
Abstract
Skin cancer incidences are rising worldwide, and one of the major causative factors is excessive exposure to solar ultraviolet radiation (UVR). Annually, ∼5 million skin cancer patients are treated in USA, mostly with nonmelanoma skin cancer (NMSC), which is also frequent in other Western countries. As sunscreens do not provide adequate protection against deleterious effects of UVR, additional and alternative chemoprevention strategies are urgently needed to reduce skin cancer burden. Over the last couple of decades, extensive research has been conducted to understand the molecular basis of skin carcinogenesis, and to identifying novel agents which could be useful in the chemoprevention of skin cancer. In this regard, several natural nontoxic compounds have shown promising efficacy in preventing skin carcinogenesis at initiation, promotion, and progression stages and are considered important in better management of skin cancer. Consistent with this, we and others have studied and established the notable efficacy of natural flavonolignan silibinin against UVB-induced skin carcinogenesis. Extensive preclinical animal and cell culture studies report strong anti-inflammatory, antioxidant, DNA damage repair, immune modulatory, and antiproliferative properties of silibinin. Molecular studies have identified that silibinin targets pleotropic signaling pathways including mitogenic, cell cycle, apoptosis, autophagy, p53, NF-κB, etc. Overall, the skin cancer chemopreventive potential of silibinin is well supported by comprehensive mechanistic studies, suggesting its greater use against UV-induced cellular damages and photocarcinogenesis.
Similar content being viewed by others

Introduction
The skin is continuously exposed to sunlight [primarily ultraviolet radiation (UVR)] and other toxicants that eventually cause skin cancer, which is the most common malignancy worldwide [1–3]. Among all the factors, UVR is the most important etiological factor in skin cancer development and accounts for about 50–90 % of total reported skin cancer cases [4, 5]. In addition, increased popularity of outdoor activities and tanning devices as well as rapid depletion of the ozone layer have also contributed to enhanced UVR exposure [6]. Therefore, not surprising, incidences of skin cancer are continuously rising. Epidemiological analysis reveals that annually in the USA alone, ∼5 million skin cancer patients are treated, and that between 40 and 50 % of Americans are susceptible to develop skin cancer at least once by the age of 65. These statistics clearly suggest that skin cancer is a major health issue.
In general, skin cancers can be broadly classified into two groups: (a) cutaneous melanoma and (b) nonmelanoma skin cancer (NMSC); the latter is further classified into (i) basal cell carcinoma (BCC) and (ii) squamous cell carcinoma (SCC). Cutaneous melanoma is aggressive form of skin cancer, and despite the fact that only 5–10 % of total diagnosed skin cancer cases are melanoma, it causes approximately 75 % of all skin cancer-related deaths because of its high metastatic potential. It is estimated that approximately 90 % of melanomas are caused by exposure to UVR [2, 7]. Regarding NMSC, majority of the cases are BCC which is more common than all other human malignancies combined. Almost all BCC occurs on excessively UVR exposed body sites, especially the uncovered ones such as face, ear pinna, neck, scalp, shoulders, and back [8]. Other than UVR, exposure to arsenic, radiation, chronic inflammatory conditions in skin, and burns, scars, infections complications or even tattoos are also the contributing factors for BCC. Similar to BCC, the primary cause for SCC is also the excessive exposure to solar UVR [9]. Both BCC and SCC could be easily treated when diagnosed early and are rarely fatal, but in a small percentage of cases, BCC and SCC have shown metastatic potential and cause death. Moreover, BCC and SCC development as well as their treatment/s could be painful and disfiguring.
Since excessive exposure to solar UVR is the main reason for skin cancer, photocarcinogenesis is an area of extensive research. UVR consists of UVA (320–400 nm), UVB (280–320 nm), and UVC (200–280 nm), where UVB and UVC are most effective in causing skin cancer; however, majority of UVC radiation is filtered by atmospheric ozone layer, and therefore, it has not much biological relevance to skin cancer [7, 10]. Majority of solar UVR reaching the earth surface is UVA (90–99 %), and only 1–10 % is composed of UVB. Earlier studies with mouse skin models showed the complete skin carcinogenic potential of both UVA and UVB radiations [11, 12]. However, higher exposure doses are needed for UVA-caused photocarcinogenesis in mice when compared to UVB in terms of both dose and duration, and also has a longer tumor latency period, which is attributed to its weak tumor initiating potential [12]. Therefore, UVB exposure is the most clinically relevant form of UVR for the development of skin cancers. The skin tumor initiating potential of UVB has been attributed to its strong absorption by DNA in skin keratinocytes resulting in DNA lesions including cyclobutane pyrimidine dimers (CPDs) and 6-4 photoproducts, which are major contributors to photocarcinogenesis [13, 14]. Furthermore, UVR generates reactive oxygen species (ROS) in the skin and induces oxidative DNA damage such as 8-hydroxy-2′-deoxyguanosine (8-OHdG) [15, 16]. UVB-generated ROS can also act as tumor promoter by activating cellular mitogenic signaling pathways promoting proliferation and inflammation [17, 18•]. The importance of DNA damaging potential of UVB can be highlighted by the fact that almost 60 % SCCs carry UVB signature mutations in tumor suppressor gene p53, such as CC→TT and C→T transitions [14, 19, 20]. UVB exposure also causes immunosuppression, photoaging, and sunburn depending on the duration as well as levels of exposure [21]. UV-induced signature mutations (CC→TT and C→T transitions) in PTCH1 (in the mitogenic sonic hedgehog pathway) and p53 genes represent the most significant pathogenic events in BCC and found in almost 50 % of sporadic BCC [22, 23]. Recent research showed that there are other genes also which are mutated in BCC such as STAT5B (signal transduction and activation of transcription), CRNKL-1 (crooked neck pre-MRNA splicing factor 1), and that telomerase reverse transcriptase (TERT) promoter also contributed toward development of this malignancy [24, 25].
For skin cancer prevention, efforts have been made to improve awareness about risk factors including minimizing sunlight exposure especially during mid-day hours, effective use of sunscreens, wearing protective clothing when outdoors and by avoiding the use of UV tanning devices. However, the current options and strategies have been largely ineffective as evident by continuous increase in skin cancer incidences, the related morbidity, and medical care expenses. Moreover, few studies have even shown that sunscreen use enhances melanoma risk, further demanding novel and alternative measures to lower skin cancer risk [26]. Many epidemiological studies have shown that modification in lifestyle factors including dietary patterns could be helpful in the prevention of two thirds of cancer cases [27–29]. As the complete skin carcinogenic potential of UVB radiation has been attributed to its potential to directly damage DNA and other cellular macromolecules as well as oxidative stress generation, nontoxic natural compounds that (a) could reduce solar UVR-caused DNA damage and/or (b) possess antioxidant, anti-inflammatory, and/or immune modulatory effect could be useful against photocarcinogenesis [10, 13, 30–32]. In this regard, many nontoxic natural compounds including silibinin, green tea extract, black tea extract, epigallocatechin 3-gallate, etc. have shown their strong efficacy against photocarcinogenesis at initiation, promotion, and complete carcinogenesis stages [13, 32–37]. The present review is focused on the effectiveness and associated mechanism(s) of silibinin against UVB-induced skin carcinogenesis.
Silibinin and Photocarcinogenesis Prevention
Silibinin is the main bioactive flavonolignan present in milk thistle extract and is a dietary supplement for its hepatoprotective activity [38, 39]. The milk thistle plant (Silybum marianum L., Family Asteraceae) is currently cultivated in several countries for commercial production of milk thistle extract [13], mostly silymarin that contains 40 % (w/w) silibinin as the major anticancer agent [40]. Several studies have reported strong antioxidant property of silibinin and its capability to scavenge and neutralize free radicals and ROS [10, 13, 32, 41, 42]. Furthermore, several in vivo studies have shown that silibinin is free of potential adverse effects in both animal and human studies, which is a desired component for cancer chemopreventive agents [13, 43–45].
The most preferred and frequently used animal model to study UVR-caused skin cancer is SKH-1 hairless mouse skin which parallels both histologically and pathological features of human NMSCs [46]. Furthermore, the multistages of photocarcinogenesis in this mouse model have been well defined and characterized. Following UVB exposure, exposed mouse skin showed epidermal hyperplasia, and further, UVB irradiation leads to the development of benign papilloma which further progresses to SCC [13]. These characteristics in this mouse model closely followed the development of NMSC in humans [47]. Therefore, this model has been used extensively to conduct mechanistic as well as preventive studies related to photocarcinogenesis [46]. Our studies in this model have clearly shown that silibinin is a strong chemopreventive agent against photocarcinogenesis and produces its activity irrespective of application on the dorsal side of mouse skin either before or after UVR exposure or given through diet. Mechanistic and biomarker analyses showed that silibinin activity against photocarcinogenesis was by inhibiting cell proliferation, mitogenic signaling, and induction of apoptosis [43, 44, 48–50]. Furthermore, it was reported that dietary feeding of silibinin at 1 % dose (w/w) for 2 weeks prior to a single UVB exposure strongly inhibits CPD formation, cell proliferation, sunburn cells, but increases p53 and its downstream molecule p21/Cip1 in irradiated SKH-1 hairless mouse skin [43]. The observed prevention by silibinin following dietary feeding is of utmost importance in chemoprevention of skin cancer. In bioavailability studies, we found that following oral dosing of silibinin (50 mg/kg body weight) to mice, it peaks at 1-h postadministration in the skin [51]. In the same study, silibinin was also detected in the liver, lung, stomach, pancreas, and prostate, indicating the appreciable bioavailability of this phytochemical [51]. Recent studies by others and us have also established the anticarcinogenic potential of silibinin against several other neoplasms such as the liver, lung, colon, and prostate, when administered orally, confirming its broad spectrum anticancer effects [10, 45, 52–56]. Overall, these findings make silibinin a potent and promising chemopreventive agent against UVB-induced skin cancer with minimal toxicity and remarkable efficacy.
Molecular Mechanism(s) of Skin Cancer Prevention by Silibinin
Due to strong preventive effects of silibinin against photocarcinogenesis, efforts have been made to understand the molecular mechanism/s underlying its chemopreventive efficacy. It is evident from the published studies by us and others that silibinin targets several molecular pathways in mouse skin which are described in detail below.
Silibinin Inhibits UVB-Caused DNA Damage
As mentioned earlier, UVB is absorbed by epidermal DNA causing various photoproduct formation including CPDs, 6-4 photoproducts, DNA-DNA/protein crosslinks, cytosine photohydrates, DNA strand breaks, etc. [13, 57]. Among these, formation of CPDs is quite frequent and important, and therefore, it is the well-studied form of UVB-caused skin epidermal DNA damage. Due to the importance of genomic integrity, there are various DNA repair mechanisms in normal cells that continuously monitor and repair majority of the DNA damage efficiently, but those DNA repair mechanisms could be ineffective in the case of severe damage to DNA which results in introduction of mutations in key genes following replication of damaged DNA (due to faulty repair) [58]. The most common mutations are CC→TT and C→T transitions which often are considered as UVB signature mutations [14, 19]. The cells harboring these mutations are referred as initiated cells, which can lie dormant for a long period of time. Our earlier studies with SKH-1 hairless mice and JB6 epidermal cells have shown strong inhibition of UVB-caused CPD formation [43, 48, 59••, 60••] together with an accelerated removal of CPDs in JB6 cells by silibinin [59••]. Dietary silibinin also showed similar inhibitory results toward UVB-caused CPD formation in mouse skin. These results strongly correlate with silibinin-mediated decrease in UVB-induced skin cancer incidence and a strong reduction in number of tumors in SKH-1 hairless mouse skin as summarized above.
Exposure of UVR to murine and or human epidermal cells causes a reduction in the levels of antioxidant enzymes and generates ROS, which leads to oxidative modification in DNA, proteins, and lipids [61]. The ROS-mediated DNA strand breaks, base modifications, and cross-links could lead to errors during replication causing genomic instability that is considered an important prerequisite for cancer development [62, 63]. Under oxidative stress conditions, lipids get oxidized and generate lipid peroxidation products, such as reactive aldehydes, which could react with DNA and form lesions, and hence, initiate cell transformation [64]. The strong protective efficacy of silibinin against ROS-induced cellular damages has been attributed to its strong antioxidant potential. In addition, silibinin has been shown to increase glutathione-S-transferase (GST) and quinine reductase (QR) activities in the skin which are involved in the removal of cellular reactive moieties [51]. Thus, it has been suggested that silibinin could also prevent DNA damage via decreasing oxidative stress.
Genomic stability is essential for cell survival, and in response to DNA damage, either the progression of cell cycle is halted for an adequate time to repair DNA damage when the damage is mild or reparable, or induce apoptotic death when the DNA damage is severe or irreparable. These DNA damage-related cellular responses are multifaceted and involve a complex signaling network in which p53 plays a crucial role [58]. Studies have shown that following DNA damage, p53 is activated rapidly, and this activation is predominantly through posttranslational modifications and interactions with other cellular cofactors [58, 65]. Following UVB exposure, p53 is phosphorylated at specific serine sites resulting in its stabilization and accumulation. Upon stabilization, p53 translocates to the nucleus and subsequently transactivates specific target genes involved in the pathways of DNA damage repair, cell cycle progression, and/or apoptosis-related signaling [66, 67]. Due to its importance, not surprisingly, the DNA binding domain of p53 is found to be a hot spot of mutations during the cancer development as observed in several malignancies including skin cancer [9, 68]. In fact, 50–60 % NMSC cases in humans harbor UVB signature mutations in p53 gene, which is comparable to murine skin tumors [9, 69]. In this regard, we and others have reported that UVB exposure to mouse skin and cultured epidermal cells caused a moderate to extensive apoptosis based on the UVB dose along with an accumulation of p53 protein [13, 44, 48, 60••, 70–73]. Importantly, silibinin further increased p53 level in SKH-1 hairless mouse skin epidermis, irrespective of its application on the dorsal side of mouse skin either before or after UVR exposure or given through diet. [44, 48]. Silibinin also increased p53 level in UVB exposure caused skin tumors [44]. In these studies, p21/Cip1, a downstream target of p53, was also found to be upregulated which contributed toward cell cycle arrest by inhibiting cyclin-dependent kinase (CDK)-cyclin complex [48]. Similarly, in cell culture experiments with epidermal JB6 and HaCaT cells, we reported that silibinin treatment further increases UVB-induced phosphorylation at serine 15 site which causes stabilization of p53 and is related to apoptosis [70, 71]. However, at lower UVB dose, silibinin inhibited apoptotic cell death in HaCaT cells [71]. Silibinin treatment also facilitated DNA damage repair by activating a p53 downstream target, namely, growth arrest and DNA damage-inducible protein alpha (GADD45α), in both JB6 cells and SKH-1 hairless mouse skin [60••]. Furthermore, another study by us showed that silibinin conferred protection against DNA damage and apoptosis in epidermal cells both in cell culture and murine skin by modulating IL-12 level indicating immune modulatory potential of silibinin [59••]. Overall, these findings clearly suggested that silibinin prevents UVB-induced DNA damage as well as potentiates the DNA repair machinery, as major mechanisms of its efficacy against photocarcinogenesis during tumor initiation stage.
Dual Efficacy of Silibinin Toward UVB-induced Proapoptotic and Antiapoptotic Machineries in Mouse Skin
Excessive exposure to UVB radiation could cause severe DNA damage which leads to removal of damaged cells via apoptotic death pathway by modulation of various proapoptotic and antiapoptotic proteins, dependent or independent of p53 involvement manner [74, 75]. Induction of apoptosis in DNA-damaged cells is of high importance, as its failure not only predisposes cells harboring DNA damage to the carcinogenesis, but also induces resistance to therapeutic interventions. Though apoptotic cell death following stress condition is widely regarded as a protective mechanism, excessive apoptosis of epidermal cells (sunburn) may compromise skin layer which acts as the first barrier for humans against adverse effects of various environmental toxins [76]. As discussed in section “Silibinin Inhibits UVB-Caused DNA Damage”, in the case of mild damage to DNA, silibinin protects normal epidermal cells from apoptosis, while following severe damage, silibinin further potentiates apoptotic death, suggesting its dual action against UVB-induced photodamage. In our previous short-term study, silibinin treatment reduced both apoptotic and sunburn cell populations in SKH-1 hairless mouse skin after UVB exposure at 180-mJ/cm2 dose, either once or 5 days [43]. However, in long-term study of 25 weeks, silibinin treatment further increased the apoptosis induced by UVB in both skin tumors and chronically UVB-exposed uninvolved mouse skin [44]. In the same study, it was also found that silibinin treatment caused activation of caspase 3 with a concomitant decrease in survivin levels in UVB-induced skin tumors and these pathways might be important contributors in silibinin-induced apoptotic cell death [44]. These results suggest that following UVB exposure, depending on the level of DNA damage, silibinin acts as DNA damage sensor and induces or inhibits apoptotic cell death.
Several in vitro cell culture studies have also reported a series of variable apoptotic effects of silibinin following UVB exposure which were dependent on the severity of DNA damage. Silibinin, at physiologically achievable doses, strongly induced apoptosis in A431 cells [72]. Importantly, silibinin inhibited apoptotic death in HaCaT cells (human immortalized keratinocytes) following UVB irradiation at lower doses (5 and 30 mJ/cm2), but enhanced apoptotic cell death following UVB exposure at a higher dose of 120 mJ/cm2 [71]. Molecular analyses revealed that silibinin treatment results in protection against UVB-induced activation of caspase 9 and PARP cleavage compared to UVB alone group, irrespective of its treatment before or immediately after UVB exposure [71]. Similar silibinin treatments were also found to alter the Bcl2 family of proteins (both proapoptotic and antiapoptotic proteins) as well as the interaction and localization of Bcl2, Bax, Bak, Bad, Bcl-xL, and cytochrome c [71]. Furthermore, survivin level was restored following silibinin treatment post-UVB irradiation. Recently, it was reported that silibinin pretreatment resulted in inhibition of UVB-caused apoptosis in HaCaT cells by downregulation of Fas-associating protein with death domain (FADD) and inhibition of procaspase-8 cleavage [77]. In addition, silibinin inhibited the UVB-induced apoptosis in A431 cells by promoting autophagy [78]. Further studies revealed that UVB activates IGFR1-PI3K-Akt signaling axis in A431 cells inhibiting autophagy, and silibinin downregulates this signaling axis and promotes autophagy, and thus opposed the proapoptotic effects of UVB [78, 79]. The induction of autophagy by silibinin arrested the cell growth transiently and promoted the repair of damaged DNA, and finally rescued the UVB-exposed cells from apoptosis. Overall, above summarized findings convincingly suggest the dual effectiveness of silibinin in inhibiting or inducing UVB-caused apoptosis in both animal and cell culture models.
Unlimited replicative or proliferative potential is considered as the essential characteristics of cancer cells including skin cancer. In this regard, the increased protein levels or interactions of cyclins and CDKs, together with a decrease in CDK inhibitors (CDKIs) expression, have been shown to play a casual role in UVB-caused skin carcinogenesis [80, 81]. Cell cycle analysis showed that majority of epidermal cells such as JB6 and HaCaT are in G1 phase which is further prolonged following UVB-induced DNA damage [60••, 71], via an increase in p21/Cip1 protein that could be dependent or independent of p53 involvement [71]. p21/Cip1 is one of the first cell cycle regulatory protein molecules upregulated in cells following DNA damage by various stress conditions including UVB, to halt the progression of cell cycle and thus proliferation of the cells harboring damaged DNA [82]. It has been observed that following chronic exposure to UVR, the protein levels of the cell cycle regulatory molecules are altered in the SKH-1 hairless mouse skin and silibinin was found to reduce UVB-induced expression of CDKs 2 and 4, cyclins B1, A, E, and D, Cdc2, Cdc25c and to up regulate the expression of CDKIs (p21/Cip1 and p27/Kip1) [44, 83]. Our cell culture findings also showed silibinin’s inhibitory activity on cell cycle progression following UVB-exposure by regulating CDKs and CDKIs [71, 84]. Overall, these studies suggest silibinin’s effects on cell cycle regulators which might be an important mechanism toward its antiproliferative action.
Silibinin Inhibits UVB-Caused Inflammation in Mouse Skin
Chronic inflammation is a critical component and prerequisite for tumor progression in skin carcinogenesis [6, 85, 86]. Skin protects from both injury and infection; whereas immune cells play a critical role in both wound healing and tissue repair, the inflammatory reactions initiated by these cells are essential in various skin pathological conditions as well as skin cancer. These immune cells secrete various growth factors, cytokines, prostaglandins, and chemotactic polypeptides, and thus lead to the condition of chronic inflammation in the skin which further contributes toward UV-induced skin carcinogenesis [3, 85, 87]. Clinical manifestations of UVB-induced skin inflammation are erythema, edema, and hyperplastic epithelial responses induced by infiltration and accumulation of inflammatory cells. Recent research suggests that UVB positively regulates autophagy in dermal inflammatory cells and inhibits their apoptotic death; however, it negatively regulates autophagy in epidermal cells and promotes their apoptosis. The apoptosis of epidermal cells results in secretion of inflammatory cytokines, mediating pro-inflammation reaction among dermal inflammatory cells, and induces chemotaxis and recruitment of neutrophils and mast cells, leading to skin inflammation [88•].
Several studies have shown the important roles of arachidonic acid, prostaglandins, cyclooxygenases (COX), lipoxygenase, tumor necrosis factor-α (TNF α), inducible nitric oxide synthase (iNOS), and several transcription factors including AP-1, NF-κB, and STAT3 in inflammation [85]. The arachidonic acid pathway is at the core of inflammatory response. In this pathway, COX enzymes are responsible for the formation of prostaglandins (PGE2, PGF2α, and PGD2), prostacyclin, and thromboxane, while lipoxygenase generates 5-HPETE which is converted to leukotrienes. COX2 is overexpressed in several cancers and considered an attractive drug target [89]. We reported previously a strong increase in the protein levels of COX2 levels in both the skin and skin tumors in mice following sustained exposure to a dose of UVB that was physiologically relevant to human exposure [50]. UV-caused increase in COX2 expression is known to induce PGE2 that is the major COX product involved in photocarcinogenesis [90]. Animal studies have clearly shown the roles of PGE2 receptors EP1, EP2, and EP4 in UVB-induced skin carcinogenesis [91]. Not surprisingly, a population-based case-control analysis in UK provides evidence that patients predisposed to NMSC might benefit from chemoprevention with nonsteroidal anti-inflammatory drugs (NSAIDs) [92•]. PGE2 has also been shown to promote inflammation, immunosuppression, tumor cell proliferation, antiapoptosis, and tumor invasion, and thus, contributing significantly toward the development of skin cancer [85, 86, 93]. Silibinin has also been shown to strongly inhibit UVB-induced COX2 and iNOS levels in the skin as well as skin tumors in SKH-1 hairless mice, irrespective of its topical application before or after UVB exposure or given in diet [50]. Furthermore, several transcription factors (AP-1, NF-κB, and STAT3) are known to regulate inflammatory cytokines, and UVB exposure has been reported to strongly activate these transcription factors. Consistent with its effects on COX2 and iNOS, similar silibinin treatments also inhibited UVB-caused NF-κB and STAT3 activation in both skin and skin tumors [50]; silibinin also strongly inhibited the activation of transcription factor AP-1 [84]. Together, these findings clearly suggested that silibinin inhibits inflammatory response in mouse skin following UVB exposure by inhibiting the expression and activity of COX2, and the level of pro-inflammatory cytokines through inhibition of AP-1, NF-κB, and STAT3 transcription factors, indicating a strong anti-inflammatory action of silibinin, which might be contributing toward its photocarcinogenesis preventive potential.
Silibinin Inhibits UVB-Caused Activation of Mitogenic and Survival Signaling in Mouse Skin
One of the hallmarks of tumor promotion and malignant transformation is the activation of mitogen-activated protein kinases (MAPKs) by both chemical tumor promoters and UVB [93, 94]. The UVB-caused activation of mitogenic signaling is mediated via extracellular regulated kinase 1/2 (ERK1/2), p38 kinase and c-jun-NH2-kinase 1/2 (JNK1/2) MAPKs [94]. These serine/threonine kinases upon activation phosphorylate and subsequently activate various downstream target molecules and regulate various cellular events including proliferation and apoptosis [95, 96]. While ERK1/2 activation is involved in cell proliferation and survival, p38 and JNK1/2 exert both antiapoptotic and proapoptotic functions dependent on stimuli and cellular context [97]. Furthermore, ERK1/2 activation also plays a critical role in UV and other tumor promoters caused cell transformation and tumor promotion [85, 98]. Akt also acts as a critical cell survival and antiapoptotic signaling molecule and plays an important role in both promotion and progression stages of carcinogenesis [99]. The UVB-mediated activation of both MAPKs and Akt leads to the activation of AP-1 and NF-κB, and these two transcription factors regulate the expression of an array of genes required for growth and proliferation including inflammatory cytokines, and further help in maintaining chronic inflammatory conditions, to promote photocarcinogenesis [85, 100].
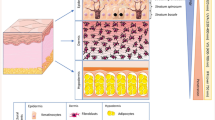
Our in vitro studies have shown that silibinin causes the inhibition of growth and death in A431 cells by significantly inhibiting the activation of ERK1/2 [72]. Conversely, in this study, silibinin activated p38 and JNK1/2 MAPKs which was associated with enhanced apoptotic cell death [72]. Similarly, silibinin also modulated these MAPKs in UVB-irradiated JB6 cells [84]. Our in vivo studies clearly suggested a dual role of MAPKs in silibinin efficacy against photocarcinogenesis. When exposure was done acutely at a single UVB dose (180 mJ/cm2), ERK1/2, p38 and JNK1/2 MAPKs and Akt were activated in the skin of SKH-1 hairless mice, which was inhibited significantly by silibinin irrespective of its topical application before or immediately after UVB irradiation or fed in diet [49]. Similar inhibitory effects of silibinin on MAPKs and Akt were observed in a chronic UVB exposure protocol when SKH-1 hairless mice were exposed with UVB dose of 180 mJ/cm2/day for 5 days [49, 83]. Conversely, in UVB-induced skin tumors in SKH-1 hairless mice, similar silibinin treatment further increased the UVB-induced activation of ERK1/2, p38 and JNK1/2 MAPKs in tumor samples at 25 weeks; however, the Akt expression was strongly decreased by silibinin [83]. These results clearly showed that silibinin inhibited various oncogenic signaling pathways required for tumor progression; however, it remains to be ascertained whether silibinin is also effective against chronic UVB-induced sustained genetic abnormalities at progression stage of skin carcinogenesis [101, 102] (Fig. 1). Overall, these findings suggest that the modulation of mitogenic signaling with silibinin is an important mechanism of its efficacy against UVB-induced skin cancer.

Effect of silibinin on UVB-induced skin damage and carcinogenesis
Pharmacokinetics and Safety of Silibinin
The oral bioavailability of silibinin is poor due to its poor solubility in water; therefore, silibinin complexes have been developed for improved absorption. The commercially available silibinin complexes are IdB 1016 (silipide) and silybin-phytosome (siliphos), which are formulation of silibinin with phosphatidylcholine, and have shown improved bioavailability compared to silibinin [103–105]. Barzaghi et al. determined the plasma silibinin levels in nine healthy volunteers following administration of single oral dose of silipide (equivalent to 360 mg silibinin) and found that free silibinin concentration in plasma reached a peak of 298 ± 96 ng/ml achieved at 1.6 ± 1 h [103]. In another study by the same group, nine healthy volunteers received silipide (equivalent to 120 mg silibinin) twice daily for 8 consecutive days. The terminal half-life of silipide in plasma on day 1 was 2.6 ± 1 h with no change on day 8. Urinary profile revealed that less than 3 % of the administered dose was excreted via urine, with a significant proportion of the dose probably being excreted in the bile [103]. In a similar study in humans, 12 healthy volunteers received a single oral dose of silipide (equivalent to 80 mg silibinin) and the free silibinin concentration reached a peak of 141 ± 32 ng/ml in plasma which was achieved at 2.4 h [104].
Due to the promising efficacy of silibinin against various malignancies in preclinical animal models, silibinin was also tested in several clinical trials. In one such trial in 2006, patients with confirmed colorectal adenocarcinoma received daily dose of silibinin (in the form of silipide) at 360, 720, or 1440 mg, and after 7 days, blood and colorectal and liver tissues were collected [106]. Further analysis showed that silibinin levels were between 0.3 and 4.0 μmol/l in blood, between 28 and 141 nmol/gm in colorectal tissue and between 1.0 and 2.5 nmol/gm in hepatic tissue [106]. These doses of silibinin were not associated with any adverse effect and termed as safe [106]. In another phase I/II clinical trial of silibinin in prostate cancer patients, authors concluded that silibinin doses up to 13 gm per day were well tolerated in patients without much side effects [107, 108]. Plasma analysis showed that the half-life of silibinin in plasma was short and ranged between 1.79 and 4.99 h, which was consistent with earlier studies with healthy volunteers, with the peak levels close to 100 μM [108]. These studies clearly suggested that there is minimal safety concerns related to silibinin consumption in humans, and bioavailability at physiologically pertinent concentrations could be attained by using formulation with phosphatidylcholine. Though, so far, no such trial has been conducted in skin cancer patients, the nontoxic nature of silibinin and its strong efficacy against UVB-induced skin carcinogenesis in preclinical animal models strongly supports its evaluation in skin cancer patients.
Conclusion
The studies elaborated above clearly show that nontoxic silibinin possesses potent efficacy against all stages of UVB-induced skin carcinogenesis through its various attributes including antioxidant, DNA damage repair, immunomodulation, anti-inflammatory, and antiproliferative actions (Fig. 1). Importantly, the chemopreventive potential of silibinin against photocarcinogenesis is strongly supported by comprehensive mechanistic rationale; therefore, its greater use is recommended against UV-induced cellular damages and photocarcinogenesis. In future, silibinin efficacy should also be tested in therapeutic settings where skin tumors have already progressed and it will be interesting to examine whether silibinin could inhibit UVB-induced sustained genetic abnormalities resulting in oncogenes activation and tumor suppressor gene inactivation (Fig. 1). Considering the fact that skin cancer incidences are increasing worldwide and sunscreens offer only a partial protection, silibinin offers a unique and novel chemopreventive option to better manage and reduce skin cancer burden.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Proksch E, Brandner JM, Jensen JM. The skin: an indispensable barrier. Exp Dermatol. 2008;17:1063–72.
Lo JA, Fisher DE. The melanoma revolution: from UV carcinogenesis to a new era in therapeutics. Science. 2014;346:945–9.
Hensler S, Mueller MM. Inflammation and skin cancer: old pals telling new stories. Cancer J. 2013;19:517–24.
Moan J, Grigalavicius M, Baturaite Z, Dahlback A, Juzeniene A. The relationship between UV exposure and incidence of skin cancer. Photodermatol Photoimmunol Photomed. 2014;31(1):26–35.
Leiter U, Eigentler T, Garbe C. Epidemiology of skin cancer. Adv Exp Med Biol. 2014;810:120–40.
Elmets CA, Ledet JJ, Athar M. Cyclooxygenases: mediators of UV-induced skin cancer and potential targets for prevention. J Investig Dermatol. 2014;134:2497–502.
Mancebo SE, Wang SQ. Skin cancer: role of ultraviolet radiation in carcinogenesis. Rev Environ Health. 2014;29:265–73.
Emmert S, Schon MP, Haenssle HA. Molecular biology of basal and squamous cell carcinomas. Adv Exp Med Biol. 2014;810:234–52.
Ratushny V, Gober MD, Hick R, Ridky TW, Seykora JT. From keratinocyte to cancer: the pathogenesis and modeling of cutaneous squamous cell carcinoma. J Clin Investig. 2012;122:464–72.
Deep G, Agarwal R. Chemopreventive efficacy of silymarin in skin and prostate cancer. Integr Cancer Ther. 2007;6:130–45.
Fu J, Bassi DE, Zhang J, Li T, Cai KQ, Testa CL, et al. Enhanced UV-induced skin carcinogenesis in transgenic mice overexpressing proprotein convertases. Neoplasia. 2013;15:169–79.
Mukhtar H, Elmets CA. Photocarcinogenesis: mechanisms, models and human health implications. Photochem Photobiol. 1996;63:356–7.
Singh RP, Agarwal R. Mechanisms and preclinical efficacy of silibinin in preventing skin cancer. Eur J Cancer. 2005;41:1969–79.
Protic-Sabljic M, Tuteja N, Munson PJ, Hauser J, Kraemer KH, Dixon K. UV light-induced cyclobutane pyrimidine dimers are mutagenic in mammalian cells. Mol Cell Biol. 1986;6:3349–56.
Bickers DR, Athar M. Oxidative stress in the pathogenesis of skin disease. J Investig Dermatol. 2006;126:2565–75.
Cadet J, Douki T, Pouget JP, Ravanat JL, Sauvaigo S. Effects of UV and visible radiations on cellular DNA. Curr Probl Dermatol. 2001;29:62–73.
Watson RE, Gibbs NK, Griffiths CE, Sherratt MJ. Damage to skin extracellular matrix induced by UV exposure. Antioxid Redox Signal. 2014;21:1063–77.
Wagener FA, Carels CE, Lundvig DM. Targeting the redox balance in inflammatory skin conditions. Int J Mol Sci. 2013;14:9126–67. A comprehensive review explaning the involvement of ROS with chronic skin inflammation and the mechanism(s) responsible for maintainance of redox balance in skin. It has also been suggested that maintaining the redox balance with anti-oxidants is an attractive target for prevention of photocarcinogenesis.
Mitchell DL, Nairn RS. The biology of the (6-4) photoproduct. Photochem Photobiol. 1989;49:805–19.
Pfeifer GP, You YH, Besaratinia A. Mutations induced by ultraviolet light. Mutat Res. 2005;571:19–31.
Schwarz T, Schwarz A. Molecular mechanisms of ultraviolet radiation-induced immunosuppression. Eur J Cell Biol. 2011;90:560–4.
Lacour JP. Carcinogenesis of basal cell carcinomas: genetics and molecular mechanisms. Br J Dermatol. 2002;146 Suppl 61:17–9.
Reichrath J, Rass K. Ultraviolet damage, DNA repair and vitamin D in nonmelanoma skin cancer and in malignant melanoma: an update. Adv Exp Med Biol. 2014;810:208–33.
Jayaraman SS, Rayhan DJ, Hazany S, Kolodney MS. Mutational landscape of basal cell carcinomas by whole-exome sequencing. J Investig Dermatol. 2014;134:213–20.
Griewank KG, Murali R, Schilling B, Schimming T, Moller I, Moll I, et al. TERT promoter mutations are frequent in cutaneous basal cell carcinoma and squamous cell carcinoma. PLoS One. 2013;8:e80354.
Weinstock MA. Do sunscreens increase or decrease melanoma risk: an epidemiologic evaluation. J Investig Dermatol Symp Proc Soc Investig Dermatol Inc Eur Soc Dermatol Res. 1999;4:97–100.
Ames BN, Gold LS, Willett WC. The causes and prevention of cancer. Proc Natl Acad Sci U S A. 1995;92:5258–65.
Potter JD. Cancer prevention: epidemiology and experiment. Cancer Lett. 1997;114:7–9.
Katta R, Desai SP. Diet and dermatology: the role of dietary intervention in skin disease. J Clin Aesthet Dermatol. 2014;7:46–51.
Afaq F, Adhami VM, Mukhtar H. Photochemoprevention of ultraviolet B signaling and photocarcinogenesis. Mutat Res. 2005;571:153–73.
Afaq F, Katiyar SK. Polyphenols: skin photoprotection and inhibition of photocarcinogenesis. Mini-Rev Med Chem. 2011;11:1200–15.
Singh RP, Agarwal R. Cosmeceuticals and silibinin. Clin Dermatol. 2009;27:479–84.
Katiyar SK. Green tea prevents non-melanoma skin cancer by enhancing DNA repair. Arch Biochem Biophys. 2011;508:152–8.
Nichols JA, Katiyar SK. Skin photoprotection by natural polyphenols: anti-inflammatory, antioxidant and DNA repair mechanisms. Arch Dermatol Res. 2010;302:71–83.
Saha P, Das S. Elimination of deleterious effects of free radicals in murine skin carcinogenesis by black tea infusion, theaflavins & epigallocatechin gallate. Asian Pac J Cancer Prev APJCP. 2002;3:225–30.
Korac RR, Khambholja KM. Potential of herbs in skin protection from ultraviolet radiation. Pharmacogn Rev. 2011;5:164–73.
Meeran SM, Mantena SK, Elmets CA, Katiyar SK. Epigallocatechin-3-gallate prevents photocarcinogenesis in mice through interleukin-12-dependent DNA repair. Cancer Res. 2006;66:5512–20.
Luper S. A review of plants used in the treatment of liver disease: part 1. Altern Med Rev J Clin Ther. 1998;3:410–21.
Kren V, Walterova D. Silybin and silymarin–new effects and applications. Biomed Pap Med Fac Univ Palacky Olomouc Czech. 2005;149:29–41.
Gyorgy I, Antus S, Blazovics A, Foldiak G. Substituent effects in the free radical reactions of silybin: radiation-induced oxidation of the flavonoid at neutral pH. Int J Radiat Biol. 1992;61:603–9.
Tewari-Singh N, Jain AK, Inturi S, Agarwal C, White CW, Agarwal R. Silibinin attenuates sulfur mustard analog-induced skin injury by targeting multiple pathways connecting oxidative stress and inflammation. PLoS One. 2012;7:e46149.
Narayanapillai S, Agarwal C, Tilley C, Agarwal R. Silibinin is a potent sensitizer of UVA radiation-induced oxidative stress and apoptosis in human keratinocyte HaCaT cells. Photochem Photobiol. 2012;88:1135–40.
Gu M, Dhanalakshmi S, Singh RP, Agarwal R. Dietary feeding of silibinin prevents early biomarkers of UVB radiation-induced carcinogenesis in SKH-1 hairless mouse epidermis. Cancer Epidemiol Biomark Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol. 2005;14:1344–9.
Mallikarjuna G, Dhanalakshmi S, Singh RP, Agarwal C, Agarwal R. Silibinin protects against photocarcinogenesis via modulation of cell cycle regulators, mitogen-activated protein kinases, and Akt signaling. Cancer Res. 2004;64:6349–56.
Singh RP, Dhanalakshmi S, Tyagi AK, Chan DC, Agarwal C, Agarwal R. Dietary feeding of silibinin inhibits advance human prostate carcinoma growth in athymic nude mice and increases plasma insulin-like growth factor-binding protein-3 levels. Cancer Res. 2002;62:3063–9.
Bowden GT. Prevention of non-melanoma skin cancer by targeting ultraviolet-B-light signalling. Nat Rev Cancer. 2004;4:23–35.
Athanassiadou AM, Lazaris AC, Patsouris E, Tsipis A, Chelidonis G, Aroni K. Significance of cyclooxygenase 2, EZH-2 polycomb group and p53 expression in actinic keratosis and squamous cell carcinomas of the skin. Am J Dermatopathol. 2013;35:425–31.
Dhanalakshmi S, Mallikarjuna GU, Singh RP, Agarwal R. Silibinin prevents ultraviolet radiation-caused skin damages in SKH-1 hairless mice via a decrease in thymine dimer positive cells and an up-regulation of p53-p21/Cip1 in epidermis. Carcinogenesis. 2004;25:1459–65.
Gu M, Dhanalakshmi S, Mohan S, Singh RP, Agarwal R. Silibinin inhibits ultraviolet B radiation-induced mitogenic and survival signaling, and associated biological responses in SKH-1 mouse skin. Carcinogenesis. 2005;26:1404–13.
Gu M, Singh RP, Dhanalakshmi S, Agarwal C, Agarwal R. Silibinin inhibits inflammatory and angiogenic attributes in photocarcinogenesis in SKH-1 hairless mice. Cancer Res. 2007;67:3483–91.
Zhao J, Agarwal R. Tissue distribution of silibinin, the major active constituent of silymarin, in mice and its association with enhancement of phase II enzymes: implications in cancer chemoprevention. Carcinogenesis. 1999;20:2101–8.
Sangeetha N, Aranganathan S, Panneerselvam J, Shanthi P, Rama G, Nalini N. Oral supplementation of silibinin prevents colon carcinogenesis in a long term preclinical model. Eur J Pharmacol. 2010;643:93–100.
Singh RP, Tyagi A, Sharma G, Mohan S, Agarwal R. Oral silibinin inhibits in vivo human bladder tumor xenograft growth involving down-regulation of survivin. Clin Cancer Res Off J Am Assoc Cancer Res. 2008;14:300–8.
Raina K, Blouin MJ, Singh RP, Majeed N, Deep G, Varghese L, et al. Dietary feeding of silibinin inhibits prostate tumor growth and progression in transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 2007;67:11083–91.
Yan Y, Wang Y, Tan Q, Lubet RA, You M. Efficacy of deguelin and silibinin on benzo(a)pyrene-induced lung tumorigenesis in A/J mice. Neoplasia. 2005;7:1053–7.
Bousserouel S, Bour G, Kauntz H, Gosse F, Marescaux J, Raul F. Silibinin inhibits tumor growth in a murine orthotopic hepatocarcinoma model and activates the TRAIL apoptotic signaling pathway. Anticancer Res. 2012;32:2455–62.
Marrot L, Meunier JR. Skin DNA photodamage and its biological consequences. J Am Acad Dermatol. 2008;58:S139–48.
Jiang L, Sheikh MS, Huang Y. Decision making by p53: life versus death. Mol Cell Pharmacol. 2010;2:69–77.
Narayanapillai S, Agarwal C, Deep G, Agarwal R. Silibinin inhibits ultraviolet B radiation-induced DNA-damage and apoptosis by enhancing interleukin-12 expression in JB6 cells and SKH-1 hairless mouse skin. Mol Carcinog. 2014;53:471–9. This study showed the immunodulatory potential of silibinin in UVB-exposed mouse skin and human epidermal cells. The major finding was that silibinin activates the DNA repair following UVB exposure by enhancing the levels of IL-12.
Roy S, Deep G, Agarwal C, Agarwal R. Silibinin prevents ultraviolet B radiation-induced epidermal damages in JB6 cells and mouse skin in a p53-GADD45alpha-dependent manner. Carcinogenesis. 2012;33:629–36. This study suggests that the DNA repair enhancing potential of silibinin against UVB-induced photodamage is dependent on p53-mediated upregaulation of GADD45α transcription factor, a regualtor of DNA repair mechanism.
Pinnell SR. Cutaneous photodamage, oxidative stress, and topical antioxidant protection. J Am Acad Dermatol. 2003;48:1–19. quiz 20-2.
Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996;313(Pt 1):17–29.
Ziech D, Franco R, Pappa A, Panayiotidis MI. Reactive oxygen species (ROS)–induced genetic and epigenetic alterations in human carcinogenesis. Mutat Res. 2011;711:167–73.
Marnett LJ. Oxy radicals, lipid peroxidation and DNA damage. Toxicology. 2002;181–182:219–22.
Farnebo M, Bykov VJ, Wiman KG. The p53 tumor suppressor: a master regulator of diverse cellular processes and therapeutic target in cancer. Biochem Biophys Res Commun. 2010;396:85–9.
Ouhtit A, Muller HK, Davis DW, Ullrich SE, McConkey D, Ananthaswamy HN. Temporal events in skin injury and the early adaptive responses in ultraviolet-irradiated mouse skin. Am J Pathol. 2000;156:201–7.
Ouhtit A, Muller HK, Gorny A, Ananthaswamy HN. UVB-induced experimental carcinogenesis: dysregulation of apoptosis and p53 signalling pathway. Redox Rep Commun Free Radic Res. 2000;5:128–9.
Olivier M, Hussain SP, Caron de Fromentel C, Hainaut P, Harris CC. TP53 mutation spectra and load: a tool for generating hypotheses on the etiology of cancer. IARC Sci Publ. 2004;157:247–70.
Giglia-Mari G, Sarasin A. TP53 mutations in human skin cancers. Hum Mutat. 2003;21:217–28.
Dhanalakshmi S, Agarwal C, Singh RP, Agarwal R. Silibinin up-regulates DNA-protein kinase-dependent p53 activation to enhance UVB-induced apoptosis in mouse epithelial JB6 cells. J Biol Chem. 2005;280:20375–83.
Dhanalakshmi S, Mallikarjuna GU, Singh RP, Agarwal R. Dual efficacy of silibinin in protecting or enhancing ultraviolet B radiation-caused apoptosis in HaCaT human immortalized keratinocytes. Carcinogenesis. 2004;25:99–106.
Mohan S, Dhanalakshmi S, Mallikarjuna GU, Singh RP, Agarwal R. Silibinin modulates UVB-induced apoptosis via mitochondrial proteins, caspases activation, and mitogen-activated protein kinase signaling in human epidermoid carcinoma A431 cells. Biochem Biophys Res Commun. 2004;320:183–9.
Benjamin CL, Ullrich SE, Kripke ML, Ananthaswamy HN. p53 tumor suppressor gene: a critical molecular target for UV induction and prevention of skin cancer. Photochem Photobiol. 2008;84:55–62.
Mullauer L, Gruber P, Sebinger D, Buch J, Wohlfart S, Chott A. Mutations in apoptosis genes: a pathogenetic factor for human disease. Mutat Res. 2001;488:211–31.
Ponten F, Berne B, Ren ZP, Nister M, Ponten J. Ultraviolet light induces expression of p53 and p21 in human skin: effect of sunscreen and constitutive p21 expression in skin appendages. J Investig Dermatol. 1995;105:402–6.
Polefka TG, Meyer TA, Agin PP, Bianchini RJ. Effects of solar radiation on the skin. J Cosmet Dermatol. 2012;11:134–43.
Li LH, Wu LJ, Tashiro S, Onodera S, Uchiumi F, Ikejima T. Silibinin prevents UV-induced HaCaT cell apoptosis partly through inhibition of caspase-8 pathway. Biol Pharm Bull. 2006;29:1096–101.
Liu W, Otkur W, Li L, Wang Q, He H, Ye Y, et al. Autophagy induced by silibinin protects human epidermoid carcinoma A431 cells from UVB-induced apoptosis. J Photochem Photobiol B Biol. 2013;123:23–31.
Liu W, Otkur W, Li L, Wang Q, He H, Zang L, et al. Interference of silibinin with IGF-1R signalling pathways protects human epidermoid carcinoma A431 cells from UVB-induced apoptosis. Biochem Biophys Res Commun. 2013;432:314–9.
Pflaum J, Schlosser S, Muller M. p53 family and cellular stress responses in cancer. Front Oncol. 2014;4:285.
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70.
Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–14.
Gu M, Singh RP, Dhanalakshmi S, Mohan S, Agarwal R. Differential effect of silibinin on E2F transcription factors and associated biological events in chronically UVB-exposed skin versus tumors in SKH-1 hairless mice. Mol Cancer Ther. 2006;5:2121–9.
Singh RP, Dhanalakshmi S, Mohan S, Agarwal C, Agarwal R. Silibinin inhibits UVB- and epidermal growth factor-induced mitogenic and cell survival signaling involving activator protein-1 and nuclear factor-kappaB in mouse epidermal JB6 cells. Mol Cancer Ther. 2006;5:1145–53.
Rundhaug JE, Fischer SM. Molecular mechanisms of mouse skin tumor promotion. Cancer. 2010;2:436–82.
Simper MS, Rundhaug JE, Mikulec C, Bowen R, Shen J, Lu Y, et al. The tumor promoting activity of the EP4 receptor for prostaglandin E in murine skin. Mol Oncol. 2014;8(8):1626–39.
Mikulec CD, Rundhaug JE, Simper MS, Lubet RA, Fischer SM. The chemopreventive efficacies of nonsteroidal anti-inflammatory drugs: the relationship of short-term biomarkers to long-term skin tumor outcome. Cancer Prev Res. 2013;6:675–85.
Wang Q, Ye Y, Liu W, Jiang S, Tashiro S, Onodera S, et al. Dual effects of silibinin treatment on autophagy-regulated dermal apoptosis retardation and epidermal apoptosis up-regulation in UVB-induced skin inflammation. J Asian Nat Prod Res. 2012;14:688–99. In this study, the role of balance between cell survival and apoptosis in dermis and epidermis in UVB-induced skin inflammation and the effect of autophagy on the balance were elucidated. It has also been reported that dual effects of silibinin on the balance between autophagy and apoptosis of epidermal and dermal cells lead to protection from UVB-induced inflammation.
Gasparini G, Longo R, Sarmiento R, Morabito A. Inhibitors of cyclo-oxygenase 2: a new class of anticancer agents? Lancet Oncol. 2003;4:605–15.
Chun KS, Langenbach R. A proposed COX-2 and PGE(2) receptor interaction in UV-exposed mouse skin. Mol Carcinog. 2007;46:699–704.
Rundhaug JE, Simper MS, Surh I, Fischer SM. The role of the EP receptors for prostaglandin E2 in skin and skin cancer. Cancer Metastasis Rev. 2011;30:465–80.
Reinau D, Surber C, Jick SS, Meier CR. Nonsteroidal anti-inflammatory drugs and the risk of nonmelanoma skin cancer. Int J Cancer J Int Cancer. 2014. A population-based case-control analysis which provides evidence that patients predisposed to NMSCs might benefit from chemoprevention with NSAIDs.
Bachelor MA, Bowden GT. UVA-mediated activation of signaling pathways involved in skin tumor promotion and progression. Semin Cancer Biol. 2004;14:131–8.
Bode AM, Dong Z. Mitogen-activated protein kinase activation in UV-induced signal transduction. Sci STKE Signal Trans Knowl Environ. 2003;2003:RE2.
Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–49.
Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–2.
Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–31.
Ming M, Han W, Maddox J, Soltani K, Shea CR, Freeman DM, et al. UVB-induced ERK/AKT-dependent PTEN suppression promotes survival of epidermal keratinocytes. Oncogene. 2010;29:492–502.
Strozyk E, Kulms D. The role of AKT/mTOR pathway in stress response to UV-irradiation: implication in skin carcinogenesis by regulation of apoptosis, autophagy and senescence. Int J Mol Sci. 2013;14:15260–85.
Marks F, Furstenberger G, Muller-Decker K. Tumor promotion as a target of cancer prevention. Recent Results Cancer Res. 2007;174:37–47.
Boukamp P. Non-melanoma skin cancer: what drives tumor development and progression? Carcinogenesis. 2005;26:1657–67.
Kaufmann WK, Carson CC, Omolo B, Filgo AJ, Sambade MJ, Simpson DA, et al. Mechanisms of chromosomal instability in melanoma. Environ Mol Mutagen. 2014;55:457–71.
Barzaghi N, Crema F, Gatti G, Pifferi G, Perucca E. Pharmacokinetic studies on IdB 1016, a silybin- phosphatidylcholine complex, in healthy human subjects. Eur J Drug Metab Pharmacokinet. 1990;15:333–8.
Gatti G, Perucca E. Plasma concentrations of free and conjugated silybin after oral intake of a silybin-phosphatidylcholine complex (silipide) in healthy volunteers. Int J Clin Pharmacol Ther. 1994;32:614–7.
Wen Z, Dumas TE, Schrieber SJ, Hawke RL, Fried MW, Smith PC. Pharmacokinetics and metabolic profile of free, conjugated, and total silymarin flavonolignans in human plasma after oral administration of milk thistle extract. Drug Metab Dispos Biol Fate Chem. 2008;36:65–72.
Hoh C, Boocock D, Marczylo T, Singh R, Berry DP, Dennison AR, et al. Pilot study of oral silibinin, a putative chemopreventive agent, in colorectal cancer patients: silibinin levels in plasma, colorectum, and liver and their pharmacodynamic consequences. Clin Cancer Res Off J Am Assoc Cancer Res. 2006;12:2944–50.
Flaig TW, Glode M, Gustafson D, van Bokhoven A, Tao Y, Wilson S, et al. A study of high-dose oral silybin-phytosome followed by prostatectomy in patients with localized prostate cancer. Prostate. 2010;70:848–55.
Flaig TW, Gustafson DL, Su LJ, Zirrolli JA, Crighton F, Harrison GS, et al. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Investig New Drugs. 2007;25:139–46.
Acknowledgments
Original studies with silibinin efficacy as skin cancer chemopreventive agent are supported by NCI R01 grant CA140368.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Rahul Kumar and Gagan Deep declare that they have no conflict of interest.
Rajesh Agarwal declares a financial interest in Protechsure Scientific, Inc. which is developing skin products containing silibinin.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Cancer Chemoprevention
Rights and permissions
About this article

Cite this article
Kumar, R., Deep, G. & Agarwal, R. An Overview of Ultraviolet B Radiation-Induced Skin Cancer Chemoprevention by Silibinin. Curr Pharmacol Rep 1, 206–215 (2015). https://doi.org/10.1007/s40495-015-0027-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40495-015-0027-9