCA1high (Ly6a) VEGRF3− (FLT4) surface phenotype and the sinusoidal niche, identified by VEcadherin+ CD31+ Endomucin+ SCA1low VEGRF3+ [28••, 31, 32]. In the context of SCF, it has been suggested the arteriole cells are responsible for the maintenance of the HSC [33]; however, it is still unclear whether these two subcategories play different instructive roles in HSC function in regard to other known or as of yet discovered pro-HSC factors [8, 17, 28••, 33,34,35].
The role of the BM endothelium in regulating the hematopoietic system first emerged in the context of hematopoietic recovery after myelosuppression [31, 35]. Studies in mice revealed that 5-fluorouracil (5-FU) treatment led to an increase in soluble KITL and plasma vascular endothelial growth factor A (VEGF-A) levels along with the expansion of Tie2-positive neovessels in the adult BM. Moreover, inhibition of Tie2 signaling contributed to impaired neoangiogenesis, leading to a delay in hematopoietic recovery [35]. The critical importance of vessel maintenance to functionally support HSCs was further demonstrated by the conditional deletion of Vegfr2 in adult mice, which was shown to block regeneration of sinusoidal ECs in sublethally irradiated animals, thus preventing hematopoietic reconstitution [31]. Furthermore, inhibition of VEGFR2 using antibodies prevented regeneration of damaged sinusoidal ECs, thereby interfering with the engraftment of HSCs and leading to hematopoietic failure in lethally irradiated mice [31].
Pioneering work from our group demonstrated that endothelial cells have the capacity to release angiocrine factors to support the in vitro self-renewal and in vivo reconstitution of long-term (LT) HSC pool after myeloablation [36••]. Specifically, activation of Akt-mTOR pathways in endothelial cells upregulated stem cell active angiocrine factors that supported the expansion of HSCs. Conversely, MAPK signaling in endothelial cells shifted the balance towards HSC differentiation, thus suggesting that the endothelial niche could support both self-renewal and lineage-specific differentiation of HSCs [37]. The expression of Notch ligands Jagged-1 and Jagged-2 by BM-derived endothelial cells (BMECs) also regulates HSC proliferation and quiescence [37, 38•, 39]. Endothelial-specific deletion of Jagged-1 using VE-cadherin-Cre mice resulted in a significant decrease in the number of phenotypic LT-HSCs at steady state, and in inhibited hematopoietic regeneration after sublethal irradiation [38•]. Notably, Jagged-2 expression in BMECs was dispensable for maintaining the capacity of hematopoietic stem and progenitor cells (HSPCs) to repopulate under steady-state conditions, but contributed to their recovery in response to myelosuppressive injury, by activating Notch 2 [39]. Recently, the activation of Notch pathway has also been described as a positive regulator of vascular growth in adult bone. In particular, a new capillary subtype, defined as a rare CD31highEndomucinhigh or “Type H” endothelium has been implicated in osteogenesis and in chondrocyte maturation [32, 40]. This endothelial cell subset was enriched in the bone metaphysis and at the endosteal surface and marked the distal end of CD31+Endomucin− arterioles. However, an independent multivariate flow cytometry analysis of adult BM utilizing intravital injection of VE-cadherin and endothelial reporter mice demonstrated that both arterioles and sinusoids were CD31high, and that all VEGFR3+ sinusoids and a subset of arterioles were Endomucinhigh [41]. Similarly, a novel endoglin (CD105)-expressing endothelial cell subpopulation was described in human BM upon regeneration after chemotherapeutic injury. These CD105-expressing endothelial cells were a rare fraction of CD31+ endothelial cells in steady-state adult BM but increased significantly (13–19-fold change increase) upon administration of 5-FU. These findings confirmed a relative increase of this subset in the regenerative phase after myeloablation, thus leading to designate these endothelial cells “human regeneration-associated ECs” (hRECs). hRECs harbor several similarities to the recently identified Type H endothelium in mice, including localization to the bone surface and reduced frequencies during aging [32, 40, 42].
Overall, these findings are particularly noteworthy in relation to the myelosuppressive preparative regimens for hematopoietic stem cell transplantation (HSCT). Indeed, pre-conditioning regimens for blood malignancies and autoimmune disorders can cause systemic endothelial damage and have been associated with HSCT-related complications, including acute graft-versus-host disease, through endothelial apoptosis and pro-inflammatory cytokine production [43, 44, 45•]. Chronic inflammatory signaling is a renewed factor implicated in impaired HSC repopulation potential [46,47,48]. Therefore, strategies aimed at limiting pro-inflammatory responses that affect HSC maintenance and BM endothelial niche integrity may be an attractive option to preserve long-term HSC function following hematopoietic injury. To this aim, we have recently identified the canonical NF-kB pathway as an extrinsic mediator of HSC function within the adult BM endothelial niche. Endothelial-specific inhibition of canonical NF-kB signaling using a dominant negative IkB-SS construct expressed under Tie2 promoter elements resulted in a profound increase in HSC self-renewal and regenerative potential [49]. More notably, the infusion of exogenous [50••, 51] NF-kB-inhibited BMECs in irradiated mice resulted in a significant radio-protective effect that mitigated hematopoietic damage [49]. Furthermore, we have demonstrated that co-infusion of young BMECs with whole BM transplants can shepherd the transplant to the BM microenvironment resulting in an increase in the engraftment potential of a limited number of donor hematopoietic cells, as well as restore the functionality of transplanted aged donor cells [52••]. Taken together, these findings underline the considerable potential of therapeutic approaches targeting the endothelial niche, either by the direct modulation of niche-specific signaling or by infusion of ex vivo expanded cellular therapeutics.
Aging of the Bone Marrow Niche and its Effects on Hematopoiesis
It is generally believed that aging results in the progressive decrease of stem cell reservoirs; although, when looking at the hematopoietic system, aging entails complex and somehow counterintuitive alterations. While the absolute number of phenotypically defined HSCs increases with age, aged HSCs exhibit decreased self-renewal and reconstitution capacity [53, 54, 55•, 56,57,58,59]. In particular, aged LT-HSC show a myeloid-biased differentiation potential compared with young HSC, and diminished lymphoid potential [54, 55•, 56, 57].
Aged HSCs express elevated levels of genes associated with nitric oxide (NO)-mediated signal transduction, stress response, inflammation [53], as well as with myeloid lineage differentiation, cell cycle [60, 61] and proliferation (including TGFβ and ERK/MAPK pathways) [56, 60]. In contrast, downregulated genes often include those involved in the preservation of genomic integrity, such as chromatin remodeling, DNA repair [53] and DNA replication. Cycling aged HSCs in mice have shown elevated levels of replication stress associated with cell cycle defects [62], reduced cell polarity, and adhesion to the BM niche [63, 64]. In all, most observations in regard to the aging of the hematopoietic system have focused on cell intrinsic changes in HSCs which can portend to leukemic transformation, but whether BM niche age-associated alterations further exacerbate or even are sufficient to initiate blood disorders is still unclear.
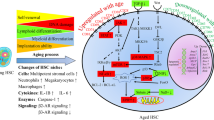
Recent evidence has demonstrated that cell extrinsic alteration can support the onset of HSC aging phenotypes. Indeed, it has been shown that the myeloid lineage skewing of aged HSCs is associated to increased secretion of the pro-inflammatory CC-chemokine ligand 5 (CCL5, also known as RANTES) by aged stroma [65]. Similarly, reduced expression of secreted matrix protein osteopontin (OPN) in aged stroma confers aging-associated phenotypes to HSCs, including loss of cell polarity and impaired engraftment potential [66]. Additionally, it has been demonstrated that loss of BM innervation by the sympathetic nervous system can promote premature aging of young HSCs [67]. In line with these findings, our group has recently demonstrated that in both in vitro and in vivo settings, aged BMECs can instruct an aged phenotype in young HSCs, whereas young BMECs can partially rejuvenate aged HSC function [52••]. These data suggest that aged BMECs could be primed to negatively affect HSC function, but that exposure to angiocrine factors from young BMECs can aid in improving aged HSC function. Indeed, it has been demonstrated that aged BMECs have compromised function in regulating hematopoietic niche cells and that it is possible to rejuvenate their functional readout by mitigating the activation of endothelial Notch signaling; however, these studies were unable to restore the full functional capacity of HSCs [42]. These findings strongly suggest that deleterious changes in aged BM microenvironment inefficiently sustain HSC homeostasis which may lead to aged-related hematopoietic disorders (Fig. 1), providing intriguing models to speculate that a “pre-malignant” niche might induce aged HSCs to undergo malignant transformation into tumor cells.

Representative diagram of the instructive role of the BM vascular niche towards HSC regulation, demonstrating that the activation of Akt signaling in young ECs aids HSC function. As aging occurs, the activation state of EC shifts towards pro-inflammatory pathways, including MAPK and NF-kB signaling, leading to impaired HSC function, while promoting leukemia cell expansion
The Leukemic Bone Marrow Niche
As previously discussed, a growing body of evidence demonstrates the critical interactions between healthy HSCs and their niche. Several groups have explored the concept of niche-initiated disease in animal models, showing that alterations of specific BM microenvironment components are sufficient to initiate or set the stage for leukemia. For the scope of this review, we will discuss in more detail experimental models of age-related myeloid disorders, including myeloproliferative neoplasms (MPNs), myelodysplastic syndrome (MDS), and acute myelogenous leukemia (AML). While several studies provided remarkable proof-of-concept in mice, it is still unclear whether a single alteration within the BM microenvironment is sufficient to trigger leukemia in humans, although few groups have recently made indirect observations that support this hypothesis. One of the first striking model of niche-initiated disease was the mesenchymal osteoprogenitor-specific deletion of the miRNA processing endonuclease Dicer1 under the Osterix promoter, which induced impaired osteoblast differentiation [68]. Abnormal osteoprogenitor cells were thus implicated in defective HSC function and in key features of human MDS, including peripheral cytopenia, dysgranulopoiesis, dysplastic megakaryocytes, and the emergence of AML [68]. Interestingly, the osteoprogenitor-specific deletion of Sbds, the gene mutated in Shwachman-Bodian-Diamond Syndrome, a condition characterized by BM failure and leukemia predisposition, largely phenocopied Dicer1 deletion [68]. Consistent with this experimental model, decreased expression of DICER1 was detected in mesenchymal stromal cell-derived osteoprogenitors from MDS patients compared to healthy individuals, along with a reduction of SBDS [69].
The altered differentiation of myeloid and lymphoid progenitors was described in mice with osteoblast-specific constitutive activation of β-catenin, leading to the development of AML [70]. Notably, β-catenin stimulated the expression of Jagged-1 in osteoblasts, triggering aberrant Notch signaling in HSC progenitors [70]. These observations were relevant to human disease, as nuclear localization of β-catenin in osteoblasts was found in a cohort of patients with AML or MDS, associated with concurrent Notch signaling activation in hematopoietic cells [70].
Mutations of the protein tyrosine phosphatase SHP2 (encoded by PTPN11), a positive regulator of the RAS pathway, in mesenchymal stem/progenitor cells and osteoprogenitors, but not in differentiated osteoblasts or endothelial cells induced MPN [71]. Conversely, the loss of signal-induced proliferation-associated gene 1 (Sipa1), a RAP1 GTPase-activating protein expressed mainly by mesenchymal stem/progenitor cells and endothelial cells induced significant alterations in the BM niche prior to the initiation of MDS/MPN in mice [72]. Importantly, transplantation of normal Sipa1+/+ hematopoietic cells in Sipa1−/− recipients was followed by neoplastic transformation in MDS/MPN, recapitulating the same disease features observed in aged Sipa1−/− mice, including anemia, increased granulocytes, pronounced B-lymphopenia, and splenomegaly [72]. Sipa1-deficient niche cells displayed enrichment of G protein signal pathways, including Ras and Rap1, and of pro-inflammatory cytokines such as transforming growth factor-β (TGFβ) and tumor necrosis factor-α (TNFα) [72]. Overall, the global dysregulation of inflammatory cytokines in the aged/pre-tumoral niche appears to be a critical precondition in the pathogenesis of myeloid disorders.
In another model, loss of canonical Notch signaling in BM stromal and endothelial cells induced significant alterations of hematopoiesis. Specifically, the conditional inhibition of the Notch pathway transcriptional repressor recombination signal-binding protein for immunoglobulin kappa J region (RBPJk) in Mx1-expressing cells induced a MPN-like disease, driven by the constitutive upregulation of miR-155 and its downstream activation of NF-kB signaling. Mx1-RBPJ−/− mice displayed a significant increase in the levels of inflammatory cytokines G-CSF and TNFα in BM MSCs and ECs [73••]. Notably, endothelial-specific loss of Notch signaling in Tie2-RBPJ−/− mice led to myeloid cell expansion and inflammatory cytokines similar to those observed in Mx1-RBPJ−/− mice, despite the moderate reduction of RBPJ in endothelial cells (30%) [73••]. Circulating endothelial progenitor cells isolated from patients with MPN were reported to display the same mutation signature detectable in the malignant hematopoietic clone, including the activating mutation of the Janus kinase 2, JAK2V617F [74]. These findings are particularly interesting in the light of recent observations made in Tie2-JAK2V617F mice, where JAK2V617F was specifically expressed in hematopoietic cells and endothelial cells [75, 76•]. JAK2V617F-expressing vascular niche promoted the expansion of JAK2V617F leukemia-initiating cells over JAK2WT HSCs in competitive BM transplants, and upregulated the expression of angiocrine factors CXCL12 and SCF compared to wild-type ECs [76•]. Taken together, the indirect observations in human leukemia seem to corroborate the hypothesis that alterations within the non-hematopoietic compartment are sufficient to aid the emergence of LSC. A question of great interest is whether BM niche cells harbor the same “first hit” mutations as pre-leukemic HSCs do, a claim particularly intriguing in the case of the vascular niche, in light of the close ancestral relationship between ECs and HSCs during development. Very fascinating aspects of the LSC-niche relationship have latterly emerged, including the composition and biophysical properties of the extracellular matrix (ECM), and the leukemic cell response to hypoxia within the BM microenvironment. CD98, an ECM protein mediating several integrin-associated adhesive signals, was recently showed to enable the propagation of AML cells [77]. Loss of CD98 after tamoxifen treatment of Cd98hcfl/fl;Rosa26-CreER mice prevented leukemia cells to form stable interactions with blood vessels in vivo, as well as in in vitro co-cultures of cKit+ leukemia cells with human umbilical vein ECs (HUVECs). Moreover, antibody-mediated blocking of integrin ligand VCAM-1 in endothelial cells had no effect on the adhesion of CD98-deficient cKit+ leukemia cells, while it significantly reduced the attachment of wild-type cKit+ cells, suggesting that CD98 is required for VCAM-1/integrinVLA-4-mediated interactions between leukemia cells and blood vessels [77]. These results are consistent with previous work from our group, demonstrating that leukemic cells cultured in direct cellular contact with primary human ECs [78] were enriched in LSCs, and exhibited a more aggressive AML phenotype when transplanted in mice [79•]. Stimulation of primary ECs with VEGF-A led to increased LSCs expansion, augmented viability after chemotherapy treatment with Ara-C, and increased adhesion to the endothelium, partially mediated by VLA-4 [79•]. Other groups recently substantiated the role of the vascular niche in human AML patient-derived xenografts (PDX), using unconditioned recipient mice to prevent irradiation-induced toxicity in the BM vasculature. AML PDX displayed an increased number of ECs associated with arterioles (CD31+ Sca1high), a loss of ECs associated with sinusoids (CD31+Sca1low), and increased leakiness in the BM [80••]. Gene set enrichment analysis (GSEA) of ECs upon AML engraftment underlined several altered pathways, including angiogenesis, and response to hypoxia. Interestingly, Nox4, a NADPH oxidase involved in the response to hypoxia via production of reactive oxygen species (ROS) and release of NO, was upregulated in ECs, consistent with increased levels of NO in the BM of AML xenografts compared with non-transplanted mice or mice engrafted with normal HSCs [80••].
Conclusions
Understanding the instructive role of the BM niche in normal hematopoiesis is a stepping stone for the development of new therapies in aging and blood malignancies. Future studies will benefit from taking advantage of novel in vivo imaging and functional assays, as well as from a more systematic analysis of non-hematopoietic cells from diagnostic biopsies. Mouse models in the last decade have suggested that a niche cell-associated sensitivity in the emergence of different neoplasms may exist and revealed how LSCs hijack the HSC marrow environment to gain survival benefits. BM inflammation and alteration of vasculature permeability have emerged as key factor in the initiation and development of hematological malignancies. Future work is required to further address the cell type-specific functions and the associated signaling pathways and reciprocal metabolic interactions among the niche components. Advances in this regard will provide us with putative targets to reverse age-dependent alterations of HSC number and function and to predict critical factors involved leukemogenesis.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
de Bruijn MFTR, Speck NA, Peeters MCE, Dzierzak E. Definitive hematopoietic stem cells first develop within the major arterial regions of the mouse embryo. EMBO J. 2000;19:2465–74. https://doi.org/10.1093/emboj/19.11.2465.
Jaffredo T, Gautier R, Eichmann A, Dieterlen-Lièvre F. Intraaortic hemopoietic cells are derived from endothelial cells during ontogeny. Development. 1998;125:4575–83. https://doi.org/10.1016/j.tcm.2006.02.005.
De Bruijn MFTR, Ma X, Robin C, et al. Hematopoietic stem cells localize to the endothelial cell layer in the midgestation mouse aorta. Immunity. 2002;16:673–83. https://doi.org/10.1016/S1074-7613(02)00313-8.
Bertrand JY, Chi NC, Santoso B, Teng S, Stainier DYR, Traver D. Hematopoietic stem cells derive directly from aortic endothelium during development. Nature. 2010;464:108–11. https://doi.org/10.1038/nature08738.
Morrison SJ, Hemmati HD, Wandycz AM, Weissman IL. The purification and characterization of fetal liver hematopoietic stem cells. Proc Natl Acad Sci. 1995;92:10302–6. https://doi.org/10.1073/pnas.92.22.10302.
Coşkun S, Chao H, Vasavada H, Heydari K, Gonzales N, Zhou X, et al. Development of the fetal bone marrow niche and regulation of HSC quiescence and homing ability by emerging osteolineage cells. Cell Rep. 2014;9:581–90. https://doi.org/10.1016/j.celrep.2014.09.013.
Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010;33:387–99. https://doi.org/10.1016/j.immuni.2010.08.017.
Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–88. https://doi.org/10.1016/j.immuni.2006.10.016.
Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, MacArthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–34. https://doi.org/10.1038/nature09262.
Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ. Bone marrow adipocytes as negative regulators of the hematopoietic microenvironment. Nature. 2009;460:259–63. https://doi.org/10.1038/nature08099.
• Greenbaum A, Hsu YMS, Day RB, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495:227–30. https://doi.org/10.1038/nature11926 Demostrates that expression of CXCL-12 from stromal cells in the perivascular region, including endothelial cells and mesenchymal progenitors, supports HSC.
• Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495:231–5. https://doi.org/10.1038/nature11885 Demostrates that stem and progenitor cells occupy different niches within the bone marrow, HSC the perivascular niche, while early progenitor cells the endosteal niche.
Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, et al. Nonmyelinating schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. 2011;147:1146–58. https://doi.org/10.1016/j.cell.2011.09.053.
Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124:407–21. https://doi.org/10.1016/j.cell.2005.10.041.
Pierce H, Zhang D, Magnon C, Lucas D, Christin JR, Huggins M, et al. Cholinergic signals from the CNS regulate G-CSF-mediated HSC mobilization from bone marrow via a glucocorticoid signaling relay. Cell Stem Cell. 2017;20:648–658.e4. https://doi.org/10.1016/j.stem.2017.01.002.
Chow A, Lucas D, Hidalgo A, Méndez-Ferrer S, Hashimoto D, Scheiermann C, et al. Bone marrow CD169 + macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med. 2011;208:261–71. https://doi.org/10.1084/jem.20101688.
Pinho S, Marchand T, Yang E, Wei Q, Nerlov C, Frenette PS. Lineage-biased hematopoietic stem cells are regulated by distinct niches. Dev Cell. 2018;44:634–641.e4. https://doi.org/10.1016/j.devcel.2018.01.016.
Bowers E, Slaughter A, Frenette PS, Kuick R, Pello OM, Lucas D. Granulocyte-derived TNFα promotes vascular and hematopoietic regeneration in the bone marrow. Nat Med. 2016;127:119–23. https://doi.org/10.1038/nm.4448.
Pierini A, Nishikii H, Baker J, Kimura T, Kwon HS, Pan Y, et al. Foxp3+regulatory T cells maintain the bone marrow microenvironment for B cell lymphopoiesis. Nat Commun. 2017;8:1–13. https://doi.org/10.1038/ncomms15068.
Hirata Y, Furuhashi K, Ishii H, Li HW, Pinho S, Ding L, et al. CD150high bone marrow Tregs maintain hematopoietic stem cell quiescence and immune privilege via adenosine. Cell Stem Cell. 2018;22:445–453.e5. https://doi.org/10.1016/j.stem.2018.01.017.
Calvi LM, Adams G, Weibrecht KW, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–6. https://doi.org/10.1038/nature02041.1.
Zhan J, Niu C, Ye L, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–41. https://doi.org/10.1038/nature02064.1.
Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, et al. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 2002;109:625–37. https://doi.org/10.1016/S0092-8674(02)00754-7.
Kiel MJ, Yilmaz ÖH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–21. https://doi.org/10.1016/j.cell.2005.05.026.
Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. 2015;526:126–30. https://doi.org/10.1038/nature15250.
Wei Q, Frenette PS. Niches for hematopoietic stem cells and their progeny. Immunity. 2018;48:632–48. https://doi.org/10.1016/j.immuni.2018.03.024.
Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, et al. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol. 2013;15:533–43. https://doi.org/10.1038/ncb2730.
•• Kunisaki Y, Bruns I, Scheiermann C, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502:637–43. https://doi.org/10.1038/nature12612 Provides evidences that quiescent HSCs associate specifically with small arterioles, associated with NG2 + pericytes, which are critical to maintaing HSC quiescence.
Kfoury Y, Scadden DT. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell. 2015;16:239–53. https://doi.org/10.1016/j.stem.2015.02.019.
Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–87. https://doi.org/10.1016/j.cell.2011.08.039.
Hooper AT, Butler JM, Nolan DJ, Kranz A, Iida K, Kobayashi M, et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2 mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell. 2009;6:263–74. https://doi.org/10.1016/j.stem.2009.01.006.
Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. 2014;507:323–8. https://doi.org/10.1038/nature13145.
Xu C, Gao X, Wei Q, Nakahara F, Zimmerman SE, Mar J, et al. Stem cell factor is selectively secreted by arterial endothelial cells in bone marrow. Nat Commun. 2018;9:2449. https://doi.org/10.1038/s41467-018-04726-3.
Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature. 2016;532:323–8. https://doi.org/10.1038/nature17624.
Kopp HG, Avecilla ST, Hooper AT, Shmelkov SV, Ramos CA, Zhang F, et al. Tie2 activation contributes to hemangiogenic regeneration after myelosuppression. Blood. 2005;106:505–13. https://doi.org/10.1182/blood-2004-11-4269.
•• Butler JM, Nolan DJ, Vertes EL, et al. Endothelial cells are essential for the self-renewal and repopulation of notch-dependent hematopoietic stem cells. Cell Stem Cell. 2010;6:251–64. https://doi.org/10.1016/j.stem.2010.02.001 Provides evidence by using angiogenic mouse models that endothelium-derived angiocrine growth factors support in vitro self-renewal and in vivo repopulation of authentic long-term HSCs.
Kobayashi H, Butler JM, Donnell RO, et al. Angiocrine factors from Akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells. Nat Cell Biol. 2010;12:1046–56. https://doi.org/10.1038/ncb2108.
• Poulos MG, Guo P, Kofler NM, et al. Endothelial Jagged-1 is necessary for homeostatic and regenerative hematopoiesis. Cell Rep. 2013;4:1022–34. https://doi.org/10.1016/j.celrep.2013.07.048 Implicates that Jagged-1, supplied by the bone marrow endothelium, regulates steady-state and regenerative hematopoiesis through a Notch- dependent signaling.
Guo P, Poulos MG, Palikuqi B, Badwe CR, Lis R, Kunar B, et al. Endothelial jagged-2 sustains hematopoietic stem and progenitor reconstitution after myelosuppression. J Clin Invest. 2017;127:4242–56. https://doi.org/10.1172/JCI92309.
Ramasamy SK, Kusumbe AP, Wang L, Adams RH. Endothelial notch activity promotes angiogenesis and osteogenesis in bone. Nature. 2014;507:376–80. https://doi.org/10.1038/nature13146.
Ramalingam P, Poulos MG, Butler JM. Regulation of the hematopoietic stem cell lifecycle by the endothelial niche. Curr Opin Hematol. 2017;24:289–99. https://doi.org/10.1097/MOH.0000000000000350.
Kusumbe AP, Ramasamy SK, Itkin T, Mäe MA, Langen UH, Betsholtz C, et al. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature. 2016;532:380–4. https://doi.org/10.1038/nature17638.
Cooke KR, Jannin A, Ho V. The contribution of endothelial activation and injury to end-organ toxicity following allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2008;14:23–32. https://doi.org/10.1016/j.bbmt.2007.10.008.
Palomo M, Diaz-Ricart M, Carbo C, Rovira M, Fernandez-Aviles F, Martine C, et al. Endothelial dysfunction after hematopoietic stem cell transplantation: role of the conditioning regimen and the type of transplantation. Biol Blood Marrow Transplant. 2010;16:985–93. https://doi.org/10.1016/j.bbmt.2010.02.008.
• Mir E, Palomo M, Rovira M, et al. Endothelial damage is aggravated in acute GvHD and could predict its development. Bone Marrow Transplant. 2017;52:1317–25. https://doi.org/10.1038/bmt.2017.121 Provides evidences that humoral factors present in patients developing acute graft-versus-host disease after allogenic HSC transplantation provoke damage to endothelial cells, shifting their phenotype to a more proinflammatory and prothrombotic one.
Zhao JL, Ma C, O’Connell RM, Mehta A, DiLoreto R, Heath JR, et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress- induced hematopoiesis. Cell Stem Cell. 2014;14:445–59. https://doi.org/10.1016/j.stem.2014.01.007.
Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang SY, et al. Chronic interleukin-1 drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol. 2016;18:607–18. https://doi.org/10.1038/ncb3346.
Mirantes C, Passegué E, Pietras EM. Pro-inflammatory cytokines: emerging players regulating HSC function in normal and diseased hematopoiesis. Exp Cell Res. 2014;329:248–54. https://doi.org/10.1016/j.yexcr.2014.08.017.
Poulos MG, Ramalingam P, Gutkin MC, Kleppe M, Ginsberg M, Crowley MJP, et al. Endothelial-specific inhibition of NF-κB enhances functional haematopoiesis. Nat Commun. 2016;7:1–15. https://doi.org/10.1038/ncomms13829.
•• Butler JM, Gars EJ, James DJ, et al. Development of a vascular niche platform for expansion of repopulating human cord blood stem and progenitor cells. Blood. 2012;120:1344–8. https://doi.org/10.1182/blood-2011-12-398115 Demonstrates that primary human endothelial cells provide a cellular platform for the expansion of normal hematopoietic stem and progenitor cells, providing a rationale for the use of endothelium-tailored strategies in hematologic disorders.
Poulos MG, Crowley MJP, Gutkin MC, Ramalingam P, Schachterle W, Thomas JL, et al. Vascular platform to define hematopoietic stem cell factors and enhance regenerative hematopoiesis. Stem Cell Reports. 2015;5:881–94. https://doi.org/10.1016/j.stemcr.2015.08.018.
•• Poulos MG, Ramalingam P, Gutkin MC, et al. Endothelial transplantation rejuvenates aged hematopoietic stem cell function. J Clin Invest. 2017;127:4163–78. https://doi.org/10.1172/JCI93940 Demonstrates that aging of endothelial cells is sufficient to drive hematopoietic aging phenotypes in young HSCs, and that infusion of young endothelial cells improves aged HSC engraftment and mitigate irradiation-induced damage to the vascualr niche.
Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5:1750–62. https://doi.org/10.1371/journal.pbio.0050201.
Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. J Exp Med. 2000;192:1273–80. https://doi.org/10.1084/jem.192.9.1273.
• Rossi DJ, Bryder D, Zahn JM, et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci. 2005;102:9194–9. https://doi.org/10.1073/pnas.0503280102 Implicates that HSC aging is accompanied by increased stem cell self-renewal, differential capacity to generate committed myeloid and lymphoid progenitors, and the expression of several genes involved in leukemic transformation.
Pang WW, Price E a, Sahoo D, et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci. 2011;108:20012–7. https://doi.org/10.1073/pnas.1116110108.
Miller JP, Allman D. The decline in B lymphopoiesis in aged mice reflects loss of very early B-lineage precursors. J Immunol. 2003;171:2326–30. https://doi.org/10.4049/jimmunol.171.5.2326.
Woolthuis CM, Mariani N, Verkaik-Schakel RN, Brouwers-Vos AZ, Schuringa JJ, Vellenga E, et al. Aging impairs long-term hematopoietic regeneration after autologous stem cell transplantation. Biol Blood Marrow Transplant. 2014;20:865–71. https://doi.org/10.1016/j.bbmt.2014.03.001.
Liang Y, Van Zant G, Szilvassy S. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood. 2005;106:1479–87. https://doi.org/10.1182/blood-2004-11-4282.
Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014;14:673–88. https://doi.org/10.1016/j.stem.2014.03.002.
Kowalczyk MS, Tirosh I, Heckl D, Rao TN, Dixit A, Haas BJ, et al. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 2015;25:1860–72. https://doi.org/10.1101/gr.192237.115.
Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014;512:198–202. https://doi.org/10.1038/nature13619.
Köhler A, Schmithorst V, Filippi M-D, et al. Altered cellular dynamics and endosteal location of aged early hematopoietic progenitor cells revealed by time-lapse intravital imaging in long bones altered cellular dynamics and endosteal location of aged early hematopoietic progenitor cells revealed by. Blood. 2009;114:290–8. https://doi.org/10.1182/blood-2008-12-195644.
Florian MC, Dörr K, Niebel A, Daria D, Schrezenmeier H, Rojewski M, et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell. 2012;10:520–30. https://doi.org/10.1016/j.stem.2012.04.007.
Ergen AV, Boles NC, Goodell MA. Rantes / Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood. 2012;119:2500–9. https://doi.org/10.1182/blood-2011-11-391730.
Guidi N, Sacma M, Ständker L, et al. Osteopontin attenuates aging-associated phenotypes of hematopoietic stem cells. EMBO J. 2017;36:1463. https://doi.org/10.15252/embj.201796968.
Maryanovich M, Zahalka AH, Pierce H, Pinho S, Nakahara F, Asada N, et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat Med. 2018;24:782–91. https://doi.org/10.1038/s41591-018-0030-x.
Raaijmakers MHGP, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464:852–7. https://doi.org/10.1038/nature08851.
Santamaria C, Muntion S, Roson B, Blanco B, Lopez-Villar O, Carrancio S, et al. Impaired expression of DICER, DROSHA, SBDS and some microRNAs in mesenchymal stromal cells from myelodysplastic syndrome patients. Haematologica. 2012;97:1218–24. https://doi.org/10.3324/haematol.2011.054437.
Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, et al. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature. 2014;506:240–4. https://doi.org/10.1038/nature12883.
Dong L, Yu W, Zheng H, et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature. 2016;539:304–8. https://doi.org/10.1038/nature20131.
Xiao P, Dolinska M, Sandhow L, Kondo M, Johansson AS, Bouderlique T, et al. Sipa1 deficiency–induced bone marrow niche alterations lead to the initiation of myeloproliferative neoplasm. Blood Adv. 2018;2:534–48. https://doi.org/10.1182/bloodadvances.2017013599.
•• Wang L, Zhang H, Rodriguez S, et al. Notch-dependent repression of miR-155 in the bone marrow niche regulates hematopoiesis in an NF-κB-dependent manner. Cell Stem Cell. 2014;15:51–65. https://doi.org/10.1016/j.stem.2014.04.021 Demonstrates that loss of canonical Notch signaling upregulates miR-155 in BM endothelial cells, leading to miR-155- mediated activation of proinflammatory NF-kB and development of myeloproliferative-like disease.
Teofili L, Martini M, Iachininoto MG, Capodimonti S, Nuzzolo ER, Torti L, et al. Endothelial progenitor cells are clonal and exhibit the JAK2 V617F mutation in a subset of thrombotic patients with Ph-negative myeloproliferative neoplasms. Blood. 2011;117:2700–7. https://doi.org/10.1182/blood-2010-07-297598.
Lin CHS, Kaushansky K, Zhan H. JAK2V617F-mutant vascular niche contributes to JAK2V617F clonal expansion in myeloproliferative neoplasms. Blood Cells Mol Dis. 2016;62:42–8. https://doi.org/10.1016/j.bcmd.2016.09.004.
• Zhan H, Lin CHS, Segal Y, Kaushansky K. The JAK2V617F-bearing vascular niche promotes clonal expansion in myeloproliferative neoplasms. Leukemia. 2018;32:462–9. https://doi.org/10.1038/leu.2017.233 Provides evidences that JAK2 V617F -bearing vascular niche promotes the expansion of the JAK2 V617F HSCs compared to JAK2 WT HSCs, and upregulates the expression of CXCL12 and SCF , suggesting that a “malignant” niche may favor leukemia cells over normal HSCs.
Bajaj J, Konuma T, Lytle NK, Kwon HY, Ablack JN, Cantor JM, et al. CD98-mediated adhesive signaling enables the establishment and propagation of acute myelogenous leukemia. Cancer Cell. 2016;30:792–805. https://doi.org/10.1016/j.ccell.2016.10.003.
Seandel M, Butler JM, Kobayashi H, Hooper AT, White IA, Zhang F, et al. Generation of a functional and durable vascular niche by the adenoviral E4ORF1 gene. Proc Natl Acad Sci. 2008;105:19288–93. https://doi.org/10.1073/pnas.0805980105.
• Poulos MG, Gars EJ, Gutkin MC, et al. Activation of the vascular niche supports leukemic progression and resistance to chemotherapy. Exp Hematol. 2014;42:976–86. https://doi.org/10.1016/j.exphem.2014.08.003 Demonstrates that endothelial cells allow the propagation of primitive and aggressive leukemic clones, and that VEGF-A activated endothelium decrease the efficacy of chemotherapeutic agents to target leukemic cells.
•• Passaro D, Di Tullio A, Abarrategi A, et al. Increased vascular permeability in the bone marrow microenvironment contributes to disease progression and drug response in acute myeloid leukemia. Cancer Cell. 2017;32:324–41. https://doi.org/10.1016/j.ccell.2017.08.001 Demonstrates that leukemia causes bone marrow vascular abnormalities and that failure to restore normal vasculature after induction chemotherapy is associated with a poor prognosis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Elisa Lazzari and Jason M. Butler declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on In Vitro and In Vivo Models in Stem Cell Biology
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lazzari, E., Butler, J.M. The Instructive Role of the Bone Marrow Niche in Aging and Leukemia. Curr Stem Cell Rep 4, 291–298 (2018). https://doi.org/10.1007/s40778-018-0143-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-018-0143-7