
Abstract
Introduction
Secukinumab and adalimumab are approved for adults with active psoriatic arthritis (PsA). In the absence of direct randomized controlled trial (RCT) data, matching-adjusted indirect comparison can estimate the comparative effectiveness in anti-tumor necrosis factor (TNF)-naïve populations.
Methods
Individual patient data from the FUTURE 2 RCT (secukinumab vs. placebo; N = 299) were adjusted to match baseline characteristics of the ADEPT RCT (adalimumab vs. placebo; N = 313). Logistic regression determined adjustment weights for age, body weight, sex, race, methotrexate use, psoriasis affecting ≥ 3% of body surface area, Psoriasis Area and Severity Index score, Health Assessment Questionnaire Disability Index score, presence of dactylitis and enthesitis, and previous anti-TNF therapy. Recalculated secukinumab outcomes were compared with adalimumab outcomes at weeks 12 (placebo-adjusted), 16, 24, and 48 (nonplacebo-adjusted).
Results
After matching, the effective sample size for FUTURE 2 was 101. Week 12 American College of Rheumatology (ACR) response rates were not significantly different between secukinumab and adalimumab. Week 16 ACR 20 and 50 response rates were higher for secukinumab 150 mg than for adalimumab (P = 0.017, P = 0.033), as was ACR 50 for secukinumab 300 mg (P = 0.030). Week 24 ACR 20 and 50 were higher for secukinumab 150 mg than for adalimumab (P = 0.001, P = 0.019), as was ACR 20 for secukinumab 300 mg (P = 0.048). Week 48 ACR 20 was higher for secukinumab 150 and 300 mg than for adalimumab (P = 0.002, P = 0.027), as was ACR 50 for secukinumab 300 mg (P = 0.032).
Conclusions
In our analysis, patients with PsA receiving secukinumab were more likely to achieve higher ACR responses through 1 year (weeks 16–48) than those treated with adalimumab. Although informative, these observations rely on a subgroup of patients from FUTURE 2 and thus should be considered interim until the ongoing head-to-head RCT EXCEED can validate these findings.
Funding
Novartis Pharma AG.
Similar content being viewed by others


Introduction
For patients with active psoriatic arthritis (PsA), international recommendations such as those of the European League Against Rheumatism (EULAR) [1, 2] and the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) [3] advise the use of biologic disease-modifying antirheumatic drugs (bDMARDs) when a patient’s response to conventional synthetic DMARDs (csDMARDs; bDMARD and csDMARD defined according to EULAR) is inadequate. Historically, bDMARDs have targeted tumor necrosis factor (TNF) but now also include antibodies targeting interleukin 12/23 and interleukin-17A (IL-17A) [4].
At least seven bDMARDs, mainly targeting TNF, are now available for use in patients with PsA. Thus, to help optimize treatment plans for this long-term chronic condition, appropriately powered head-to-head (H2H) randomized controlled trials (RCTs) comparing the efficacy and safety of different bDMARDs are required. One such trial is EXCEED, directly comparing the long-term (52 weeks) efficacy and safety of the fully human anti-IL-17A secukinumab with the anti-TNF adalimumab [5]. Until data from this trial become available, clinicians and health technology assessment bodies may need to resort to adjusted indirect comparisons of these medicines as the best available evidence to inform treatment decisions.
One means of bridging this evidence gap is through the use of inferential analyses such as network meta-analysis [6, 7]. Such techniques are useful when there is a common comparator arm between RCTs (or in general a connected network of studies [6]) and similar study populations, but the methodology is limited by further cross-trial differences, potential lack of common comparators (or connected networks), sensitivity to modeling assumptions, and disparities in definitions of outcome measures [6, 7]. Matching-adjusted indirect comparison (MAIC) incorporates individual patient data (IPD) to address several of the limitations that arise in comparisons based only on aggregate data; thus, it can simulate more closely how treatments may have performed if compared directly [8,9,10].
MAIC uses IPD from one or multiple studies for one treatment to match clinically relevant baseline aggregate characteristics from a published study of another treatment. The patient characteristics from the IPD are adjusted, using a frequently used form of propensity score matching, so that the mean baseline characteristics match those of the aggregate data. This process results in a reduced effective sample size (ESS) for the IPD arms. Outcomes from common comparator arms such as placebo can be used to validate the matching. After matching, the mean of the recalculated matched IPD is compared with the observed mean for the aggregate data from the published study [8].
The results from MAIC in PsA and other conditions have been reported in peer-reviewed publications [11,12,13], and MAIC has been acknowledged as a valid methodological tool by health-technology assessment agencies [10, 14]. MAIC is evolving and becoming a useful complementary technique to meta-analysis in providing comparative effectiveness evidence, especially when such information is unavailable from direct clinical trial comparisons [8]. MAIC has previously been applied to assess the comparative effectiveness of adalimumab versus etanercept [11] and adalimumab versus etanercept or infliximab in patients with psoriasis and PsA, respectively [13]. A short- to midterm MAIC analysis (24 weeks) between adalimumab and secukinumab in patients with PsA was recently published [12].
Our study sought to address the evidence gap in the comparative effectiveness of up to 1 year biologic treatment of IL-17A versus anti-TNF in patients with PsA. Therefore, MAIC was used to compare adalimumab and secukinumab based on common primary and secondary outcome measures from the ADEPT [15,16,17,17] and FUTURE 2 [18] trials.
Methods
Systematic Literature Review
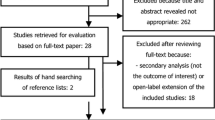
A systematic literature review (SLR) was conducted in September 2014 and updated on November 6, 2015 to identify all relevant clinical evidence for the use of secukinumab and relevant comparators in the treatment of adult patients with PsA. This article is based on previously conducted studies and does not involve any new studies of human or animal subjects. The SLR eligibility criteria are outlined in Table S1, and the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) flow chart is shown in Fig. S1, both in the supplementary material. Following full-text screening, 29 trials were suitable for inclusion according to the eligibility criteria. For the purpose of this analysis, adalimumab is considered the comparator of interest to secukinumab, because of its equivalent efficacy to other anti-TNFs and widespread clinical use [19].
Of the 29 trials, 19 included neither secukinumab nor adalimumab and were excluded from this MAIC. The remaining ten studies are shown in Table S2 in the supplementary material with the reasons for exclusion. Of these ten trials, six investigated secukinumab (CLEAR [20], ERASURE [21], FUTURE 1 [22], FIXTURE [21], FUTURE 2 [18], and McInnes et al. [23]), and the remaining four studied adalimumab (ADEPT [15], Behrens et al. [24], Genovese et al. [25], and van Kuijk et al. [26]). Three of these trials, CLEAR, ERASURE, and FIXTURE, were then excluded because they included patients with moderate-to-severe psoriasis, of whom only a subgroup had concomitant PsA. The study by Behrens et al. [24] was excluded because it was an observational study. Genovese et al. [25] was excluded because the randomized placebo-controlled stage lasted only until week 12, and McInnes et al. [23] was excluded because it was a phase 2 study. Van Kuijk et al. [26] was excluded because the study was restricted to synovial biopsy analyses. Finally, the phase 3 study FUTURE 1 [22] was excluded because only one of two licensed subcutaneous maintenance doses of secukinumab was investigated (150 mg) and it used an intravenous loading method.
Source Data
FUTURE 2 (NCT01752634) was a phase 3, double-blind RCT in adults with active PsA (Fig. 1) [18, 27,28,29]. Patients were randomized to subcutaneous secukinumab (300, 150, or 75 mg) or placebo, once per week from baseline to week 4 and every 4 weeks thereafter [18]. The primary endpoint was the proportion of patients achieving a 20% or greater improvement in the American College of Rheumatology (ACR 20) response criteria at week 24 [30]. Patients receiving concomitant corticosteroid or methotrexate (MTX) medication or who had a previous inadequate response or inability to tolerate up to three different anti-TNF therapies (anti-TNF-IR) could enroll. These biologic-experienced patients made up 35% of the total study population. At week 16, patients were assessed as either responders (≥ 20% improvement from baseline in swollen and tender joint counts) or nonresponders. Placebo-treated patients were rerandomized in a 1:1 ratio to secukinumab 150 or 300 mg every 4 weeks from week 16 (nonresponders) and week 24 (responders).

FUTURE 2 and ADEPT trial designs relative to MAIC analysis. aPatients who had a ≥ 20% improvement compared with baseline in TJC and SJC. bPatients who had a < 20% improvement compared with baseline in TJC and SJC. c153 patients were randomized; however, two patients were not given adalimumab. Analyses were performed on 151 patients who received adalimumab. dPatients who completed the 24-week double-blind period were eligible for the open-label extension study (adalimumab 40 mg EOW). Patients who failed to demonstrate a ≥ 20% improvement compared with baseline in TJC and SJC (study week 36) were allowed to increase the adalimumab dosage to 40 mg weekly. ePatients who did not demonstrate a ≥ 20% improvement compared with baseline in ACR 20 could receive rescue therapy (corticosteroids and/or csDMARDs). The time points (weeks from first subcutaneous injection) at which postmatching outcome comparisons were made are indicated. ACR outcomes were compared at week 12 (placebo-adjusted) and at weeks 16, 24, and 48/52 (nonplacebo-adjusted). The placebo-adjusted phase comparison was valid only until week 12 (shown by yellow rectangle) because of the rescue therapy design component of ADEPT. Numbers in gray denote the ITT populations. All FUTURE 2 secukinumab outcomes were ITT to week 52. ADEPT adalimumab ITT outcome data were maintained until week 48 using published data [16]. Open-label extension adalimumab week 36 dose increase to weekly adalimumab (n = 15 and n = 23 in the original adalimumab and placebo groups, respectively) was classified as NRI. ACR American College of Rheumatology, csDMARD conventional synthetic DMARD, DMARD disease-modifying antirheumatic drug, EOW every other week, ITT intent-to-treat, MAIC matching-adjusted indirect comparison, mTSS modified total Sharp score, NRI nonresponder imputation, R randomization, SJC swollen joint count, TJC tender joint count
ADEPT was a phase 3, double-blind RCT in adults with active PsA (Fig. 1) [15]. Patients were randomized to receive subcutaneous adalimumab 40 mg or placebo every other week. The primary endpoints were the proportion of patients achieving an ACR 20 response at week 12 and the change in modified total Sharp score of structural damage on radiographs of the hands and feet at week 24. Patients could enroll if they were receiving concomitant MTX medication as long as the dose was stable for at least 3 months and was not higher than 30 mg per week. Patients who had previously used any form of anti-TNF biologic were excluded. After week 12, patients whose swollen and tender joint counts did not decrease by at least 20% on two consecutive visits could receive rescue therapy with corticosteroids or csDMARDs [15]. All patients who completed the initial 24 weeks of treatment were eligible to receive adalimumab 40 mg as part of an open-label extension study [15, 16].
MAIC Methodology
The MAIC methodology is graphically summarized in Fig. 2.

MAIC methodology using a hypothetical example. Two treatments of interest have been identified (by SLR): one treatment is examined in study A and another treatment in study B. The researcher has access to IPD for study B but only aggregate published data for study A. Step 1: relevant clinical baseline parameters are selected for matching following consultation with clinical and statistical experts. Step 2: the similarity of IPD baseline characteristics with those of the aggregate study A characteristics (color-coded in this example: red, high through to green, low) will dictate how influential that IPD will be within the matching process. Step 3: matching is performed by application of weights to each IPD (derived by logistic regression) using a matching algorithm similar to propensity score matching. The method of moment was applied using the quasi-Newton optimization “BFGS” implemented in the R function optim, as recommended [9, 10]. IPD with a closer match to the aggregate study A baseline characteristics are “upweighted,” while those with a poor match are “downweighted.” In this example, this leads to an ESS of 4 (rounded to the nearest integer to avoid confusion) using the equation shown. Step 4: study B mean IPD population baseline characteristics match the mean of study A and outcomes can now be compared directly between the two studies. The same weights are used to recalculate each IPD outcome, and then the mean recalculated study B IPD outcomes are directly compared with the published aggregate study A outcomes using appropriate statistical tests. aSome IPD when highly incompatible with the target trial population are given weights that are extremely small (weighting is on a quantitative scale) and effectively act as a zero weight. ESS effective sample size, IPD individual patient data, MAIC matching-adjusted indirect comparison, SLR systematic literature review
Selection of Baseline Characteristics for Matching
Matching variables were selected for their potential influence on key efficacy outcomes on joints and skin; there was no specific matching for baseline safety parameters. The clinical relevance of potential matching variables was discussed among the authors, who include clinical experts in the treatment of active PsA, as well as health economics and comparative effectiveness research experts. In addition, correlation analyses of patient baseline characteristics with ACR 20, 50, and 70 responses achieved at week 48 in FUTURE 2 informed the choice of matching variables (Tables S3–S5 in the supplementary material). This strategy is in line with a recent National Institute for Health and Care Excellence (NICE) guidance on MAIC methodology (NICE Decision Support Unit technical support document 18 [NICE DSU TSD 18]) [10], which recommends justifying the choice of matching parameter by clinical expert advice and/or empirical identification of all prognostic variables and effect modifiers in the weighting model (depending on whether anchored [all effect modifiers] or unanchored [all effect modifiers and prognostic variables] comparisons are being made). Although NICE is geared to a UK payer perspective, its MAIC guidelines were constructed by a group of globally acknowledged academic experts in comparative statistics and represent the only methodologically advanced guidelines published to date.
Two scenarios were developed that differed in the combination of matching variables included. First, a principal analysis replicated the baseline characteristics of a previous MAIC between the same two trials [12], including established prognostic variables, but updated to include parameters identified by logistic regression analysis as having the greatest impact on ACR criteria outcomes (prognostic variables or effect modifiers). One additional baseline parameter, biologic-experienced (i.e., previous inadequate response or intolerance to anti-TNF exposure), was identified as a key variable to have an impact on responses (ACR 20 odds ratio [95% confidence interval (CI)], 0.431 [0.265, 0.701]; P = 0.0007) and therefore included in the principal analysis (Table 1). Similarly, data from the trial ACCLAIM, an open-label study, have shown that patients previously exposed to a biologic therapy have a lower response with adalimumab than those who are biologic-naïve [31]. The sensitivity analysis (Table S6 in the supplementary material) used a more comprehensive set of matching variables and included all of those used in the principal analysis plus three more clinically relevant baseline characteristics to increase the matching stringency: PsA disease duration (time since diagnosis), swollen joint count (SJC), and C-reactive protein (CRP) levels.
Matching and Adjusting IPD to Published Aggregate Data
IPD from the pooled secukinumab arms of the FUTURE 2 trial (75, 150, and 300 mg) were weighted to match the selected patient baseline characteristics for the adalimumab arm of ADEPT. The adalimumab data were reported aggregates taken from the trial publications. Only secukinumab 150 and 300 mg outcomes were compared with adalimumab outcomes because these are the doses licensed in PsA. The methodology was based on Signorovitch et al. [11], subsequent studies [8, 13, 32], and NICE DSU TSD 18 [9, 10]. SAS version 9.4 and R version 3.2.1 were used for the analysis. The regression results were used to weight patients in FUTURE 2, using the method of moments (mean only) so that each patient’s weight corresponded to his or her relative propensity for enrolling in FUTURE 2 versus ADEPT. After this matching process, the weighted mean baseline characteristics of the FUTURE 2 population matched those reported for ADEPT, and the sample size of FUTURE 2 was reduced to a lower ESS.
Comparing Outcomes Using Recalculated Patient Data
The weights were used to recalculate outcomes for each IPD, and these were used to estimate the comparative effectiveness of secukinumab and adalimumab [8].
Analyses
Missing Data Handing
In ADEPT, all published outcomes were from the intention-to-treat (ITT) population. All missing binary outcome data (ACR 20, 50, and 70) were handled using nonresponder imputation (NRI) [15, 16], while missing patient-reported outcomes (PROs; continuous) were handled using last observation carried forward (LOCF) methodology. In FUTURE 2 [18], outcomes were from the ITT population. All missing binary outcome data (ACR 20, 50, and 70) were derived using NRI, while all missing PRO data (continuous) were derived using LOCF to match the available data from ADEPT. It is worth highlighting that our analysis did not use penalties at week 24 for early non-responders, thereby removing ‘bias’ when comparing our results with those for adalimumab and making our approach different from the numbers reported in the main FUTURE 2 publications.
Outcomes
Outcomes selected for comparison were in line with the Outcomes Measures in Rheumatology (OMERACT) [33, 34] and GRAPPA [35] recommendations on outcome measures that should be included in PsA clinical trials [33, 34].
-
ACR response rates: ACR 20, 50, and 70 response rates were assessed at weeks 12, 16, 24, and 48 in both trials.
-
PRO scores: mean change from baseline in Health Assessment Questionnaire Disability Index (HAQ-DI), Patient Global Assessment (PGA), pain assessment, and the Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F) score were included. Outcomes were reported at weeks 12, 24, and 48 in ADEPT so comparisons were feasible at these time points; no data were available for week 16 in ADEPT.
Placebo-Adjusted and Nonplacebo-Adjusted Outcome Comparisons
The study designs of the trials (Fig. 1) specified that patients randomized to placebo could receive active treatment from week 16 in FUTURE 2 or rescue therapy (corticosteroids or csDMARDs) from week 12 in ADEPT; hence, unbiased placebo-adjusted treatment comparisons were not possible after week 12. After week 12, outcomes from the adalimumab arm in ADEPT were directly compared with outcomes from the recalculated secukinumab arm of FUTURE 2. This is similar to a comparison of outcomes of two single-arm trials. In these situations, the use of MAIC may be the only way to adjust for cross-trial differences and should be preferred over naïve unadjusted comparisons. Commonly used in observational studies [36], this approach has been successfully applied to previous MAIC analyses of RCTs, such as when placebo comparisons were not available or valid [37], to overcome placebo crossover, or extrapolation beyond study end [32, 38].
A week 12, placebo-adjusted comparison was also made. In addition to providing meaningful short-term placebo-adjusted data, the placebo arms also act as a form of matching control, because if the populations have been properly matched (assuming no variables that impact on placebo responses that cannot be controlled through matching), there should be limited cross-trial differences in placebo arm responses.
Pairwise Comparisons
For ACR outcomes, relative risk (RR; see Fig. S2 in the supplementary material) values were estimated as the ratio of response rates (Table S7 in the supplementary material), and P values (two-sided) for indirect treatment comparison using RR were derived from the corresponding Z-statistic. For placebo-adjusted comparisons, RRs and corresponding standard errors were calculated using the Bucher method [7]. Odds ratios (ORs; see Fig. S2 in the supplementary material) were also calculated for ACR responses (Table S7 in the supplementary material). The commonly used threshold of P < 0.05 was considered as a threshold for statistical significance (i.e., the incompatibility of observed data with the null hypothesis position that there is no difference between secukinumab and adalimumab for the outcome being compared). In acknowledgment of the recent American Statistical Association statement on P values and their preference to avoid such a threshold in clinical research [39, 40], our data were also analyzed using a more modern definition of the strength of evidence that P values can provide [41, 42] (0.1 > P < 0.001 as increasing evidence, P ≤ 0.001 as strong evidence against the null hypothesis; Table S8 in the supplementary material). For nonplacebo-adjusted comparisons, standard errors for RR values were estimated based on the information provided by a fictitious 2 × 2 contingency table that shows outcomes in the adalimumab arm of the ADEPT trial and outcomes in the recalculated secukinumab arm of FUTURE 2 (with the ESS used as the sample size for FUTURE 2). For the analysis at week 12 (placebo-adjusted), RRs (and ORs) for the secukinumab arm versus the placebo arm in the reweighted FUTURE 2 population were derived from a logistic regression model by using generalized estimating equations with robust standard errors as suggested in previous work [9,10,11]. Generalized estimating equations were fitted using PROC GENMOD in SAS.
For PRO scores, a 95% CI around mean change scores of patients in the adalimumab arm of the ADEPT trial was estimated using the normal approximation. The P values for the difference in mean change scores between secukinumab and adalimumab were calculated using a Z-statistic that divides the difference in mean change scores by the combined standard error.
Psoriasis Area and Severity Index Outcomes
We did not report comparative Psoriasis Area and Severity Index (PASI) data, as the baseline characteristic data for the ADEPT trial psoriasis subgroup were not available. This meant that uncontrolled imbalances could persist post-matching. Indeed, the high absolute differences in PASI at baseline between studies lend further weight to this limitation. It is also worth noting that PASI outcome data were collected only in a subgroup of patients in both studies (patients with psoriasis affecting ≥ 3% body surface area), thus missing data in the non-PASI-matched population will further compound this study bias.
Methotrexate Subgroup Analysis
The long-term efficacy of anti-TNFs has been linked to concomitant MTX use [43]. MTX prescribed in combination with an anti-TNF may prolong anti-TNF survival [44] and dampen the generation of neutralizing antibodies to anti-TNFs [45,46,47,48]. Data from clinical registry studies suggest that the role of MTX in promoting persistence varies between different anti-TNFs [49].
Therefore, the impact of concomitant MTX on IL-17A versus anti-TNF treatment was assessed by a subgroup analysis in which patients were divided by concomitant MTX use at baseline. The issues surrounding the PASI data (above) do not affect the MTX subgroup analysis, as these data were collected for all patients (recorded as receiving or not receiving concomitant MTX) at the included timepoints.
Results
Principal Analysis
Matching Baseline Characteristics
Table 1 shows the baseline characteristics of patients from the FUTURE 2 secukinumab (pooled 75, 150, and 300 mg arms, n = 299 before matching) and placebo (n = 98 before matching) arms before and after matching (150 and 300 mg only [75 mg was not used in this comparison] and placebo) to the ADEPT adalimumab (n = 151) and placebo (n = 162) arms. Before matching, the study populations of FUTURE 2 and ADEPT were heterogeneous with FUTURE 2 including more difficult-to-treat patients. One of the key differences between the patient populations was that patients treated with secukinumab were either anti-TNF-naïve (65.2%) or anti-TNF-IR (34.8%), whereas all patients treated with adalimumab were anti-TNF-naïve (100%). After matching IPD from FUTURE 2 to ADEPT, all patients treated with secukinumab (or placebo) were anti-TNF-naïve (100%). Another key prematch dissimilarity between trials was baseline PASI score and a more severe HAQ-DI, indicating higher functional disability of the patient population in FUTURE 2.
Achieving homogeneity between the two populations reduced the sample size; the ESSs for FUTURE 2 after matching were 36 (secukinumab 150 mg), 38 (secukinumab 300 mg), and 27 (placebo).
It has been proposed that outcomes from common comparator arms such as placebo can be used to validate the matching process, i.e., a good match should lead to equivalent or near-equivalent placebo arm responses [8, 50]. Placebo arm ACR 20 (week 12) responses were 14.2% (ADEPT) and 26.7% (FUTURE 2); ACR 50 responses were 3.7% (ADEPT) and 4.3% (FUTURE 2). Given that ACR 50 is a significantly more stringent outcome than ACR 20, the near equivalence of the placebo response between ADEPT and recalculated FUTURE 2 suggests a good match.
ACR Response Rates
Figure 3 and Table 2 show the ACR 20, 50, and 70 response rates in the principal analysis. At week 12, there were no statistically significant differences in any ACR placebo-adjusted response rates between secukinumab and adalimumab. Week 16 ACR 20 and 50 response rates were significantly higher for secukinumab 150 mg than for adalimumab [RR, 1.34 (95% CI: 1.05, 1.70); P = 0.017 and RR, 1.54 (95% CI: 1.03, 2.30); P = 0.033, respectively], as was the ACR 50 response rate for secukinumab 300 mg over adalimumab [RR, 1.54 (95% CI: 1.04, 2.29); P = 0.030]. Week 24 ACR 20 and 50 response rates were significantly higher for secukinumab 150 mg than for adalimumab [RR, 1.42 (95% CI: 1.15, 1.75); P = 0.001 and RR, 1.50 (95% CI: 1.07, 2.10); P = 0.019, respectively], as was the ACR 20 response rate for secukinumab 300 mg over adalimumab [RR, 1.27 (95% CI: 1.00, 1.62); P = 0.048]. The week 48 ACR 20 response rate was significantly higher for secukinumab 150 mg than for adalimumab [RR, 1.41 (95% CI: 1.14, 1.76); P = 0.002], as were the ACR 20 and 50 response rates for secukinumab 300 mg over adalimumab [RR, 1.31 (95% CI: 1.03, 1.66); P = 0.027 and RR, 1.41 (95% CI: 1.03, 1.92); P = 0.032, respectively].

ACR comparisons: principal analysis. P values are derived from relative risk values. Error bars show 95% confidence intervals. Numbers above bars are the absolute mean predicted responses (ADEPT) and the predicted mean responses (FUTURE 2). Yellow background indicates that comparison was placebo-adjusted, white background indicates that comparison was nonplacebo-adjusted. ACR 20/50/70 20%/50%/70% or greater improvement in the American College of Rheumatology response criteria, ADA adalimumab, ESS effective sample size, SEC secukinumab
PRO Scores
Table 3 shows the principal analysis comparison of HAQ-DI, PGA, pain assessment, and FACIT-F scores. At week 12, there were no significant differences in any continuous outcome data between secukinumab and adalimumab. No ADEPT data were available for week 16. At week 24, treatment with secukinumab 150 mg resulted in significantly greater improvements in HAQ-DI, PGA, and pain scores relative to adalimumab (– 0.53 vs. – 0.40, P = 0.046; – 30.2 vs. – 21.1, P = 0.005; and – 30.4 vs. – 24.0, P = 0.039, respectively). Adalimumab therapy resulted in a significantly greater improvement in FACIT-F score relative to secukinumab 300 mg (7.1 vs. 4.5, P = 0.021), although there was no difference relative to secukinumab 150 mg. At week 48, treatment with secukinumab 150 mg resulted in a significantly greater improvement in HAQ-DI score, while secukinumab 300 mg had a significantly greater improvement in PGA score relative to adalimumab (– 0.54 vs. – 0.40, P = 0.037 and – 29.4 vs. – 22.4, P = 0.016, respectively).
Methotrexate Subgroup Analysis
Concomitant MTX subgroup data from ADEPT were available only at weeks 24 and 48. After matching, the sample sizes at week 24 for the subgroup receiving MTX (at baseline) were: adalimumab, n = 77; secukinumab 150 mg, ESS = 19; and secukinumab 300 mg, ESS = 17, while the sample sizes for the subgroup not receiving MTX (at baseline) were: ADEPT, n = 74; secukinumab 150 mg, ESS = 17; and secukinumab 300 mg, ESS = 22.
Figure 4 and Table 4 show the ACR response rates for the principal analysis MTX subgroup comparison. At week 24, for patients receiving MTX at baseline, ACR 20 response rates were higher with secukinumab 150 mg [RR, 1.55 (95% CI: 1.18, 2.05); P = 0.002] than with adalimumab. For patients not receiving MTX at baseline, ACR 20 response rates were higher with secukinumab 300 mg than with adalimumab [RR, 1.33 (95% CI: 1.00, 1.77); P = 0.048]. At week 48, for patients receiving MTX at baseline, ACR 20 response rates were higher with secukinumab 150 mg [RR, 1.37 (95% CI: 1.06, 1.76); P = 0.015] than with adalimumab. For patients not receiving MTX at baseline, ACR 20 [RR, 1.53 (95% CI: 1.11, 2.11); P = 0.010], ACR 50 [RR, 1.78 (95% CI: 1.18, 2.67); P = 0.006] and ACR 70 [RR, 1.88 (95% CI: 1.12, 3.17); P = 0.017] response rates were higher with secukinumab 300 mg than with adalimumab.

Methotrexate subgroup analysis: MAIC-predicted ACR response rates at weeks 24 and 48 in patients (a) receiving and (b) not receiving methotrexate at baseline. P values are derived from RR values. Error bars show 95% confidence intervals. Numbers above bars show the absolute mean predicted responses (ADEPT) and the predicted mean responses (FUTURE 2). ACR 20/50/70 20%/50%/70% or greater improvement in the American College of Rheumatology response criteria, ADA adalimumab, ESS effective sample size, MAIC matching-adjusted indirect comparison, RR relative risk, SEC secukinumab
Sensitivity Analysis
Matching Baseline Characteristics
The sensitivity analysis matched for the same parameters as the principal analysis, with the addition of three variables (PsA disease duration, SJC, and CRP), as shown in Table S6 in the supplementary material. The ESSs in FUTURE 2 after matching were 15 (secukinumab 150 mg), 25 (secukinumab 300 mg), and 20 (placebo).
Placebo arm ACR 20 (week 12) response rates were 14.2% (ADEPT) and 19.3% (secukinumab 150 mg, FUTURE 2); and ACR 50 responses were 3.7% (ADEPT) and 0.3% (FUTURE 2). The ACR 20 data suggest that this match was even closer than that of the principal analysis. There appears to be a lower ACR 50 placebo response with secukinumab 150 mg relative to adalimumab, although the low ESS and low values suggest this is a less reliable observation than the ACR 20 comparison.
ACR Response Rates
Results were broadly consistent with those of the principal analysis (Table 2), in that ACR 20 and 50 response rates were significantly higher for at least one secukinumab dose than for adalimumab at weeks 16, 24, and 48. In addition, at the week-12 placebo-adjusted time point, both doses of secukinumab showed a significantly higher probability of achieving an ACR 50 response than adalimumab.
PRO Scores
The sensitivity analysis also confirmed the higher outcomes for secukinumab over adalimumab in HAQ-DI, PGA, and pain scores, as seen in the principal analysis. In addition, week-12 placebo-adjusted secukinumab 300 mg showed a significantly greater improvement in HAQ-DI scores from baseline, and there was no longer any sign of higher outcomes for adalimumab in any of the FACIT-F scores (Table S9 in the supplementary material).
Methotrexate Subgroup Analysis
As shown in Table 4, the results were consistent with the principal analysis. A consistent statistically significantly higher probability of ACR 20 response rates was seen with secukinumab, relative to adalimumab, at weeks 24 and 48 (nonplacebo-adjusted).
Discussion
Our study used a methodologically valid MAIC [10, 51] to assess the comparative effectiveness of secukinumab versus adalimumab in patients with active PsA. Although several indirect comparison methodologies exist, MAIC was chosen because it allows for a greater degree of adjustment for cross-trial population differences than analyses that use only aggregate data. When populations are well matched [8], it is possible to compare long-term data beyond the placebo-controlled phase. This is necessary for chronic conditions such as PsA because the short-term placebo-controlled phase of most RCTs provides only limited data to inform the mid- to long-term treatment choices; indeed, several oncology studies have applied MAIC successfully to long-term patient survival data [37, 38].
In the absence of H2H RCT data, MAIC is a useful surrogate method that is, relative to an RCT, faster, cheaper, and capable of providing timely comparative evidence to relevant stakeholders. Nevertheless, observational data generated by MAIC should be viewed as interim to the reporting of a “gold-standard” H2H RCT, such as the ongoing EXCEED trial [5].
Our study included both binary (ACR 20, 50, and 70) and continuous (PRO scores) outcomes in line with the OMERACT and GRAPPA recommendations [33, 34]. The choice of matching variables was finalized after consultation with clinical experts in the spondyloarthritides and statistical fields, supplemented with empirical data identifying key effect-modifying variables [10]. Further baseline characteristics considered potentially clinically relevant were included in a sensitivity analysis. We selected adalimumab as the comparator because of its equivalent efficacy to other anti-TNFs and widespread clinical use [19]. Anti-TNFs are established and recommended treatment options for active PsA [52,53,54,55]. Although there have been no H2H RCTs between them, indirect retrospective data suggest that they have similar efficacy and safety profiles [55, 56]. Meta-analyses have repeatedly shown no real differences in terms of ACR outcomes among anti-TNFs [54, 57,58,59,60].
Our MAIC indicated that patients treated with secukinumab had at least an equivalent, or greater likelihood (depending on time point and outcome) of experiencing an improvement in joint signs and symptoms than individuals treated with adalimumab. In the principal analysis, both bDMARDs had comparable ACR responses at week 12 (placebo-adjusted), whilst we observed several statistically significant higher responses for secukinumab from week 16 onwards using a nonplacebo-adjusted comparison. The strongest evidence supporting a higher ACR response with secukinumab was apparent at weeks 24 and 48 for the 150-mg dose. The sensitivity analysis replicated the findings of the principal analysis. Taken together, our analyses provide a consistent body of evidence suggesting that patients with active PsA treated with secukinumab (150 or 300 mg) are more likely to achieve an ACR response through 1 year (weeks 16–48).
In the principal analysis of PRO data, we observed significantly improved week-24 HAQ-DI, PGA, and pain assessments and week-48 HAQ-DI scores for patients treated with secukinumab 150 mg, and a significantly improved PGA score for secukinumab 300 mg relative to those treated with adalimumab. Improvement in pain is considered by PsA patients to be the most important domain [61]. The improvements estimated for patients treated with secukinumab 300 mg were similar to those for individuals treated with adalimumab, for all time points and all PRO scores, except PGA.
In both of our concomitant MTX subgroups, we observed that patients receiving secukinumab had a greater likelihood of experiencing an improvement in joint signs and symptoms, measured by ACR response criteria at weeks 24 and 48, than did individuals treated with adalimumab. These findings, confirmed by the sensitivity analysis, suggest that the higher long-term outcomes for secukinumab compared with adalimumab for ACR response are independent of MTX use. However, as separate baseline matching was not performed in subgroups defined by MTX use, we cannot exclude the possibility that these populations may differ between the treatment groups.
The data reported in our study differ from a recently published MAIC analysis that compared IPD of the ADEPT trial with pooled FUTURE 1 and 2 data [12]. Their primary analysis showed no statistically significant differences in ACR responses between secukinumab and adalimumab at week 24 (placebo-adjusted) [12]. However, there are five major methodological limitations in this analysis. First, the matching parameters did not include previous anti-TNF exposure, the single most important observable baseline parameter. This is shown by regression analysis (Table S4 in the supplementary material), by anti-TNF IR subgroup analysis of FUTURE 2 [62], and by an earlier analysis of adalimumab [31]. Second, the analysis was based on a placebo-adjusted outcome comparison at week 24. This may lead to bias [9, 63] because by week 24, the respective placebo arms of both studies had undergone significant population loss at cumulatively unequal rates as a consequence of different study designs (Fig. 1). Third, we consider the use of pooled FUTURE 1 and FUTURE 2 study aggregate data a questionable strategy for short-term comparisons owing to their differences in loading administration. Fourth, PASI comparisons were made without being able to match the PASI patient subgroup. For the same reasons, we opted not to report PASI outcomes in this publication, as described in the Methods section above. Fifth, the other MAIC analysis did not use a linear predictor scale, for instance ORs or RRs, which is recommended by MAIC methodology guidelines [10]. Overall, our study goes beyond the scope of the previously published MAIC [12] in two main aspects: by providing comparative effectiveness data for up to 1 year at multiple time points and for multiple clinical endpoints and PROs, and by adjusting for previous anti-TNF exposure. Despite the above-mentioned limitations, the results reported in the sensitivity analysis of the previously published MAIC [12] showed the same trend and are in agreement with our study, i.e., higher ACR 20 and 50 outcomes in anti-TNF-naïve patients receiving secukinumab compared with adalimumab. Moreover, two recent publications indicate favorable sustainability and safety data for secukinumab [27, 64].
Our MAIC has limitations, both intrinsic to the methodology and specific to this analysis. Although observed patient variables at baseline can be matched, it is not possible to control for unobserved or unreported variables. Although both studies were not contemporaneous, a common issue in comparative analysis, we have made significant efforts to accommodate differences in study design such as our matching of appropriate missing data-handling and imputation methods. More specific to our study, the relatively small ESS used in all of the analyses, driven by limited overlap between trial populations, must be taken into account when interpreting our findings. A majority of the FUTURE 2 patients were effectively lost after matching (particularly in the sensitivity and MTX subgroup analyses), and as such, the findings of this MAIC rely upon a subset of trial participants. Finally, due to differences in the study trials regarding the time point from which placebo-treated patients can receive rescue therapy, an unbiased placebo-adjustment was not possible after week 12. Therefore, we present nonplacebo-adjusted comparisons that, while providing a legitimate means to compare long-term data, do not permit adjustment for unobserved differences in trial populations or study design. Given these limitations, our MAIC analysis is strongest during the placebo-matched period, is hypothesis-generating only, and needs to be confirmed via the ongoing EXCEED H2H RCT of secukinumab versus adalimumab in patients with active PsA.
Conclusions
This MAIC showed evidence suggesting that patients with active PsA treated with secukinumab and matched to the ADEPT study population have a significantly higher probability of achieving ACR 20 and 50 response rates through 1 year than those receiving adalimumab, an effect that appears to be independent of concomitant MTX medication. To substantiate these hypothesis-generating interim findings, the next level of evidence will be determined by the ongoing H2H RCT EXCEED [5].
Change history
01 June 2018
Under Methods section, heading Analyses: Pairwise Comparisons, one of the probability value was published incorrectly in a sentence. The correct sentence should read as follows.
References
Gossec L, Smolen JS, Ramiro S, de Wit M, Cutolo M, Dougados M, et al. European League Against Rheumatism (EULAR) recommendations for the management of psoriatic arthritis with pharmacological therapies: 2015 update. Ann Rheum Dis. 2016;75(3):499–510.
Gossec L, Smolen JS, Gaujoux-Viala C, Ash Z, Marzo-Ortega H, van der Heijde D, et al. European League Against Rheumatism recommendations for the management of psoriatic arthritis with pharmacological therapies. Ann Rheum Dis. 2012;71(1):4–12.
Coates LC, Kavanaugh A, Mease PJ, Soriano ER, Laura Acosta-Felquer M, Armstrong AW, et al. Group for research and assessment of psoriasis and psoriatic arthritis 2015 treatment recommendations for psoriatic arthritis. Arthritis Rheumatol. 2016;68(5):1060–71.
Tintle SJ, Gottlieb AB. Psoriatic arthritis for the dermatologist. Dermatol Clin. 2015;33(1):127–48.
NCT02745080. Efficacy of secukinumab compared to adalimumab in patients with psoriatic arthritis (EXCEED 1). 2016. https://clinicaltrials.gov/ct2/show/NCT02745080. Accessed 1 Dec 2017.
Dias S, Sutton AJ, Ades AE, Welton NJ. Evidence synthesis for decision making 2: a generalized linear modeling framework for pairwise and network meta-analysis of randomized controlled trials. Med Decis Making. 2013;33(5):607–17.
Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. J Clin Epidemiol. 1997;50(6):683–91.
Signorovitch JE, Sikirica V, Erder MH, Xie J, Lu M, Hodgkins PS, et al. Matching-adjusted indirect comparisons: a new tool for timely comparative effectiveness research. Value Health. 2012;15(6):940–7.
Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submission to NICE. 2016. http://www.nicedsu.org.uk. Accessed 1 Dec 2017.
Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for population-adjusted indirect comparisons in health technology appraisal. Med Decis Making. 2018;38(2):200–11.
Signorovitch JE, Wu EQ, Yu AP, Gerrits CM, Kantor E, Bao Y, et al. Comparative effectiveness without head-to-head trials: a method for matching-adjusted indirect comparisons applied to psoriasis treatment with adalimumab or etanercept. Pharmacoeconomics. 2010;28(10):935–45.
Strand V, Betts KA, Mittal M, Song J, Skup M, Joshi A. Comparative effectiveness of adalimumab versus secukinumab for the treatment of psoriatic arthritis: a matching-adjusted indirect comparison. Rheumatol Ther. 2017;4(2):349–62.
Kirson NY, Rao S, Birnbaum HG, Kantor E, Wei RS, Cifaldi M. Matching-adjusted indirect comparison of adalimumab vs. etanercept and infliximab for the treatment of psoriatic arthritis. J Med Econ. 2013;16(4):479–89.
Thom H, Jugl SM, Palaka E, Jawla S. Matching adjusted indirect comparisons to assess comparative effectiveness of therapies: usage in scientific literature and health technology appraisals. Value Health. 2016;19:A100–1.
Mease PJ, Gladman DD, Ritchlin CT, Ruderman EM, Steinfeld SD, Choy EH, et al. Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: results of a double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2005;52(10):3279–89.
Gladman DD, Mease PJ, Ritchlin CT, Choy EH, Sharp JT, Ory PA, et al. Adalimumab for long-term treatment of psoriatic arthritis: forty-eight week data from the adalimumab effectiveness in psoriatic arthritis trial. Arthritis Rheum. 2007;56(2):476–88.
Mease PJ, Ory P, Sharp JT, Ritchlin CT, Van den Bosch F, Wellborne F, et al. Adalimumab for long-term treatment of psoriatic arthritis: 2-year data from the Adalimumab Effectiveness in Psoriatic Arthritis Trial (ADEPT). Ann Rheum Dis. 2009;68(5):702–9.
McInnes IB, Mease PJ, Kirkham B, Kavanaugh A, Ritchlin CT, Rahman P, et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2015;386(9999):1137–46.
Armuzzi A, Lionetti P, Blandizzi C, Caporali R, Chimenti S, Cimino L, et al. Anti-TNF agents as therapeutic choice in immune-mediated inflammatory diseases: focus on adalimumab. Int J Immunopathol Pharmacol. 2014;27(1 Suppl):11–32.
Thaci D, Blauvelt A, Reich K, Tsai TF, Vanaclocha F, Kingo K, et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: CLEAR, a randomized controlled trial. J Am Acad Dermatol. 2015;73(3):400–9.
Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, et al. Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med. 2014;371(4):326–38.
Mease PJ, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, et al. Secukinumab inhibition of interleukin-17A in patients with psoriatic arthritis. N Engl J Med. 2015;373(14):1329–39.
McInnes IB, Sieper J, Braun J, Emery P, van der Heijde D, Isaacs JD, et al. Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann Rheum Dis. 2014;73(2):349–56.
Behrens F, Kohm M, Arndt U, Wittig B, Greger G, Thaci D, et al. Impact of methotrexate on anti-TNF treatment in psoriatic arthritis? An in-depth analysis of a large prospective observational study with adalimumab. Ann Rheum Dis. 2014;73:723.
Genovese MC, Mease PJ, Thomson GT, Kivitz AJ, Perdok RJ, Weinberg MA, et al. Safety and efficacy of adalimumab in treatment of patients with psoriatic arthritis who had failed disease modifying antirheumatic drug therapy. J Rheumatol. 2007;34(5):1040–50.
van Kuijk AW, Gerlag DM, Vos K, Wolbink G, de Groot M, de Rie MA, et al. A prospective, randomised, placebo-controlled study to identify biomarkers associated with active treatment in psoriatic arthritis: effects of adalimumab treatment on synovial tissue. Ann Rheum Dis. 2009;68(8):1303–9.
McInnes IB, Mease PJ, Ritchlin CT, Rahman P, Gottlieb AB, Kirkham B, et al. Secukinumab sustains improvement in signs and symptoms of psoriatic arthritis: 2 year results from the phase 3 FUTURE 2 study. Rheumatology. 2017;56(11):1993–2003.
Kavanaugh A, McInnes IB, Mease PJ, Hall S, Chinoy H, Kivitz AJ, et al. Efficacy of subcutaneous secukinumab in patients with active psoriatic arthritis stratified by prior tumor necrosis factor inhibitor use: results from the randomized placebo-controlled FUTURE 2 study. J Rheumatol. 2016;43(9):1713–7.
Mease P, McInnes IB. Secukinumab: a new treatment option for psoriatic arthritis. Rheumatol Ther. 2016;3(1):5–29.
Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, et al. American College of Rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum. 1995;38(6):727–35.
Gladman DD, Investigators AS, Sampalis JS, Illouz O, Guerette B. Responses to adalimumab in patients with active psoriatic arthritis who have not adequately responded to prior therapy: effectiveness and safety results from an open-label study. J Rheumatol. 2010;37(9):1898–906.
Signorovitch J, Swallow E, Kantor E, Wang X, Klimovsky J, Haas T, et al. Everolimus and sunitinib for advanced pancreatic neuroendocrine tumors: a matching-adjusted indirect comparison. Exp Hematol Oncol. 2013;2(1):32.
Gladman DD, Mease PJ, Strand V, Healy P, Helliwell PS, Fitzgerald O, et al. Consensus on a core set of domains for psoriatic arthritis. J Rheumatol. 2007;34(5):1167–70.
Tillett W, Eder L, Goel N, De Wit M, Gladman DD, FitzGerald O, et al. Enhanced patient involvement and the need to revise the core set—report from the Psoriatic Arthritis Working Group at OMERACT 2014. J Rheumatol. 2015;42(11):2198–203.
Orbai AM, Mease PJ, de Wit M, Kalyoncu U, Campbell W, Tillett W, et al. Report of the GRAPPA-OMERACT Psoriatic Arthritis Working Group from the GRAPPA 2015 Annual Meeting. J Rheumatol. 2016;43(5):965–9.
Austin PC. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivar Behav Res. 2011;46(3):399–424.
Swallow E, Song J, Yuan Y, Kalsekar A, Kelley C, Mu F, et al. Daclatasvir + sofosbuvir versus standard of care for hepatitis C genotype 3: a matching-adjusted indirect comparison. J Comp Eff Res. 2016;5(2):129–39.
Dearden L, Majer I, Heeg B, Liwing J, Sandstrom K, Diels J. Comparison of mean overall survival (OS) and radiographic progression free survival (RPFS) based on matching adjusted indirect comparison of abiraterone acetate and enzalutamide for the treatment of castration-resistant prostate cancer in chemotherapy naive patients. Value Health. 2014;17(7):A616.
Wasserstein RL, Lazar NA. The ASA’s statement on p-values: context, process, and purpose. Am Stat. 2016;70(2):129–33.
Best AM 3rd, Greenberg BL, Glick M. From tea tasting to t test: a P value ain’t what you think it is. J Am Dent Assoc. 2016;147(7):527–9.
Sterne JA, Davey Smith G. Sifting the evidence—what’s wrong with significance tests? BMJ. 2001;322(7280):226–31.
Goodman S, Greenland S. Why most published research findings are false: problems in the analysis. PLoS Med. 2007;4(4):e168.
Kang EJ, Kavanaugh A. Psoriatic arthritis: latest treatments and their place in therapy. Ther Adv Chronic Dis. 2015;6(4):194–203.
Behrens F, Canete JD, Olivieri I, van Kuijk AW, McHugh N, Combe B. Tumour necrosis factor inhibitor monotherapy vs combination with MTX in the treatment of PsA: a systematic review of the literature. Rheumatology. 2015;54(5):915–26.
Zisapel M, Zisman D, Madar-Balakirski N, Arad U, Padova H, Matz H, et al. Prevalence of TNF-alpha blocker immunogenicity in psoriatic arthritis. J Rheumatol. 2015;42(1):73–8.
van Kuijk AW, de Groot M, Stapel SO, Dijkmans BA, Wolbink GJ, Tak PP. Relationship between the clinical response to adalimumab treatment and serum levels of adalimumab and anti-adalimumab antibodies in patients with psoriatic arthritis. Ann Rheum Dis. 2010;69(3):624–5.
Kavanaugh A, McInnes IB, Mease P, Krueger GG, Gladman D, van der Heijde D, et al. Clinical efficacy, radiographic and safety findings through 5 years of subcutaneous golimumab treatment in patients with active psoriatic arthritis: results from a long-term extension of a randomised, placebo-controlled trial (the GO-REVEAL study). Ann Rheum Dis. 2014;73(9):1689–94.
Murdaca G, Spano F, Contatore M, Guastalla A, Penza E, Magnani O, et al. Immunogenicity of infliximab and adalimumab: what is its role in hypersensitivity and modulation of therapeutic efficacy and safety? Expert Opin Drug Saf. 2016;15(1):43–52.
Mease PJ, Collier DH, Saunders KC, Li G, Kremer JM, Greenberg JD. Comparative effectiveness of biologic monotherapy versus combination therapy for patients with psoriatic arthritis: results from the Corrona registry. RMD Open. 2015;1(1):e000181.
Sikirica V, Findling RL, Signorovitch J, Erder MH, Dammerman R, Hodgkins P, et al. Comparative efficacy of guanfacine extended release versus atomoxetine for the treatment of attention-deficit/hyperactivity disorder in children and adolescents: applying matching-adjusted indirect comparison methodology. CNS Drugs. 2013;27(11):943–53.
Veroniki AA, Straus SE, Soobiah C, Elliott MJ, Tricco AC. A scoping review of indirect comparison methods and applications using individual patient data. BMC Med Res Methodol. 2016;16(1):47.
Ceponis A, Kavanaugh A. Treatment of psoriatic arthritis with biological agents. Semin Cutan Med Surg. 2010;29(1):56–62.
Saber TP, Veale DJ. Psoriatic arthritis management update—biotherapeutic options. J Rheumatol Suppl. 2009;83:65–8.
Ash Z, Gaujoux-Viala C, Gossec L, Hensor EM, FitzGerald O, Winthrop K, et al. A systematic literature review of drug therapies for the treatment of psoriatic arthritis: current evidence and meta-analysis informing the EULAR recommendations for the management of psoriatic arthritis. Ann Rheum Dis. 2012;71(3):319–26.
Sritheran D, Leung YY. Making the next steps in psoriatic arthritis management: current status and future directions. Ther Adv Musculoskelet Dis. 2015;7(5):173–86.
Mease PJ. Psoriatic arthritis—update on pathophysiology, assessment, and management. Bull NYU Hosp Jt Dis. 2010;68(3):191–8.
Saad AA, Symmons DP, Noyce PR, Ashcroft DM. Risks and benefits of tumor necrosis factor-alpha inhibitors in the management of psoriatic arthritis: systematic review and metaanalysis of randomized controlled trials. J Rheumatol. 2008;35(5):883–90.
Lemos LL, de Oliveira Costa J, Almeida AM, Junior HO, Barbosa MM, Kakehasi AM, et al. Treatment of psoriatic arthritis with anti-TNF agents: a systematic review and meta-analysis of efficacy, effectiveness and safety. Rheumatol Int. 2014;34(10):1345–60.
National Institute for Health and Care Excellence: TA199, etanercept, infliximab and adalimumab for the treatment of psoriatic arthritis. 2016. https://www.nice.org.uk/guidance/ta199. Accessed 1 Dec 2017.
Fenix-Caballero S, Alegre-del Rey EJ, Castano-Lara R, Puigventos-Latorre F, Borrero-Rubio JM, Lopez-Vallejo JF. Direct and indirect comparison of the efficacy and safety of adalimumab, etanercept, infliximab and golimumab in psoriatic arthritis. J Clin Pharm Ther. 2013;38(4):286–93.
Orbai AM, de Wit M, Mease P, Shea JA, Gossec L, Leung YY, et al. International patient and physician consensus on a psoriatic arthritis core outcome set for clinical trials. Ann Rheum Dis. 2017;76(4):673–80.
Kavanaugh A, McInnes IB, Mease PJ, Hall S, Chinoy H, Kivitz AJ, et al. Efficacy of subcutaneous secukinumab in patients with active psoriatic arthritis stratified by prior tumor necrosis factor inhibitor use: results from the randomized placebo-controlled FUTURE 2 Study. J Rheumatol. 2016;43(9):1713–7.
Song F, Loke YK, Walsh T, Glenny AM, Eastwood AJ, Altman DG. Methodological problems in the use of indirect comparisons for evaluating healthcare interventions: survey of published systematic reviews. BMJ. 2009;338:b1147.
Kammüller M, Tsai TF, Griffiths CE, Kapoor N, Kolattukudy PE, Brees D, et al. Inhibition of IL-17A by secukinumab shows no evidence of increased Mycobacterium tuberculosis infections. Clin Transl Immunol. 2017;6:e152.
Gladman DD, Mease PJ, Cifaldi MA, Perdok RJ, Sasso E, Medich J. Adalimumab improves joint-related and skin-related functional impairment in patients with psoriatic arthritis: patient-reported outcomes of the adalimumab effectiveness in psoriatic arthritis trial. Ann Rheum Dis. 2007;66(2):163–8.
Gottlieb AB, Langley RG, Philipp S, Sigurgeirsson B, Blauvelt A, Martin R, et al. Secukinumab improves physical function in subjects with plaque psoriasis and psoriatic arthritis: results from two randomized, phase 3 trials. J Drugs Dermatol. 2015;14(8):821–33.
Gottlieb AB, Sigurgeirsson B, Blauvelt A, Mpfofu S, Martin R, Papavassilis C. Secukinumab shows substantial improvement in both psoriasis symptoms and physical functioning in moderate-to-severe plaque psoriasis patients with psoriatic arthritis: a subanalysis of a phase 3, multicenter, double-blind, placebo-controlled study. Arthritis Rheum. 2013;65:S136–7.
Blauvelt A, Gottlieb A, Sigurgeirsson B, Papavassilis C, Martin R. Secukinumab efficacy in subjects with moderate-to-severe plaque psoriasis and concomitant psoriatic arthritis: a subanalysis of the ERASURE study. J Am Acad Dermatol. 2014;70(5):AB2.
Gottlieb A, Sigurgeirsson B, Blauvelt A, Gong Y, Papavassilis C, Mpofu S. Secukinumab reduces hsCRP levels in subjects with moderate-to-severe plaque psoriasis and concomitant psoriatic arthritis: a sub-analysis from the phase 3 erasure study. Ann Rheum Dis. 2014;73:1047–8.
Philipp S, Gottlieb AB, Langley RG, Sigurgeirsson B, Blauvelt A, Gong Y, et al. Secukinumab decreases inflammation as measured by a biomarker hsCRP in subjects with moderate-to-severe plaque psoriasis and concomitant psoriatic arthritis: subanalyses from two phase 3 studies. Exp Dermatol. 2015;24(3):E27.
Gottlieb A, Langley R, Philipp S, Martin R, Papavassilis C, Mpfofu S. Improvement in psoriasis symptoms and physical functioning with secukinumab compared with placebo and etanercept in subjects with moderate-to-severe plaque psoriasis and psoriatic arthritis: results of a subanalysis from the phase 3 fixture study. Arthritis Rheum. 2013;65(12):3322.
Sigurgeirsson B, Gottlieb AB, Langley RG, Philipp S, Martin RLM, Papavassilis C, et al. Effect of secukinumab on psoriasis symptoms and physical functioning compared with placebo and etanercept in subjects with moderate-to-severe plaque psoriasis and concomitant psoriatic arthritis: a subanalysis from the phase 3 fixture study. Scand J Rheumatol. 2014;43:64–5.
Gottlieb AB, Mease P, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, significantly reduces psoriasis burden in patients with psoriatic arthritis: results from a phase 3 randomized controlled trial. Arthritis Rheum. 2014;66(Suppl 10):S233.
Mease P, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, improves active psoriatic arthritis and inhibits radiographic progression: efficacy and safety data from a phase 3 randomized, multicenter, double-blind, placebo-controlled study. Arthritis Rheum. 2014;66(Suppl 10):S423–4.
Mease P, McInnes I, Kirkham B, Kavanaugh A, Rahman P, Van der Heijde D, et al. Secukinumab provides sustained improvements in psoriatic arthritis: 2-year efficacy and safety results from a phase 3 randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. 2015;67(Suppl 10):2576–8.
Mease P, McInnes I, Kirkham B, Kavanaugh A, Rahman P, Van der Heijde D, et al. Secukinumab improves active psoriatic arthritis and inhibits radiographic progression: results of a phase 3 randomized, multicenter, double-blind, placebo-controlled study. J Invest Dermatol. 2015;135:S29.
Strand V, Mease P, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, et al. Secukinumab, an anti-interleukin-17A monoclonal antibody, improves physical function, quality of life and work productivity in patients with active psoriatic arthritis: results from a phase 3, randomized, controlled trial. Arthritis Rheumatol. 2014;66(Suppl 10):S240–1.
van der Heijde D, Landewe RBM, Mease P, McInnes IB, Conaghan PG, Pricop L, et al. Secukinumab, a monoclonal antibody to interleukin-17A, provides significant and sustained inhibition of joint structural damage in active psoriatic arthritis regardless of prior TNF inhibitors or concomitant methotrexate: a phase 3 randomized, double-blind, placebo-controlled study. Arthritis Rheumatol. 2014;66:S424–5.
Gottlieb A, McInnes I, Mease P, Kirkham B, Kavanaugh A, Ligozio G, et al. Secukinumab improves signs and symptoms of active psoriatic arthritis: results from a phase 3 randomized, multicenter, double-blind, placebo-controlled study using a subcutaneous dosing regimen (FUTURE 2). J Invest Dermatol. 2015;135(Suppl 3):S29.
Gottlieb A, Strand V, McInnes I, Marzo-Ortega H, Kavanaugh A, Kandala S, et al. Secukinumab improves physical function, quality of life, fatigue and work productivity in patients with active psoriatic arthritis: results of a randomized, double-blind, placebo-controlled phase 3 trial (FUTURE 2). J Invest Dermatol. 2015;135:S30.
Kavanaugh A, McInnes I, Mease P, Hall S, Chinoy H, Kivitz AJ, et al. Secukinumab provides sustained improvements in the signs and symptoms of active psoriatic arthritis in anti-TNF-naive patients and those previously exposed to anti-TNF therapy: 52-week results from a randomized, double-blind, placebo-controlled phase 3 trial with subcutaneous dosing. Arthritis Rheumatol. 2015;67(Suppl 10):2890.
McInnes IB, Mease PJ, Kirkham B, Kavanaugh A, Ritchlin CT, Rahman P, et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, improves active psoriatic arthritis: 24-week efficacy and safety data from a phase 3 randomized, multicenter, double-blind, placebo-controlled study using subcutaneous dosing. Arthritis Rheumatol. 2014;66(Suppl 10):3529.
Mease P, Kirkham B, McInnes I, Kremer J, Kandala S, Pricop L, et al. Secukinumab is effective in reducing dactylitis and enthesitis using multiple measures in patients with psoriatic arthritis: results of a phase 3 randomized, multicenter, double-blind, placebo-controlled study (FUTURE 2). J Invest Dermatol. 2015;135(Suppl 3):S30.
Rahman P, Strand V, McInnes I, Marzo-Ortega H, Dokoupilova E, Churchill M, et al. Secukinumab improves physical function, quality of life, fatigue and work productivity in patients with active psoriatic arthritis in FUTURE 2, a phase 3 trial. Ann Rheum Dis. 2015;74(Suppl 2):356.
Genovese MC, Mease PJ, Thomson GT, Kivitz AJ, Perdok RJ, Weinberg MA, et al. Safety and efficacy of adalimumab in treatment of patients with psoriatic arthritis who had failed disease-modifying antirheumatic drug therapy. J Rheumatol. 2007;34:1040–50.
McInnes I, Sieper J, Braun J, Emery P, van der Heijde D, Isaacs J, et al. Secukinumab, a fully human anti-interleukin-17A antibody, improves signs and symptoms of psoriatic arthritis: a 24-week, double-blind, placebo-controlled, multicenter trial. Dermatol Ther. 2012;2:S37.
McInnes I, Sieper J, Braun J, Emery P, van der Heijde D, Isaacs J, et al. Effect of secukinumab on signs and symptoms of psoriatic arthritis: results of a 24-week multicentre, double-blind, randomized, placebo-controlled trial. Rheumatology (United Kingdom). 2012;51:iii90–1.
de Groot M, Picavet DI, van Kuijk AW, Tak PP, Bos JD, de Rie MA, et al. A prospective, randomized, placebo-controlled study to identify biomarkers associated with active treatment in psoriatic arthritis: effects of adalimumab treatment on lesional and nonlesional skin. Dermatology. 2012;225(4):298–303.
Acknowledgements
Funding
The MAIC and associated report completed by the MAPI Group were funded by Novartis Pharma AG. The original SLR was performed by RTI Health Solutions, and the update was performed by Costello Medical Consulting Ltd, both funded by Novartis Pharma AG. RTI Health Solutions conducted the regression analysis of baseline parameters in FUTURE 2, which was funded by Novartis Pharma AG. The FUTURE 2 trial was funded by Novartis Pharma AG, and the ADEPT trial was funded by Abbott Laboratories (now AbbVie Inc.). We also want to thank all investigators, patients, and sponsors who participated in the FUTURE 2 and ADEPT trials. Article processing charges were funded by Novartis Pharma AG. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published. Peter Nash, Iain B. McInnes, Philip J. Mease, Howard Thom, Kunal Gandhi, Matthias Hunger, Andreas Karabis, and Steffen M. Jugl designed the study, analyzed and interpreted the data, and decided on manuscript content and structure. Howard Thom advised on and reviewed the statistical methodology. Steffen Jugl wrote the manuscript. Shephard Mpofu advised on design, data content, interpretation, and contributed to manuscript writing. Matthias Hunger and Andreas Karabis performed the MAIC analysis and advised on methodology.
Medical Writing and/or Editorial Assistance
Writing support was provided by Martin Dalziel and Christian Eichinger, from Oxford PharmaGenesis, Oxford, UK, and was funded by Novartis Pharma AG, Basel, Switzerland.
Disclosures
Peter Nash has received grant/research support and consulting fees from Novartis. Iain B. McInnes has received consulting fees from AbbVie, Janssen, Pfizer, Novartis, and UCB. Philip J. Mease has received grant/research support from Corrona, and speakers’ bureau from Corrona, Merck, and Novartis. Howard Thom has received consulting fees from Eli Lilly, Novartis, Pfizer, and Roche. Matthias Hunger is an employee of The MAPI Group. Andreas Karabis is an employee of The MAPI Group. Kunal Gandhi is a shareholder and employee of Novartis. Shephard Mpofu is a shareholder and employee of Novartis. Steffen Jugl is a shareholder and employee of Novartis.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Data Availability
The datasets analyzed in the current study are available from the corresponding author on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to https://doi.org/10.6084/m9.figshare.5947795.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nash, P., McInnes, I.B., Mease, P.J. et al. Secukinumab Versus Adalimumab for Psoriatic Arthritis: Comparative Effectiveness up to 48 Weeks Using a Matching-Adjusted Indirect Comparison. Rheumatol Ther 5, 99–122 (2018). https://doi.org/10.1007/s40744-018-0106-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-018-0106-6