
Abstract
Periodontal disease results from an interaction between the host’s defense mechanisms and the microorganisms that constitute the dental plaque biofilm, and penetrate gingival tissue. Therefore, the progression and severity of the disease are strongly modulated by the host immune response, particularly, T cell responses. Because T cells consist of a variety of subpopulations, numerous studies have attempted to associate an impaired balance between each T cell subset and periodontal tissue destruction in periodontitis patients. Here, we overview studies examining human specimens obtained from patients with periodontitis and experiments analyzing rodent models with age-related or pathogen-induced experimental periodontitis. Human research provides valuable insights but also inconsistent results, which may be attributed to the difference in experimental approaches and lack of evaluation of disease activity. Rodent models have shown that an optimal balance between functionally different T cells is essential in the protection against periodontal tissue destruction.
Similar content being viewed by others


Introduction
Periodontal disease results from an interaction between the host’s defense mechanisms and the microorganisms that are present in dental plaque biofilms. Although bacteria play indispensable roles in the initiation of the disease, the subsequent progression and severity of the disease are strongly influenced by the host immune response. Periodontitis affects tooth-supporting bone and soft tissues and is a major cause of tooth loss. Periodontitis lesions are characterized as a predominant B cell response; however, T cells have an essential immunoregulatory role [1]. In addition, several studies have suggested the involvement of autoreactive T cells and autoantibodies in the pathogenesis of periodontitis [2–5].
Because it is well known that T cells are a heterogeneous population, and a balance between functionally different T cell subsets is crucial in immune regulation, a series of studies have attempted to associate an impaired balance between each T cell subset and periodontal disease progression. Nevertheless, it seems difficult to reveal this association because each T cell subset exhibits beneficial and detrimental properties depending on disease conditions [6].
In this review, we will discuss the presence and the role of various T cell subsets, including CD8+ cytotoxic T cells, CD4+ T helper cells (Th1, Th2, and Th17), regulatory T cells, and natural killer T (NKT) cells in the pathogenesis of periodontitis. To this end, we will give an overview of the human studies that analyzed specimens such as gingival tissues, gingival crevicular fluid, and peripheral blood obtained from periodontitis patients. In addition, we summarize the current knowledge about rodent models with age-related or experimental periodontitis induced by major periodontopathic bacteria (Table 1).
CD4+ Helper and CD8+ Cytotoxic T Cell Lineages
CD4+ and CD8+ T cells differentiate from CD4+CD8+ double-positive precursors in the thymus and exhibit distinct antigen specificities and functions. CD4+ T cells are major histocompatibility complex (MHC)-II-restricted and preprogrammed for helper functions, whereas CD8+ T cells are MHC-I-restricted and preprogrammed for cytotoxic functions. Because the ratio of CD4+ to CD8+ T cells is one of the indexes to determine the character of immune responses, its association with periodontal disease progression has been extensively examined. Some researchers analyzed the cells extracted from human gingival tissues and found decreased CD4/CD8 ratios in periodontitis lesions compared with healthy sites or peripheral blood [23, 24]. According to histologic analysis, some studies reported decreased CD4/CD8 ratios in periodontitis lesions compared with gingivitis or healthy sites [25, 26], whereas some reported increased CD4/CD8 ratios [27, 28], and others found no differences in the ratios [29, 30]. As discussed below, contradictory results obtained in human studies could be attributable to differences in methodology between studies, and the timing of sampling relative to disease activity.
A few studies have examined the impact of CD4+ and CD8+ T cells on periodontopathic bacteria-induced alveolar bone resorption in mouse models. Baker et al showed that mice lacking MHC-II-restricted CD4+ T cells, but not MHC-I-restricted CD8+ T cells, were resistant to alveolar bone loss induced by Porphyromonas gingivalis oral infection [7]. In addition, oral Aggregatibacter actinomycetemcomitans challenge of NOD/SCID mice transplanted with peripheral blood lymphocytes from periodontitis patients (HuPBL-NOD/SCID mice) demonstrated that CD4+ T cells, but not CD8+ T cells, were essential mediators of alveolar bone destruction [8]. These findings in mouse models suggest that CD4+ T cells contribute to bone destruction in periodontal disease.
CD4+ T Helper Subsets (Th1, Th2, and Th17)
Several CD4+ T cell subsets have been identified based on their distinct cytokine and transcription factor profiles [31]. Peptide affinity for MHC-II, T cell receptor activation, the expressed costimulatory molecules, and the cytokines secreted by dendritic cells collectively determine the signal strength that influences the differentiation of naïve T helper cells into Th1, Th2, Th17, or inducible regulatory T cells. Th1 and Th2 are classic T helper subsets that are characterized by the transcription factors T-bet and GATA-3, respectively. Th1 differentiation is accelerated by interleukin (IL)-12, while exposure to IL-4 drives Th2 differentiation. Th1 cells secrete IL-2 and interferon (IFN)-γ, which promote cell-mediated immunity by activating macrophages, CD8+ T cells, and NK cells.
The Th1 response is essential in immune defense against intracellular bacteria, viruses, and tumors, but possibly promotes autoimmune disease. However, the Th2 response produces IL-4, IL-5, IL-6, IL-10, and IL-13, which mediate humoral immunity by promoting B cell proliferation, differentiation, and antibody production. The Th2 response is involved in parasitic worm immunity and allergic inflammatory responses, including airway hyper-reactivity. Th17 is a recently identified T helper subset that expresses the transcription factors RORγt and RORα and produces IL-17A, IL-17 F, IL-21, IL-22, and IL-26. It has been reported that cytokines such as IL-1β, IL-6, IL-21, IL-23, and TGF-β are important for Th17 differentiation and development.
Th17 cells play a protective role against extracellular bacteria and fungi but are also related to several autoimmune and inflammatory disorders such as the inflammatory bone destructive disorder rheumatoid arthritis [32]. Importantly, although Th1 and Th2 cells have inhibitory effects on osteoclastogenesis through IFN-γ and IL-4, respectively, Th17 is defined as an osteoclastogenic T helper subset because it induces receptor activator of nuclear factor-κB ligand (RANKL) on osteoclastogenesis-supporting cells such as osteoblasts and synovial fibroblasts. Additionally, IL-17 enhances local inflammation and increases the production of inflammatory cytokines such as tumor necrosis factor (TNF)-α, IL-1, and IL-6, which further promote RANKL expression. Furthermore, Th17 cells themselves express RANKL and may participate directly in osteoclastogenesis [33, 34••].
Until the discovery of Th17, researchers had focused on the Th1/Th2 paradigm and attempted to identify either subset as causative for the progression of periodontal disease. In humans, numerous studies have supported the hypothesis that Th1 cells are associated with stable lesions and Th2 cells are associated with progressive lesions [35–40]. In contrast, several studies have demonstrated that upregulation of Th1 responses or downregulation of Th2 responses are involved in periodontal tissue destruction [41–43]. Moreover, others have shown a comparable presence of Th1 and Th2 cytokines in human periodontitis lesions [44–49].
To clarify the functional role of Th1 and Th2 in periodontal disease, several rodent models have been developed. Mice with A. actinomycetemcomitans-induced periodontitis demonstrated upregulation of Th1 cytokines (IFN-γ and IL-12) in the early stage and that of Th2 cytokines (IL-4 and IL-10) in the late stage, indicating the involvement of each T cell subset in different disease stages [9]. Destructive effects of Th1 and Th2 cytokines were suggested in an experiment that showed that IFN-γ- and IL-6-deficient mice were resistant to P. gingivalis-induced alveolar bone loss [7]. However, the pathogenic role of Th1 cells, as opposed to Th2 cells, was indicated in a study using HuPBL-NOD/SCID mice with A. actinomycetemcomitans-induced alveolar bone resorption, in which bone loss was associated with CD4+ T cells expressing IFN-γ rather than IL-4. In addition, the injection of IFN-γ into the mice further enhanced the bone loss [10]. Furthermore, adoptive transfer of antigen-specific Th1 cells but not Th2 cells enhanced alveolar bone destruction in rats injected with the A. actinomycetemcomitans 29-kDa outer membrane protein and lipopolysaccharide (LPS) into gingiva [11]. In contrast, a pathogenic role of the Th2 response was proposed in a study demonstrating that Tannerella forsythia–induced alveolar bone loss was accompanied by an increase in IL-5 but not of IFN-γ or IL-17 [12]. However, the protective roles of the Th1 and Th2 subsets were suggested in several articles, as described below. Oral gavage of P. gingivalis-derived antigen plus CpG oligodeoxynucleotides decreased their susceptibility to P. gingivalis-induced alveolar bone destruction, which was accompanied by upregulated production of Th1 and Th2 cytokines such as IFN-γ, IL-4, IL-5, and IL-6 in antigen-specific CD4+ T cells [13]. In addition, deficiency in the individual Th1 or Th2 cytokines (IFN-γ, IL-12p40, IL-4, or IL-10) resulted in enhanced, age-related alveolar bone loss [14].
It has become apparent that the pathogenesis of periodontitis cannot be fully explained through the prism of the Th1/Th2 paradigm; therefore, the discovery of Th17 has been expected to clarify the complex pathogenesis of periodontitis. Numerous reports have shown an elevated level of IL-17 in gingival tissues or gingival crevicular fluid from periodontitis lesions compared with healthy sites [50–57]. These reports also demonstrated the association of other Th17-related cytokines (eg, IL-1β, IL-6, IL-21, and IL-23) with periodontitis. In addition, immunohistochemical analysis revealed an increased infiltration of IL-17+ cells or RORC2+ (human homologue of mouse RORγt) cells in periodontal lesions [58••, 59, 60]. Furthermore, the serum concentration of IL-17 was dramatically increased in aggressive periodontitis patients compared with healthy subjects [61]. Zhao et al reported that nonsurgical periodontal therapy reduced the proportion of IL-17+ cells in peripheral blood CD4+ T cells. The authors also demonstrated decreased IL-17, increased IL-4, and no change in IFN-γ levels in gingival crevicular fluid after treatment [62•]. However, the question arises as to whether the increased Th17 cells are protective or destructive in periodontal diseases.
The protective role of Th17 cells against periodontal tissue destruction was suggested by a study using mice deficient for IL-17RA, a member of the IL-17 receptor family. The mice were susceptible to P. gingivalis-induced alveolar bone loss because of deficits in neutrophil migration [15]. However, mice lacking functional IL-17 or IL-23 are resistant to joint bone erosion induced by collagen-induced arthritis, which implies a destructive aspect of Th17 cells [63, 64]. The apparent difference in these studies may be due to the importance of Th17 cells in the control of infection (eg, periodontitis), but not in noninfectious diseases (eg, autoimmune arthritis).
Collectively, human and animal studies suggest that each T-helper subset possibly has beneficial and detrimental aspects in the pathogenesis of periodontal disease; thereby, an appropriate balance between the functionally different subsets may be important to eliminate infection and abrogate tissue destruction. The precise role of Th17 cells in physiological and pathologic conditions, including actions on innate immunity and osteoclastogenesis, should be further explored because Th17 cells and their cytokines are consistently increased in human periodontitis lesions.
Regulatory T Cells
Immunological self-tolerance is supported by several mechanisms: physical elimination (clonal deletion), functional inactivation (anergy), and T cell-mediated suppression. Regulatory T cells, which suppress the activation, proliferation, and effector functions of a wide range of immune cells, play a crucial role in maintaining immune homeostasis and in regulating immune responses in inflammatory conditions [65]. To date, several types of regulatory T cells have been described and have 2 main subsets: naturally occurring Foxp3+ Tregs, which develop in the thymus, and inducible regulatory T cells (eg, IL-10-producing Tr1 cells, TGF-β-producing cells, inducible Foxp3+ regulatory T cells), which develop in the periphery after exposure to various signals. Foxp3+ Tregs constitutively express CD25 at a high level and other Treg-related molecules, such as CTLA-4, GITR, CD122, CD44, CD11a, and CD54. Mutation of the Foxp3 gene elicits fatal autoimmune and inflammatory diseases due to the lack of functional Tregs [66, 67]. Multiple mechanisms of Foxp3+ Treg-mediated suppression have been suggested: impairment of dendritic cell function to activate effector T cells, a CTLA-4-dependent mechanism, anti-inflammatory cytokines (eg, IL-10 and TGF-β), and other mechanisms. Intriguingly, Tregs also regulate bone metabolism by directly inhibiting osteoclastogenesis, which is most likely mediated by Treg-derived cytokines such as TGF-β, IL-10, and IL-4, or CTLA-4 [20].
Therefore, Foxp3+ Tregs are suggested to play an essential role in the regulation of inflammation in periodontitis lesions where various types of effector/self-reactive T cells infiltrate. In humans, several reports have demonstrated increased infiltration of Tregs in lesions. The proportion of CD4+CD25+ T cells in lymphocytes extracted from gingival tissues was higher than that from peripheral blood in periodontitis patients [68], and Foxp3 gene expression was upregulated in periodontitis lesions compared with gingivitis lesions/healthy sites [69, 70]. In accordance with these findings, immunohistologic analysis showed an increased infiltration of CD4+CD25+ T cells and Foxp3+ cells in periodontitis lesions [58••, 59, 69, 71]. In addition, CD4+CD25+ T cells extracted from periodontitis lesions expressed Treg-related molecules such as Foxp3, CTLA-4, and GITR at a high rate [70]. Inconsistent with the many studies described above, Ernst et al reported that Foxp3+CD25+ cells were reduced in periodontitis lesions [72]. However, the reason for the discrepancy has not been clarified.
In rats, A. actinomycetemcomitans-induced experimental periodontitis did not change the proportion of Foxp3+ cells in draining node lymphocytes [16]. In contrast, mice with A. actinomycetemcomitans-induced alveolar bone loss showed an increased number of Foxp3+CD4+ T cells and elevated expression of CTLA-4, IL-10, and TGF-β in gingival tissues [17]. It is noteworthy that this study also showed that the inhibition of Tregs by anti-GITR treatment downregulated the Treg-related molecules, but upregulated IFN-γ and RANKL, and further enhanced bone loss without changing the bacterial load. These data indicate the possibility that Tregs attenuate the severity of periodontitis without impairing infection control. More recent research demonstrated that all-trans retinoic acid (ATRA) suppressed P. gingivalis-induced alveolar bone loss and inflammatory cell infiltration, which was accompanied by an increase of Foxp3+ cells and the level of IL-10 and TGF-β, and a decrease of RORγt+ cells and the level of IL-17 [18•]. In addition, the suppressive effect of Foxp3+ Tregs on inflammation-mediated bone loss was elegantly demonstrated in the latest literature. Glowacki et al reported that recruitment of Foxp3+ Tregs into gingival tissue by injection of C-C motif chemokine ligand 22 (CCL22)-releasing microparticles induced an increase of Treg-associated anti-inflammatory molecules, a decrease of proinflammatory cytokines, and a marked reduction of alveolar bone resorption in mice with A. actinomycetemcomitans-induced experimental periodontitis [19•]. These findings suggest that ATRA-induced or CCL22-mediated immune modulations may be novel therapeutic approaches to control periodontitis.
CTLA-4-mediated suppression is considered to be a central mechanism of Foxp3+ Treg function [73]. Immunohistologic analysis showed only a small number of CTLA-4+ cells in periodontitis lesions and healthy sites [74, 75]. In rats, administration of CTLA4-immunoglobulin fusion proteins (CTLA4Ig) abrogated the alveolar bone resorption that was induced by gingival injection of A. actinomycetemcomitans-derived antigens, suggesting a protective role of CTLA-4 in periodontitis [11]. Anti-inflammatory cytokines are an additional mechanism through which regulatory T cells control inflammation. IL-10 is produced by various cell populations, including regulatory T cells and T-helper subsets. IL-10 gene expression was higher in periodontitis tissues than in healthy sites or peripheral blood mononuclear cells [70, 76]. In addition, IL-10+ cells were widely distributed in periodontitis lesions [77]. The protective role of IL-10 was suggested by studies where IL-10-deficient mice displayed increased susceptibility to age-related or P. gingivalis-induced alveolar bone loss [14, 20]. The intraperitoneal injection of IL-10 into A. actinomycetemcomitans-infected HuPBL-NOD/SCID mice suppressed alveolar bone loss, which further supported the protective role of IL-10 [21]. TGF-β is another immunosuppressive cytokine with pleiotropic functions. Like IL-10, several reports show that TGF-β gene expression and TGF-β+ cells were increased in human periodontitis lesions [69, 70, 78]. However, Dutzan et al showed a negative correlation between the levels of IL-10 and TGF-β and the presence or disease activity of periodontitis in humans [57, 79]. Taken together, a large number of studies support the view that regulatory T cells are recruited into periodontitis lesions and protect against tissue destruction; therefore, impairment of their function may be linked to the progression of periodontal disease.
Interestingly, nearly all CD4+ T cell clones that were established from periodontitis tissues expressed the Foxp3 gene, and the Foxp3+CD4+ T cell clones did not exhibit a suppressive function in vitro [80, 81]. Although it is possible that the T cell clones were derived from non-Tregs that could express transient Foxp3 after activation [82], a fraction of Foxp3+ T cells in periodontitis lesions might be functionally impaired. Recent studies have shown that appropriate stimuli convert human Foxp3+ Tregs into IL-17-producing cells in the presence of IL-2 and inflammatory cytokines in vitro [83–85]. Indeed, IL-17A+Foxp3+ cells seem to be involved in periodontitis and other disorders such as psoriasis, inflammatory bowel disease, and colon cancer, although their roles are not fully elucidated [58••, 86–88].
NKT Cells
Invariant NKT (iNKT) cells are characterized by the coexpression of the natural killer cell receptor, NK1.1 (CD161), and restricted T cell receptors, Vα24Jα18 in humans and Vα14Jα18 in mice. Multiple iNKT cell populations differ in their function, location, and phenotype. iNKT cells typically recognize glycolipids that are presented by CD1d molecules, which are predominantly expressed on antigen-presenting cells (eg, dendritic cells, macrophages, and B cells) [89]. It has been reported that iNKT cells recognize antigens derived from bacteria such as Borrelia burgdorferi, Sphingomonas spp, Streptococcus pneumonia, and group B streptococcus [90–92]. In addition, some self-antigens are proposed to contribute to iNKT cell function [93]. iNKT cells rapidly respond to antigens and release substantial amounts of IFN-γ and IL-4, which subsequently activate other lymphocytes and shape the course of adaptive responses. Thus, iNKT cells play important functions in autoimmune disease, cancer, infection, and inflammation [94]
Several studies have reported the involvement of iNKT and CD1d+ cells in human periodontitis. Yamazaki et al used single-strand conformation polymorphism analysis to demonstrate that the proportion of NKT cells was elevated in periodontitis lesions compared with gingivitis lesions [95]. Immunohistologic analysis revealed that CD1d+ and iNKT cells were increased in periodontitis lesions, and a fraction of the CD1d+ cells was identified as CD19+ B cells [96]. A more recent report showed increased iNKT cells in gingival tissues with aggressive periodontitis, but not chronic periodontitis, compared with healthy biopsies [97].
The pathogenic role of NKT cells in periodontal bone destruction was suggested by experiments utilizing mice with either a deficiency or activation of NKT cells. CD1d-/- mice, which lack Vα14 NKT cells, were completely resistant to P. gingivalis-induced alveolar bone resorption. In contrast, administration of α-galactosylceramide, which activates NKT cells in a CD1d-dependent manner, further accelerated P. gingivalis-induced bone resorption [22••]. However, it is still unknown whether P. gingivalis-derived antigens were directly recognized by NKT cells in the mouse models. To apply the pathogenicity of NKT cells in mice to humans, it should be determined whether the NKT cells observed in human periodontitis lesions are sufficiently activated to skew immune responses.
Conclusions
It is clear that T cells in periodontal tissues play a crucial role in infection control and tissue destruction in periodontal disease, whereas the mechanism of immune regulation underlying the disease is highly complicated. In humans, numerous studies have revealed phenomena that occur in periodontitis lesions by using a variety of materials and methods. These studies provide valuable insights into the T cell-mediated immune regulation of the pathogenesis of periodontitis, but they have also shown inconsistent results, which may be attributed to differences in the experimental approaches and a lack of evaluation of disease activity. Because it is considered that a diseased site exhibits an active state at times, but inactive state at other times, the change of disease activity could affect the immune responses at local sampling sites. What is not addressed in clinical studies because of ethical considerations can possibly be examined in research with animal models.
Several studies on experimental periodontitis in mice with deletions of T cell-related genes have implicated that an optimal balance between functionally different T cells may be more important in the protection against periodontal tissue destruction than a strong, polarized response on either side. This framework could be applied to humans, although it should be noted that these periodontitis models do not represent all aspects of human periodontal disease. Gemmell et al proposed that T cells are homeostatic in maintaining a balance between the host and biofilm, and disease progression occurs if the homeostatic balance is disrupted by environmental influences that lead to increased pathogens or depression of the host’s defense mechanisms [1].
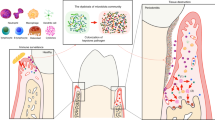
Finally, we briefly summarize the potential role of T cells in inflammatory responses and bone destruction in Fig. 1, where the T cells form a cytokine network that affects other immune and resident cells. Current approaches in periodontal treatment include the removal of bacteria and their components rather than modulating the host response. However, a deeper understanding of how T cells regulate the balance between protective and destructive immune responses may contribute to the development of new therapeutic modalities for periodontal disease in the future.

The potential role of distinct T cell subsets in inflammatory responses and alveolar bone resorption in periodontitis lesions. As a consequence of sustained infection by periodontopathic bacteria, an adaptive immune response is established. NKT cells are involved in shaping the course of the immune response. Th1, Th2, and Th17 cells contribute to infection control in different ways because of their distinct cytokine profiles. However, their action also enhances inflammatory responses that lead to periodontal tissue destruction. Particularly, Th17 cells have a high potential to facilitate osteoclastogenesis thorough the production of IL-17 to induce RANKL expression on osteoblasts, enhancement of local inflammation, and RANKL expression on themselves. However, Tregs attenuate the inflammatory responses by suppressing other immune cells and inhibit osteoclastogenesis; therefore, they could protect against tissue destruction. Enhanced inflammation may convert a fraction of Tregs to IL-17-producing cells. Mϕ macrophage, Ob osteoblast
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Gemmell E, Yamazaki K, Seymour GJ. The role of T cells in periodontal disease: homeostasis and autoimmunity. Periodontol 2000. 2007;43:14–40.
Yamazaki K, Nakajima T. Antigen specificity and T-cell clonality in periodontal disease. Periodontol 2000. 2004;35:75–100.
Tabeta K, Yamazaki K, Hotokezaka H, Yoshie H, Hara K. Elevated humoral immune response to heat shock protein 60 (hsp60) family in periodontitis patients. Clin Exp Immunol. 2000;120(2):285–93.
Hirsch HZ, Tarkowski A, Miller EJ, Gay S, Koopman WJ, Mestecky J. Autoimmunity to collagen in adult periodontal disease. J Oral Pathol. 1988;17(9–10):456–9.
Yamazaki K, Ohsawa Y, Tabeta K, Ito H, Ueki K, Oda T, et al. Accumulation of human heat shock protein 60-reactive T cells in the gingival tissues of periodontitis patients. Infect Immun. 2002;70(5):2492–501.
Garlet GP. Destructive and protective roles of cytokines in periodontitis: a re-appraisal from host defense and tissue destruction viewpoints. J Dent Res. 2010;89(12):1349–63.
Baker PJ, Dixon M, Evans RT, Dufour L, Johnson E, Roopenian DC. CD4+ T cells and the proinflammatory cytokines gamma interferon and interleukin-6 contribute to alveolar bone loss in mice. Infect Immun. 1999;67(6):2804–9.
Teng YT, Nguyen H, Gao X, Kong YY, Gorczynski RM, Singh B, et al. Functional human T-cell immunity and osteoprotegerin ligand control alveolar bone destruction in periodontal infection. J Clin Invest. 2000;106(6):R59–67.
Garlet GP, Cardoso CR, Silva TA, Ferreira BR, Ávila-Campos MJ, Cunha FQ, et al. Cytokine pattern determines the progression of experimental periodontal disease induced by Actinobacillus actinomycetemcomitans through the modulation of MMPs, RANKL, and their physiological inhibitors. Oral Microbiol Immunol. 2006;21(1):12–20.
Teng YT, Mahamed D, Singh B. Gamma interferon positively modulates Actinobacillus actinomycetemcomitans-specific RANKL+ CD4+ Th-cell-mediated alveolar bone destruction in vivo. Infect Immun. 2005;73(6):3453–61.
Kawai T, Eisen-Lev R, Seki M, Eastcott JW, Wilson ME, Taubman MA. Requirement of B7 costimulation for Th1-mediated inflammatory bone resorption in experimental periodontal disease. J Immunol. 2000;164(4):2102–9.
Myneni SR, Settem RP, Connell TD, Keegan AD, Gaffen SL, Sharma A. TLR2 signaling and Th2 responses drive Tannerella forsythia-induced periodontal bone loss. J Immunol. 2011;187(1):501–9.
Liu C, Hashizume T, Kurita-Ochiai T, Fujihashi K, Yamamoto M. Oral immunization with Porphyromonas gingivalis outer membrane protein and CpGoligodeoxynucleotides elicits T helper 1 and 2 cytokines for enhanced protective immunity. Mol Oral Microbiol. 2010;25(3):178–89.
Alayan J, Ivanovski S, Farah CS. Alveolar bone loss in T helper 1/T helper 2 cytokine-deficient mice. J Periodontal Res. 2007;42(2):97–103.
Yu JJ, Ruddy MJ, Wong GC, Sfintescu C, Baker PJ, Smith JB, et al. An essential role for IL-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires IL-17 receptor-dependent signals. Blood. 2007;109(9):3794–802.
de Brito BB, Andriankaja O, Kang J, Pacios S, Bae HJ, Li Y, et al. A.actinomycetemcomitans-induced periodontal disease promotes systemic and local responses in rat periodontium. J Clin Periodontol. 2012;39(4):333–41.
Garlet GP, Cardoso CR, Mariano FS, Claudino M, de Assis GF, Campanelli AP, et al. Regulatory T cells attenuate experimental periodontitis progression in mice. J Clin Periodontol. 2010;37(7):591–600.
Wang L, Wang J, Jin Y, Gao H, Lin X. Oral administration of all-trans retinoic acid suppresses experimental periodontitis by modulating the Th17/Treg imbalance. J Periodontol. 2013. doi:10.1902/jop.2013.130132. This study demonstrated that all-trans retinoic acid suppressed alveolar bone destruction in experimental periodontitis through modulating the Th17/Treg balance, suggesting a new therapeutic approach for the prevention of periodontitis in the context of immune modulation.
Glowacki AJ, Yoshizawa S, Jhunjhunwala S, Vieira AE, Garlet GP, Sfeir C, et al. Prevention of inflammation-mediated bone loss in murine and canine periodontal disease via recruitment of regulatory lymphocytes. Proc Natl Acad Sci U S A. 2013;110(46):18525–30. This study showed that injection of CCL22-releasing microparticles into gingival tissue suppressed inflammatory bone resorption in experimental murine periodontitis via recruitment of Tregs, suggesting that induction of balanced immune response has a therapeutic potential for the treatment of periodontitis.
Sasaki H, Okamatsu Y, Kawai T, Kent R, Taubman M, Stashenko P. The interleukin-10 knockout mouse is highly susceptible to Porphyromonas gingivalis-induced alveolar bone loss. J Periodontal Res. 2004;39(6):432–41.
Zhang X, Teng YT. Interleukin-10 inhibits gram-negative-microbe-specific human receptor activator of NF-κB ligand-positive CD4+-Th1-cell-associated alveolar bone loss in vivo. Infect Immun. 2006;74(8):4927–31.
Aoki-Nonaka Y, Nakajima T, Miyauchi S, Miyazawa H, Yamada H, Domon H, et al. Natural killer T cells mediate alveolar bone resorption and a systemic inflammatory response in response to oral infection of mice with Porphyromonas gingivalis. J Periodontal Res. 2014;49(1):69–76. This study demonstrated the pathogenic role of NKT cells in pathogen-induced experimental periodontitis by analyzing mice with either a deficiency or activation of NKT cells.
Cole KL, Seymour GJ, Powell RN. Phenotypic and functional analysis of T cells extracted from chronically inflamed human periodontal tissues. J Periodontol. 1987;58(8):569–73.
Stoufi ED, Taubman MA, Ebersole JL, Smith DJ, Stashenko PP. Phenotypic analyses of mononuclear cells recovered from healthy and diseased human periodontal tissues. J Clin Immunol. 1987;7(3):235–45.
Johannessen AC, Nilsen R, Kristoffersen T, Knudsen GE. Variation in the composition of gingival inflammatory cell infiltrates. J Clin Periodontol. 1990;17(5):298–305.
Jully JM, Bene MC, Martin G, Faure G. Immunohistological identification of cell subsets in human gingiva after local treatment for gingivitis or periodontitis. J Clin Periodontol. 1986;13(3):223–7.
Syrjanen S, Markkanen H, Syrjanen K. Inflammatory cells and their subsets in lesions of juvenile periodontitis. A family study. Acta Odontol Scand. 1984;42(5):285–92.
Modeer T, Dahllof G, Axio E, Sundqvist KG. Subpopulations of lymphocytes in connective tissue from adolescents with periodontal disease. Acta Odontol Scand. 1990;48(3):153–9.
Lappin DF, Koulouri O, Radvar M, Hodge P, Kinane DF. Relative proportions of mononuclear cell types in periodontal lesions analyzed by immunohistochemistry. J Clin Periodontol. 1999;26(3):183–9.
Reinhardt RA, Bolton RW, McDonald TL, DuBois LM, Kaldahl WB. In situ lymphocyte subpopulations from active versus stable periodontal sites. J Periodontol. 1988;59(10):656–70.
Ahlers JD, Belyakov IM. Molecular pathways regulating CD4+ T cell differentiation, anergy and memory with implications for vaccines. Trends Mol Med. 2010;16(10):478–91.
Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol. 2012;181(1):8–18.
Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203(12):2673–82.
Okamoto K, Takayanagi H. Regulation of bone by the adaptive immune system in arthritis. Arthritis Res Ther. 2011;13(3):219. This review excellently describes the functional role of each T cell subset, particularly focusing on the Th17 subset, in osteoclastogenesis and inflammatory bone destruction.
Aoyagi T, Sugawara-Aoyagi M, Yamazaki K, Hara K. Interleukin 4 (IL-4) and IL-6-producing memory T-cells in peripheral blood and gingival tissue in periodontitis patients with high serum antibody titers to Porphyromonas gingivalis. Oral Microbiol Immunol. 1995;10(5):304–10.
Gemmell E, Seymour GJ. Cytokines and T cell switching. Crit Rev Oral Biol Med. 1994;5(3–4):249–79.
Manhart SS, Reinhardt RA, Payne JB, Seymour GJ, Gemmell E, Dyer JK, et al. Gingival cell IL-2 and IL-4 in early-onset periodontitis. J Periodontol. 1994;65(9):807–13.
Reinhardt RA, McDonald TL, Bolton RW, DuBois LM, Kaldahl WB. IgG subclasses in gingival crevicular fluid from active versus stable periodontal sites. J Periodontol. 1989;60(1):44–50.
Sigusch B, Klinger G, Glockmann E, Simon HU. Early-onset and adult periodontitis associated with abnormal cytokine production by activated T lymphocytes. J Periodontol. 1998;69(10):1098–104.
Tokoro Y, Matsuki Y, Yamamoto T, Suzuki T, Hara K. Relevance of local Th2-type cytokine mRNA expression in immunocompetent infiltrates in inflamed gingival tissue to periodontal diseases. Clin Exp Immunol. 1997;107(1):166–74.
Ebersole JL, Taubman MA. The protective nature of host responses in periodontal diseases. Periodontol 2000. 1994;5:112–41.
Takeichi O, Haber J, Kawai T, Smith DJ, Moro I, Taubman MA. Cytokine profiles of T-lymphocytes from gingival tissues with pathological pocketing. J Dent Res. 2000;79(8):1548–55.
Ukai T, Mori Y, Onoyama M, Hara Y. Immunohistological study of interferon-γ- and interleukin-4-bearing cells in human periodontitis gingiva. Arch Oral Biol. 2001;46(10):901–8.
Nakajima T, Yamazaki K, Cullinan MP, Gemmell E, Seymour GJ. T-cell antigen specificity in humans following stimulation with Porphyromonas gingivalis. Arch Oral Biol. 1999;44(12):1045–53.
Prabhu A, Michalowicz BS, Mathur A. Detection of local and systemic cytokines in adult periodontitis. J Periodontol. 1996;67(5):515–22.
Wassenaar A, Reinhardus C, Thepen T, Abraham-Inpijn L, Kievits F. Cloning, characterization, and antigen specificity of T-lymphocyte subsets extracted from gingival tissue of chronic adult periodontitis patients. Infect Immun. 1995;63(6):2147–53.
Yamamoto M, Fujihashi K, Hiroi T, McGhee JR, Van Dyke TE, Kiyono H. Molecular and cellular mechanisms for periodontal diseases: role of Th1 and Th2 type cytokines in induction of mucosal inflammation. J Periodontal Res. 1997;32(1 Pt 2):115–9.
Berglundh T, Liljenberg B, Lindhe J. Some cytokine profiles of T-helper cells in lesions of advanced periodontitis. J Clin Periodontol. 2002;29(8):705–9.
Gemmell E, Grieco DA, Cullinan MP, Westerman B, Seymour GJ. The proportion of interleukin-4, interferon-gamma and interleukin-10-positive cells in Porphyromonas gingivalis-specific T-cell lines established from P. gingivalis-positive subjects. Oral Microbiol Immunol. 1999;14(5):267–74.
Honda T, Aoki Y, Takahashi N, Maekawa T, Nakajima T, Ito H, et al. Elevated expression of IL-17 and IL-12 genes in chronic inflammatory periodontal disease. Clin Chim Acta. 2008;395(1–2):137–41.
Ohyama H, Kato-Kogoe N, Kuhara A, Nishimura F, Nakasho K, Yamanegi K, et al. The involvement of IL-23 and the Th17 pathway in periodontitis. J Dent Res. 2009;88(7):633–8.
Cardoso CR, Garlet GP, Crippa GE, Rosa AL, Júnior WM, Rossi MA, et al. Evidence of the presence of T helper type 17 cells in chronic lesions of human periodontal disease. Oral Microbiol Immunol. 2009;24(1):1–6.
Lester SR, Bain JL, Johnson RB, Serio FG. Gingival concentrations of interleukin-23 and -17 at healthy sites and at sites of clinical attachment loss. J Periodontol. 2007;78(8):1545–50.
Vernal R, Dutzan N, Chaparro A, Puente J, Antonieta Valenzuela M, Gamonal J. Levels of interleukin-17 in gingival crevicular fluid and in supernatants of cellular cultures of gingival tissue from patients with chronic periodontitis. J Clin Periodontol. 2005;32(4):383–9.
Moutsopoulos NM, Kling HM, Angelov N, Jin W, Palmer RJ, Nares S, et al. Porphyromonas gingivalis promotes Th17 inducing pathways in chronic periodontitis. J Autoimmun. 2012;39(4):294–303.
Szkaradkiewicz AK, Karpinski TM, Zeidler A, Wyganowska-Swiatkowska M, Szkaradkiewicz A. Protective effect of oral Lactobacilli in pathogenesis of chronic periodontitis. J Physiol Pharmacol: Off J Pol Physiol Soc. 2011;62(6):685–9.
Dutzan N, Vernal R, Vaque JP, García-Sesnich J, Hernandez M, Abusleme L, et al. Interleukin-21 expression and its association with proinflammatory cytokines in untreated chronic periodontitis patients. J Periodontol. 2012;83(7):948–54.
Okui T, Aoki Y, Ito H, Honda T, Yamazaki K. The Presence of IL-17+/FOXP3+ Double-positive Cells in Periodontitis. J Dent Res. 2012;91(6):574–9. This is the first study to show the involvement of IL-17A + Foxp3 + cells in periodontitis using immunohistological analysis and characterization of CD4 + T cell lines that were established from gingival tissue or peripheral blood of patients.
Duarte PM, Santos VR, Dos Santos FA, de Lima Pereira SA, Rodrigues DB, Napimoga MH. Role of smoking and type 2 diabetes in the immunobalance of advanced chronic periodontitis. J Periodontol. 2011;82(3):429–38.
Adibrad M, Deyhimi P, Ganjalikhani Hakemi M, Behfarnia P, Shahabuei M, Rafiee L. Signs of the presence of Th17 cells in chronic periodontal disease. J Periodontal Res. 2012;47(4):525–31.
Schenkein HA, Koertge TE, Brooks CN, Sabatini R, Purkall DE, Tew JG. IL-17 in sera from patients with aggressive periodontitis. J Dent Res. 2010;89(9):943–7.
Zhao L, Zhou Y, Xu Y, Sun Y, Li L, Chen W. Effect of non-surgical periodontal therapy on the levels of Th17/Th1/Th2 cytokines and their transcription factors in Chinese chronic periodontitis patients. J Clin Periodontol. 2011;38(6):509–16. This manuscript showed that non-surgical periodontal therapy suppressed the Th17 cell response, but enhanced the Th2 cell response, by examining the gingival crevicular fluid and peripheral blood CD4 + T cells of periodontitis patients.
Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171(11):6173–7.
Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, et al. Divergent pro- and anti-inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198(12):1951–7.
Belkaid Y, Tarbell K. Regulatory T cells in the control of host-microorganism interactions. Annu Rev Immunol. 2009;27:551–89.
Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27(1):18–20.
Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27(1):68–73.
Takeuchi Y, Yoshie H, Hara K. Expression of interleukin-2 receptor and HLA-DR on lymphocyte subsets of gingival crevicular fluid in patients with periodontitis. J Periodontal Res. 1991;26(6):502–10.
Nakajima T, Ueki-Maruyama K, Oda T, Ohsawa Y, Ito H, Seymour GJ, et al. Regulatory T-cells infiltrate periodontal disease tissues. J Dent Res. 2005;84(7):639–43.
Cardoso CR, Garlet GP, Moreira AP, Júnior WM, Rossi MA, Silva JS. Characterization of CD4+CD25+ natural regulatory T cells in the inflammatory infiltrate of human chronic periodontitis. J Leukoc Biol. 2008;84(1):311–8.
Kim YC, Ko Y, Hong SD, Kim KY, Lee YH, Chae C, et al. Presence of Porphyromonas gingivalis and plasma cell dominance in gingival tissues with periodontitis. Oral Dis. 2010;16(4):375–81.
Ernst CW, Lee JE, Nakanishi T, Karimbux NY, Rezende TM, Stashenko P, et al. Diminished forkhead box P3/CD25 double-positive T regulatory cells are associated with the increased nuclear factor-κB ligand (RANKL+) T cells in bone resorption lesion of periodontal disease. Clin Exp Immunol. 2007;148(2):271–80.
Yamaguchi T, Wing JB, Sakaguchi S. Two modes of immune suppression by Foxp3+ regulatory T cells under inflammatory or non-inflammatory conditions. Semin Immunol. 2011;23(6):424–30.
Gemmell E, McHugh GB, Grieco DA, Seymour GJ. Costimulatory molecules in human periodontal disease tissues. J Periodontal Res. 2001;36(2):92–100.
Orima K, Yamazaki K, Aoyagi T, Hara K. Differential expression of costimulatory molecules in chronic inflammatory periodontal disease tissue. Clin Exp Immunol. 1999;115(1):153–60.
Yamazaki K, Nakajima T, Kubota Y, Gemmell E, Seymour GJ, Hara K. Cytokine messenger RNA expression in chronic inflammatory periodontal disease. Oral Microbiol Immunol. 1997;12(5):281–7.
Lappin DF, MacLeod CP, Kerr A, Mitchell T, Kinane DF. Anti-inflammatory cytokine IL-10 and T cell cytokine profile in periodontitis granulation tissue. Clin Exp Immunol. 2001;123(2):294–300.
Steinsvoll S, Halstensen TS, Schenck K. Extensive expression of TGF-β1 in chronically-inflamed periodontal tissue. J Clin Periodontol. 1999;26(6):366–73.
Dutzan N, Gamonal J, Silva A, Sanz M, Vernal R. Over-expression of forkhead box P3 and its association with receptor activator of nuclear factor-κB ligand, interleukin (IL) -17, IL-10 and transforming growth factor-β during the progression of chronic periodontitis. J Clin Periodontol. 2009;36(5):396–403.
Okui T, Ito H, Honda T, Amanuma R, Yoshie H, Yamazaki K. Characterization of CD4+ FOXP3+ T-cell clones established from chronic inflammatory lesions. Oral Microbiol Immunol. 2008;23(1):49–54.
Ito H, Honda T, Domon H, Oda T, Okui T, Amanuma R, et al. Gene expression analysis of the CD4+ T-cell clones derived from gingival tissues of periodontitis patients. Oral Microbiol Immunol. 2005;20(6):382–6.
Wang J, Ioan-Facsinay A, van der Voort EI, Huizinga TW, Toes RE. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur J Immunol. 2007;37(1):129–38.
Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood. 2008;112(6):2340–52.
Deknuydt F, Bioley G, Valmori D, Ayyoub M. IL-1β and IL-2 convert human Treg into TH17 cells. Clin Immunol. 2009;131(2):298–307.
Valmori D, Raffin C, Raimbaud I, Ayyoub M. Human RORγt+ TH17 cells preferentially differentiate from naive FOXP3+ Treg in the presence of lineage-specific polarizing factors. Proc Natl Acad Sci U S A. 2010;107(45):19402–7.
Bovenschen HJ, van de Kerkhof PC, van Erp PE, Woestenenk R, Joosten I, Koenen HJ. Foxp3+ Regulatory T Cells of Psoriasis Patients Easily Differentiate into IL-17A-Producing Cells and Are Found in Lesional Skin. J Invest Dermatol. 2011;131(9):1853–60.
Hovhannisyan Z, Treatman J, Littman DR, Mayer L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology. 2011;140(3):957–65.
Li L, Boussiotis VA. The role of IL-17-producing Foxp3+ CD4+ T cells in inflammatory bowel disease and colon cancer. Clin Immunol. 2013;148(2):246–53.
Lawson V. Turned on by danger: activation of CD1d-restricted invariant natural killer T cells. Immunology. 2012;137(1):20–7.
Kinjo Y, Tupin E, Wu D, Fujio M, Garcia-Navarro R, Benhnia MR, et al. Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat Immunol. 2006;7(9):978–86.
Mattner J, Debord KL, Ismail N, Goff RD, Cantu III C, Zhou D, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature. 2005;434(7032):525–9.
Kinjo Y, Illarionov P, Vela JL, Pei B, Girardi E, Li X, et al. Invariant natural killer T cells recognize glycolipids from pathogenic Gram-positive bacteria. Nat Immunol. 2011;12(10):966–74.
Brennan PJ, Tatituri RV, Brigl M, Kim EY, Tuli A, Sanderson JP, et al. Invariant natural killer T cells recognize lipid self antigen induced by microbial danger signals. Nat Immunol. 2011;12(12):1202–11.
Tupin E, Kinjo Y, Kronenberg M. The unique role of natural killer T cells in the response to microorganisms. Nat Rev Microbiol. 2007;5(6):405–17.
Yamazaki K, Ohsawa Y, Yoshie H. Elevated proportion of natural killer T cells in periodontitis lesions: a common feature of chronic inflammatory diseases. Am J Pathol. 2001;158(4):1391–8.
Amanuma R, Nakajima T, Yoshie H, Yamazaki K. Increased infiltration of CD1d and natural killer T cells in periodontal disease tissues. J Periodontal Res. 2006;41(1):73–9.
Nowak M, Krämer B, Haupt M, Papapanou PN, Kebschull J, Hoffmann P, et al. Activation of invariant NK T cells in periodontitis lesions. J Immunol. 2013;190(5):2282–91.
Acknowledgments
The works by the authors in this review were supported by JSPS KAKENHI. The authors thank Dr. Tomoyuki Honda for preparing the figure.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Dr. Takafumi Okui, Dr. Yukari Aoki-Nonaka, Dr. Takako Nakajima, and Dr. Kazuhisa Yamazaki each declare no potential conflicts of interest relevant to this article.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Okui, T., Aoki-Nonaka, Y., Nakajima, T. et al. The Role of Distinct T Cell Subsets in Periodontitis—Studies from Humans and Rodent Models. Curr Oral Health Rep 1, 114–123 (2014). https://doi.org/10.1007/s40496-014-0013-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40496-014-0013-z