
Abstract
Fabry disease is a rare lysosomal disorder characterized by deficient or absent α-galactosidase A activity resulting from mutations in the GLA gene. Migalastat (Galafold™), a pharmacological chaperone, stabilizes and facilitates trafficking of amenable mutant forms of α-galactosidase A enzyme from the endoplasmic reticulum to lysosomes and increases its lysosomal activity. Oral migalastat is the first pharmacological chaperone approved for treating patients [aged ≥ 18 years (USA and Canada) or ≥ 16 years in other countries] with Fabry disease who have a migalastat-amenable GLA mutation. In the FACETS trial in enzyme replacement therapy (ERT)-naive patients with GLA mutations amenable or non-amenable to migalastat, there was no significant difference between the migalastat and placebo groups for the proportion of patients achieving a ≥ 50% reduction in the number of globotriaosylceramide (GL-3) inclusions/kidney interstitial capillary (KIC) at 6 months [primary endpoint; intent-to-treat (ITT) population]. In the modified ITT population (i.e. patients with migalastat-amenable GLA mutations), relative to placebo, migalastat treatment significantly reduced the mean number of GL-3 inclusions/KIC and plasma lyso-globotriaosylsphingosine levels at 6 months. Among evaluable patients, migalastat maintained renal function and reduced cardiac mass after ≤ 24 months’ therapy. In the ATTRACT trial in ERT-experienced patients, renal function was maintained during 18 months of migalastat or ERT; however, migalastat significantly reduced cardiac mass compared with ERT. Migalastat was generally well tolerated in both of these trials. Given its convenient oral regimen and the limited therapeutic options available, migalastat is an important treatment option for Fabry disease in patients with migalastat-amenable GLA mutations.
Similar content being viewed by others


An oral pharmacological chaperone that selectively and reversibly binds to the active sites of amenable mutant forms of α-galactosidase enzyme |
Convenient every-other-day oral regimen |
Reduces substrate accumulation in renal tissue, urine and plasma, maintains renal function and reduces cardiac mass in ERT-naive and -experienced patients with migalastat-amenable GLA-mutations |
1 Introduction
Fabry disease is a rare, progressive lysosomal disorder caused by mutations in the GLA gene [1]. GLA encodes the homodimeric glycoprotein, α-galactosidase A [1], which acts in lysosomes to degrade globotriaosylceramide (GL-3) and its deacylated form, globotriaosylsphingosine (lyso-Gb3) [2, 3]. In Fabry disease, the activity of α-galactosidase A is deficient or absent, leading to progressive accumulation of glycolipids, primarily GL-3 and lyso-Gb3, in the plasma and numerous cell types throughout the body [1, 4]. This leads to a variety of clinical manifestations and phenotypes with potentially life-threatening complications [1, 4], and can have a considerable impact on a patient’s health-related quality of life [5].
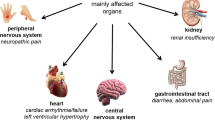
Fabry disease is broadly divided into “classic” and “late-onset” phenotypes. Patients with the classic phenotype of Fabry disease are typically male, have severely low or undetectable (< 3% of mean normal) α-galactosidase A activity and develop clinical manifestations in multiple organ systems, including the renal, cardiac, nervous and gastrointestinal systems [1, 6]. The first symptoms (e.g. chronic neuropathic pain and episodic severe pain crises) present within the first or second decades of life; symptomatic organ complications [e.g. chronic kidney disease progression to renal failure, and left ventricular hypertrophy (LVH) associated with arrhythmias, strokes and myocardial fibrosis] typically emerge in young adult patients and eventually lead to a premature death [1]. Patients with the late-onset phenotype have varied ages of onset and clinical manifestations, with typical cardiac and renal symptoms [e.g. LVH and reduced glomerular filtration rate (GFR)] presenting in the fourth to seventh decades of life [1].
Fabry disease is an X-linked disorder; the GLA gene is located on the long arm of the X chromosome [1]. Consequently, female patients with the disease are heterozygous and are affected to varying degrees [1], ranging from seemingly asymptomatic to severe [6,7,8]. Clinical presentation varies considerably, even among patients with the same genotype [9]. This phenotypic heterogeneity is thought to arise partly due to lyonization, in which one copy of the X chromosome in all cells is randomly inactivated during early embryonic development [10]. Consequently, female Fabry patients are a ‘mosaic’ of both normal cells and mutant Fabry cells in varying proportions [10]. Clinical symptoms in female Fabry patients may arise as a result of skewed X chromosome inactivation; for example, when a higher percentage of X chromosomes with the mutant GLA gene are expressed in a particular tissue [10].
The heterogeneous spectrum of Fabry clinical manifestations means that an individualized approach to patient care is warranted, based on the genotype, phenotype, family history, gender and the severity of symptoms of the patient [1]. The current therapeutic options for Fabry disease include enzyme replacement therapy (ERT) with intravenous (IV) agalsidase beta (approved in the USA, EU and elsewhere [1]) or agalsidase alfa (approved in the EU and elsewhere; not approved in the USA [1]) every two weeks, and migalastat for patients with amenable gene mutations, along with supportive care to manage symptoms [1, 5]. The two ERTs are similar, but not identical, formulations of recombinant α-galactosidase A [11], and both provide clinical benefits in patients with Fabry disease [10, 12]. However, ERT is limited by several factors, including considerable clinical variation, high costs, a frequent incidence of mild to moderate infusion-related reactions (which may arise from immunoglobulin antibody formation specific to the infused enzyme), a lack of consensus with regards to the optimal age to initiate therapy and a life-long burden of biweekly IV infusions (i.e. every two weeks) [10, 12, 13].
A novel approach to overcome some of the limitations of ERT is pharmacological chaperone therapy using oral small molecule agents (as reviewed by Parenti et al. [14]), which restores endogenous enzyme activity and degradation of GL3 and other disease substrates (Fig. 1) [14, 15]. Oral migalastat (Galafold™) is one such agent that is indicated for Fabry disease in patients with migalastat-amenable (i.e. responsive to migalastat) mutant forms of α-galactosidase A (i.e. GLA variants that retain their catalytic activity despite abnormal protein folding) [2, 3]. Migalastat is the first pharmacological chaperone to be approved for the treatment of patients [aged ≥ 18 years in the USA and Canada; aged ≥ 16 years in other countries (Sect. 6)] who have a confirmed diagnosis of Fabry disease and a migalastat-amenable GLA mutation. This article reviews the efficacy and tolerability of migalastat in this patient population, and summarizes its pharmacological properties.

Reproduced from Parenti et al. [14] with permission
The mechanism of action of pharmacological chaperones (hexagons), which act to selectively bind and stabilize otherwise unstable enzymes to enhance or partially restore folding and stability. Pharmacological chaperones allow for normal trafficking of enzymes, thus increasing enzyme activity in lysosomes.
2 Pharmacodynamic Properties of Migalastat
Migalastat, a low molecular weight iminosugar analogue of the terminal galactose residue on GL-3 [2], binds selectively and reversibly to the active sites of amenable mutant forms of α-galactosidase A enzyme [16,17,18]. This binding allows migalastat to act as a pharmacological chaperone (occurs at sub-inhibitory concentrations of the drug), thereby stabilizing migalastat-amenable mutant forms of α-galactosidase A in the endoplasmic reticulum (ER; preventing retention and degradation) and facilitating proper trafficking to lysosomes [16, 17, 19]. Once in lysosomes, migalastat dissociates from α-galactosidase A as a result of the more acidic pH (compared with a neutral pH in the ER) and higher concentration of substrates, allowing the enzyme to break down GL-3 [2, 16, 17, 19]. Following dissociation from the enzyme, migalastat is rapidly removed from the cell and excreted (Sect. 3).
In preclinical studies, migalastat increased α-galactosidase A activity in cultured lymphoblasts and fibroblasts derived from patients with Fabry disease [15, 16]. In Fabry transgenic mice expressing mutant forms of α-galactosidase A (hR301Q α-galactosidase A Tg/KO and TgM/KO mice), migalastat increased α-galactosidase A activity [16, 20, 21] and reduced GL-3 [20,21,22] and lyso-Gb3 levels [22] in major tissues. Increased α-galactosidase A activity and reduced GL-3 levels were also observed in the brain [20], indicating that migalastat may cross the blood-brain barrier.
Migalastat has been reported to have a shorter tissue half-life than α-galactosidase A in Fabry transgenic mice (typically hours vs. days) [20]. This difference allows for a less frequent dosing regimen and an opportunity to achieve a maximal pharmacological chaperone effect, with periods of optimal enzyme stabilization in the ER and trafficking to lysosomes, followed by periods of optimal substrate degradation in lysosomes without migalastat administration [20].
In a phase 1 trial in healthy volunteers, twice-daily oral migalastat hydrochloride 150 mg [hereafter referred to as migalastat 123 mg; the recommended dose (Sect. 6)] resulted in a twofold greater increase in wild-type α-galactosidase A activity in white blood cell lysates at days 4 and 7 than a 50 mg dose [23].
In phase 2 trials in patients with Fabry disease, oral migalastat 123 mg once every other day resulted in increased α-galactosidase A activity in the blood, skin and kidney [24], reduced GL-3 levels in the urine, skin and certain kidney cell types [22, 24, 25] and reduced lyso-Gb3 in plasma [22], compared with baseline. These beneficial responses were greater and/or more consistent in patients with migalastat-amenable GLA mutations than in patients with non-amenable mutations (assessed in an ex vivo lymphocyte assay [25] or an in vitro assay [22, 24]).
At ≈ 8 times the recommended dose (Sect. 6), migalastat did not prolong the QT interval to any clinically relevant extent [2].
2.1 Amenability of GLA Mutations
There are more than 1000 mutations in the GLA gene known to be associated with Fabry disease [26]; an estimated 35-50% of patients with Fabry disease have mutations that are amenable to migalastat therapy [27]. The amenability of GLA mutations to migalastat therapy is determined by a validated good laboratory practice (GLP) in vitro pharmacogenetics assay, which uses human embryonic kidney (HEK) 293 cells that have been transfected with individual GLA-containing DNA plasmids [28]. In this assay, migalastat-amenable mutations are defined as those in GLA that translate to mutant forms of α-galactosidase A and display a ≥ 1.2-fold increase in α-galactosidase A activity over baseline and an absolute increase of ≥ 3% over wild-type α-galactosidase A activity, in the presence of 10 μmol/L migalastat [28]. This amenability assay is applicable in both male and female patients and does not require patient samples [28]. Moreover, it has greater precision, consistency, rigor and quality control [27, 29] than a previously developed assay in HEK cells [30]; thus, it better predicts the amenability of mutant forms of α-galactosidase A to migalastat. However, it must be acknowledged that this assay is not intended for use in assisting with the diagnosis of Fabry disease or for predicting the pathogenicity of a genetic variant [28].
3 Pharmacokinetic Properties of Migalastat
Oral migalastat exhibits dose-proportional pharmacokinetics across a dose range of 75–1250 mg [2, 3, 23]. In healthy fasted volunteers, migalastat was rapidly absorbed, reaching maximum plasma concentrations (Cmax) within 1–6 h (median 3 h) after a single 123 mg dose [23, 31, 32]. In patients with Fabry disease receiving migalastat 123 mg once every other day for 14 days, Cmax was attained within an estimated time of 2.6 h (abstract) [33]. Food reduced the rate and extent of migalastat absorption to a clinically relevant extent (Sect. 6), with migalastat mean total exposure and Cmax reduced by 37–42 and 15 to ≈ 40% compared with the fasted state [2, 3, 34]. The absolute bioavailability of migalastat after a single 123 mg dose is ≈ 75% [2, 3].
Migalastat is extensively distributed into organs and tissues, with an apparent volume of distribution of ≈ 89 L (range 77–133 L) [2, 3]. No plasma protein binding was detected across a migalastat radiolabelled dose range of 1 to 100 μmol/L [2, 3]. No accumulation of migalastat was observed with migalastat 123 mg taken every other day [2].
After a single 123 mg dose, migalastat was rapidly removed from the plasma compartment [mean elimination half-life (t1/2) of ≈ 4 h], with an apparent clearance of 12.5 L/h [2]. In healthy volunteers, 77 and 20% of the total migalastat radiolabelled dose was recovered in the urine and faeces (total recovery of 98% within 96 h) [2, 3, 35].
Migalastat is a substrate for uridine diphosphate glucuronosyltransferase (UGT), a minor elimination pathway, based on in vivo data [2, 3]. Migalastat is not a substrate for P-glycoprotein in vitro, is unlikely to inhibit or induce cytochrome P450 enzymes, does not inhibit human efflux or uptake transporter proteins, and exhibits low affinity for sodium-glucose linked transporter 1 (SGLT1) as a substrate and inhibitor [2, 3].
The pharmacokinetics of migalastat are not altered to a clinically relevant extent by gender or race [2]. In a population pharmacokinetic analysis, the clearance of migalastat did not differ to a clinically relevant extent between patients with Fabry disease aged ≥ 65 years and those aged < 65 years [3]. In non-Fabry patients with renal impairment, exposure to migalastat and t1/2 increased with increasing degrees of renal impairment (mild, moderate and severe renal impairment were associated with 1.2-, 1.8- and 4.5-fold increases in exposure relatively to healthy controls, respectively; t1/2 was 7.7, 22.2 and 32.3 h, respectively) [31]. The use of migalastat has not been studied in patients with Fabry disease who have severe renal impairment [2, 3], or who have hepatic impairment; however, hepatic impairment is not expected to affect the pharmacokinetics of migalastat, based on the metabolism and excretion pathways [3].
4 Therapeutic Efficacy of Migalastat
The efficacy of oral migalastat 123 mg every other day in patients aged 16–74 years with genetically confirmed Fabry disease was assessed in two pivotal, randomized, multicentre, placebo-controlled (FACETS [29]) or active comparator-controlled (ATTRACT [27]) phase 3 trials and two open-label extension (OLE) trials [3]. Migalastat efficacy assessment for approval in the USA was primarily based on data from FACETS [29] due to the double-blind, placebo-controlled nature of the trial; the ATTRACT [27] trial provided the basis for approval in Europe and Japan.
In phase 3 trials, eligible patients were required to have a migalastat-amenable GLA mutation based on the initial HEK-293 assay [intent-to-treat (ITT) population; in FACETS and ATTRACT: n = 67 and 57] [27, 29]. Post-randomization, with the availability of the GLP-validated HEK assay (Sect. 2.1), participants were reclassified as having an amenable or non-amenable GLA variant to migalastat therapy; patients who had migalastat-amenable GLA mutations according to the GLP-validated HEK assay are hereafter referred to as the modified ITT (mITT) population (FACETS and ATTRACT: n = 50 and 53) [27, 29]. Other eligibility criteria included an estimated GFR (eGFR) of ≥ 30 mL/min/1.73 m2 [27, 29] and/or a urinary GL-3 level ≥ 4 times the upper limit of the normal range [29]. Patients taking angiotensin converting enzyme inhibitors or angiotensin receptor blockers (or renin inhibitors [29]) were required to have been on stable regimens for ≥ 4 weeks prior to screening [27, 29].
In the ITT population of the FACETS and ATTRACT trials, the majority of patients were female (64 and 56%) and the mean age of the patients was 43.1 and 48.9 years [27, 29]. In ATTRACT, amenable mutations of enrolled and treated patients included p.N215S and p.A143T (n = 10 and 3; all associated with the non-classic phenotype), and p.A156T, p.D322E and p.R301P (n = 6, 4 and 3, respectively; all associated with the classic phenotype) [27]. In FACETS, amenable mutations included p.I253T (n = 4; unknown phenotypic association), and p.R301Q, p.A156T, p.Y216C and p.P259R (all n = 3; all associated with the classic phenotype, except p.R301Q, which is associated with both classic and non-classic phenotypes) [29].
4.1 In Enzyme Replacement Therapy (ERT)-Naive Patients
4.1.1 FACETS
Eligible patients had never received ERT or had not received ERT for ≥ 6 months (i.e. considered ERT-naive) [29]. In the initial 6-month double-blind period, patients were randomized to receive migalastat or placebo (stage 1); all patients who completed stage 1 were eligible to receive open-label migalastat for the next 6 months (stage 2) and then for an additional year (months 12-24). The reclassification of amenability for all GLA variants, based on the GLP-validated HEK assay for the mITT population, occurred prior to unblinding of the stage 1 data [29].
Of the 45 patients in the mITT population with available renal biopsy data, 28 had a relatively low burden of substrate deposition (i.e. GL-3 inclusions < 0.3) and 17 had a more extensive burden of substrate deposition (i.e. GL-3 inclusions ≥ 0.3) at baseline [36]. In these patients, males [mean 2.29 and 1.02 GL-3 inclusions/kidney interstitial capillary (KIC) in the migalastat and placebo groups] had higher mean baseline levels of GL-3 inclusions/KIC than females (mean 0.199 and 0.284 GL-3 inclusions/KIC in the migalastat and placebo groups), indicating a higher substrate burden and more severe disease on a histologic level [36], which is in line with the aetiology of the disease (Sect. 1).
The primary endpoint was the percentage of patients in the migalastat and placebo groups of the ITT population who had a decrease from baseline of ≥ 50% in the number of GL-3 inclusions/KIC at the end of stage 1 (6 months) [29]. The number of GL-3 inclusions/KIC was assessed in 300 capillaries from each renal biopsy by three independent pathologists who were blinded to the study treatment and visit [29].
At 6 months, the primary endpoint did not significantly differ between the migalastat and placebo groups in the ITT population (Table 1) [29].
When the primary efficacy outcome was assessed in evaluable patients in the mITT population, 52 and 45% of migalastat and placebo recipients achieved a ≥ 50% reduction in the number of GL-3 inclusions/KIC at 6 months (statistical data not reported) [2]. According to subgroup analyses, the changes in the number of GL-3 inclusions/KIC were minimal after 6 months’ migalastat therapy in patients with low substrate burden at baseline [36]. However, in patients who had a high substrate burden at baseline, 78% (seven out of nine) of migalastat-treated patients achieved the primary efficacy outcome, as compared to 25% (two out of eight) of placebo-treated patients (statistical data not reported) [36]. In subgroup analyses categorized by gender, treatment with migalastat and placebo resulted in 71 and 44% of male patients (n = 7 and 9) achieving the primary efficacy outcome at 6 months, and 44 and 45% of female patients (n = 18 and 11) achieving the primary efficacy outcome at 6 months (statistical data not reported) [2, 36].
In the mITT population, migalastat was significantly more effective than placebo with regards to other efficacy endpoints, including the mean change from baseline to month 6 in the number of GL-3 inclusions/KIC (post hoc analysis) and plasma lyso-Gb3 levels (Table 1) [29]. In migalastat recipients with renal biopsy data, subgroup analyses by gender showed that males had numerically greater improvements from baseline in the mean number of GL-3 inclusions/KIC than females at the end of stage 1, with females showing minimal changes in GL-3 inclusions/KIC (mean change − 0.805 vs. − 0.034 GL-3 inclusions/KIC; statistical data not reported). In placebo recipients, males had an increase from baseline in the mean number of GL-3 inclusions/KIC (mean change + 0.229 GL-3 inclusions/KIC), whereas females had minimal change from baseline (mean change − 0.058; similar change to that in females receiving migalastat) [29].
In prespecified analyses, patients who switched from placebo to migalastat at month 6 (stage 2) experienced a significant reduction in the mean number of GL-3 inclusions/KIC (- 0.33 GL-3 inclusions/KIC; p = 0.01; n = 17) and plasma lyso-Gb3 levels (− 15.5 ng/mL; p < 0.001; n = 13) after 6 months of therapy (i.e. at month 12) [29]. The beneficial effects of 6 months’ migalastat therapy on these two surrogate endpoints during the double-blind period were maintained during a further 6 months of open-label migalastat treatment [29]. After 6 months of treatment, significant reductions from baseline in the mean volume of GL-3 inclusions/glomerular podocyte were observed in renal biopsy samples from migalastat recipients (p = 0.02; n = 8) [37]. After 12 months, GL-3 levels in glomerular podocytes, endothelial cells and mesangial cells were reduced from baseline in 22, 26 and 48% of renal biopsy samples from migalastat recipients (n = 23), respectively, with no change in GL-3 levels in the other biopsy samples [29].
At 24 months (i.e. after 18 months’ migalastat therapy in patients who switched from placebo or 24 months of continuous migalastat), there was a significant decrease from baseline in the left ventricular mass index (LVMi) in the mITT population (mean value − 7.7 g/m2; 95% CI − 15.4 to – 0.01; n = 27) [29]. Of note, the mean values for LVMi at baseline (96.5 g/m2) and at 24 months (not reported) were within or very close to the normal LVMi range for males and females (49–115 and 43–95 g/m2). Changes in LVMi correlated with changes in the intraventricular septum thickness (p = 0.006), but not with changes in left ventricular posterior wall thickness (LVPWT) [29].
In the mITT population, improvements in gastrointestinal symptoms were observed, as assessed by the Gastrointestinal Symptom Rating Scale (GSRS; comprising five domains: diarrhoea, reflux, abdominal pain, constipation and indigestion) [29]. At 6 months, migalastat recipients experienced significant (p = 0.03) improvements from baseline in diarrhoea symptoms; no significant improvements were experienced in any other domain at this time point [29]. A minimal clinically important difference (MCID) in GSRS score improvement in diarrhoea symptoms from baseline of 0.33 was experienced by 43 and 11% of migalastat and placebo recipients at 6 months [38]. In subgroup analyses of patients with symptoms at baseline, significant (p = 0.047) improvements from baseline were experienced at 6 months in reflux, but not in any other domain [29]. At 24 months, all migalastat recipients had significant improvements in diarrhoea and indigestion symptoms, including patients with symptoms at baseline (statistical data not reported) [29]. Further studies are required to confirm long-term benefits of migalastat on gastrointestinal symptoms [29].
No significant between-group differences were observed in the mITT population during the initial 6 months of the study in terms of changes in eGFR or measured GFR (mGFR), 24 h urine protein excretion or 24 h urinary GL-3 substrate levels [29]. Notably, baseline values were within normal range for GFR [29] (> 90 mL/min/1.73 m2/year [39, 40]). Among patients with non-amenable GLA mutations (n = 17), migalastat treatment had no effects on interstitial capillary GL-3 or plasma lyso-Gb3 levels [29]. Clinical outcomes (i.e. changes in GL-3 inclusions/KIC, plasma lyso-Gb3 levels and LVMi) were achieved regardless of the degree of renal function at baseline [41].
The clinical benefits of migalastat were also demonstrated in a subgroup analysis of 14 male patients in the FACETS trial with the classic Fabry phenotype [42].
4.1.2 Open-Label Extension
ERT-naive patients with Fabry disease and migalastat-amenable mutations who completed a phase 2 extension trial (FAB-CL-205) or FACETS [29] were eligible to enrol in a long-term OLE study (NCT01458119) [43]. In this study, renal outcomes were assessed via annualized changes from baseline in eGFR using the Chronic Kidney Disease Epidemiology Collaboration (eGFRCKD-EPI) or Modification of Diet in Renal Disease (eGFRMDRD) equations in 52 patients who received migalastat 123 mg once every other day for ≥ 17 months (median treatment duration 3.5–4.8 years; maximum duration 5.3 years) [43]. Overall, renal function was generally maintained during long-term treatment with migalastat in both male and female patients with migalastat-amenable GLA mutations and baseline proteinuria levels of ≤ 1000 mg/24 h (declines in mean eGFR were observed in patients with baseline proteinuria levels of > 1000 mg/24 h) [43].
With regards to OLE patients who had previously completed the FACETS trial, sustained reductions from baseline in the mean LVMi were reported after 30 or 36 months with migalastat therapy (− 17.0 g/m2; 95% CI − 26.2 to – 7.9; n = 15), including in patients with LVH at baseline [− 20.8 g/m2; 95% CI − 37.4 to – 4.1; n = 11], demonstrating the long-term effects of migalastat in reducing cardiac mass [44].
4.2 In ERT-Experienced Patients
In ATTRACT [27], eligible patients had initiated ERT ≥ 12 months before the baseline visit. During screening, patients were randomized to switch to open-label migalastat or continue biweekly ERT (agalsidase alfa 0.2 mg/kg or agalsidase beta 1.0 mg/kg) for 18 months followed by a 12-month OLE with migalastat [27].
The co-primary endpoints were the annualized changes from baseline through month 18 in eGFRCKD–EPI and mGFR by iohexol clearance (mGFRiohexol) [27]. To assess comparability between the migalastat and ERT groups, prespecified criteria comprised least-squares mean annualized rates of change in eGFRCKD–EPI and mGFRiohexol within 2.2 mL/min/1.73 m2/year and a > 50% overlap of the 95% CIs [27]. Of note, the baseline eGFRCKD–EPI and mGFRiohexol values were within or slightly below the normal range for GFR in adults (Table 2; Sect. 4.1).
At month 18, the prespecified criteria for the co-primary endpoints were met, indicating comparable efficacy between the migalastat and ERT groups in the mITT population (Table 2) [27]. The annualized changes in eGFRMDRD (a secondary endpoint) were also similar between the migalastat and ERT groups (Table 2). Subgroup analyses based on the degree of renal impairment at baseline [mGFRiohexol ≥ 30 to < 60 (n = 3) and ≥ 60 mL/min/1.73 m2/year (n = 31)] show that the mean annualized rates of change in eGFRCKD–EPI were − 3.3 and − 0.4 mL/min/1.73 m2/year (no statistical data reported), indicating stabilization of renal function [41]. However, when interpreting this data, the small number of patients with baseline mGFRiohexol < 60 mL/min/1.73 m2/year should be considered [41].
In the mITT population, migalastat therapy was associated with better cardiac outcomes than ERT [27]. In the migalastat group, the mean LVMi significantly decreased from baseline at month 18 (Table 2) [27]; mean LVMi reductions were also observed at month 30 (− 3.8 g/m2; n = 30) [44]. Conversely, in the ERT group, no significant change in LVMi was observed at month 18, potentially reflecting the smaller number of patients in this group [27]. Similar to findings from FACETS (Sect. 4.1), the mean baseline values for LVMi were within the normal range (Table 2), and the changes in LVMi at 18 months correlated with changes in intraventricular septum thickness (p = 0.003) but not LVPWT [27]. Left ventricular ejection fraction, fractional shortening, and systolic and diastolic grades were generally normal at baseline and were maintained over 18 months [27].
In evaluable patients in the mITT population, plasma lyso-Gb3 levels were maintained at the same low levels in the migalastat and ERT groups, and comparable results were observed between male and female patients [27]. Conversely, in two patients with non-amenable GLA mutations, plasma lyso-Gb3 levels increased after switching from ERT to migalastat [27].
The mean change from baseline in 24 h urine protein was approximately fourfold lower in migalastat recipients than ERT recipients after 18 months (49.2 vs. 194.5 mg) [27]. Clinical outcomes for GL-3 inclusions/KIC, plasma lyso-Gb3 levels and the LVMi were not affected by renal function at baseline [41].
4.3 Real-World Studies
The efficacy of migalastat in the real-world setting has been evaluated in an ongoing prospective single-centre study in patients with migalastat-amenable mutations of α-galactosidase A [45]. Initial follow-up after 3–6 months’ treatment in 17 evaluable patients suggested beneficial effects of migalastat on cardiac morphology, with significant reductions from baseline in the myocardial mass index (from 129.38 to 119.88 g/m2; p = 0.02). After 12 months of migalastat treatment, five evaluable patients had an increase in α-galactosidase A activity (0.21 vs. 0.13 nmol/min/mg protein at baseline; p = 0.043). Lyso-Gb3 levels in leukocytes did not alter to a significant degree after 3–6 or 12 months of treatment. Renal function was maintained after 3-6 months and GFRCKD-EPI did not significantly change after 12 months of migalastat treatment [45].
5 Tolerability of Migalastat
Migalastat was generally well tolerated in patients with Fabry disease in the two phase 3 clinical trials discussed in Sect. 4. Similar incidences of adverse events (AEs) were observed in the migalastat and placebo groups [29] and migalastat and ERT groups [27] during these studies. These AEs were mostly mild to moderate in severity and no AEs led to migalastat discontinuation. Two serious adverse events (SAEs) were considered to be possibly related to migalastat treatment in FACETS (fatigue and paraesthesia) [29]; no SAEs considered related to migalastat were reported in the ATTRACT trial [27]. In these trials, no deaths were reported and no clinically relevant effects of migalastat were observed for any clinical laboratory measurements, vital signs, physical-examination findings or ECG results [27, 29].
In FACETS, the most common adverse reactions occurring within the first 6 months in migalastat and placebo recipients (n = 34 and 33) were headache (35 and 21%), nasopharyngitis (18 and 6%), urinary tract infection (including cystitis and kidney infection; 15 and 0%), nausea (12 and 6%), pyrexia (12 and 3%), abdominal pain (9 and 3%), back pain (9 and 0%), cough (9 and 0%), diarrhoea (9 and 3%) and epistaxis (9 and 3%) [2]. Between months 6 and 12 of open-label migalastat therapy, the most frequently reported adverse events were headache (14%) and procedural pain related to renal biopsies (11%); the most frequently reported adverse events during the OLE period were proteinuria (16%; one case was deemed to be migalastat-related), headache (11%) and bronchitis (11%) [29]. These findings are consistent with those in the 18-month ATTRACT trial in which the most common AEs in migalastat and ERT recipients (n = 34 and 18) were nasopharyngitis (33 and 33%) and headache (25 and 24%) [27].
6 Dosage and Administration of Migalastat
Oral migalastat is approved for the treatment of adult patients (aged ≥ 18 years) in the USA [2] and Canada [46], as well as the long-term treatment of adult and adolescent patients (aged ≥ 16 years) in the EU [3], Japan [47], Australia [48] and elsewhere, who have a confirmed diagnosis of Fabry disease (α-galactosidase A deficiency [3, 46, 48]) and an amenable GLA mutation (based on in vitro assay data [2, 46]). In the USA, patients must have a migalastat-amenable GLA variant that is interpreted by a clinical genetics professional as being causative for Fabry disease (i.e. pathogenic or likely pathogenic) in the clinical context of the individual patient [2]. Migalastat therapy should be initiated and supervised by specialist physicians who are experienced in the diagnosis and treatment of Fabry disease [3, 46, 48].
The recommended dosage regimen of migalastat is 123 mg once every other day at the same time of day [2, 3, 46,47,48]. Migalastat must be taken on an empty stomach; food should not be consumed for at least 2 h before and 2 h after taking migalastat [2, 3, 46,47,48], although clear liquids may be consumed during this fasting period [2, 3]. Migalastat should not be taken on two consecutive days [46, 48]. In the event of a missed dose (if > 12 h have passed in the EU and USA [2, 3]), administration of migalastat should be resumed at the next scheduled dose [46, 48]. In the EU and USA, if a dose is missed and < 12 h have passed, then the missed dose of migalastat should be taken [2].
Migalastat is not indicated and should not be used in patients with Fabry disease who have non-amenable GLA mutations [2, 3, 46,47,48]. Local prescribing information and/or the migalastat amenability table website (http://www.galafoldamenabilitytable.com/) should be consulted for a list of amenable and non-amenable GLA mutations to migalastat.
Migalastat is not recommended for use in patients who have severe renal impairment (eGFR < 30 mL/min/1.73 m2) [2, 3, 46,47,48] (Sect. 3). It is also not recommended in patients with end-stage renal disease requiring dialysis in the USA [2]. Migalastat is not intended for concomitant use with ERT [3, 46, 48]. Local prescribing information should be consulted for further details, including contraindications, use in special populations and other warnings and precautions.
7 Place of Migalastat in the Management of Fabry Disease
Over recent years, continuous advancements have been made in the development of therapeutic options for patients with lysosomal disorders, including Fabry disease [14]. The current treatment options for Fabry disease are ERT and migalastat [1]. An increased understanding of the pathophysiology of lysosomal disorders has led to the development of novel targeted therapies such as pharmacological chaperones. These agents bind and stabilize mutant enzymes in the ER, allowing normal trafficking to lysosomes and ultimately increasing enzyme activity (Fig. 1) [14].
Migalastat, a small molecule pharmacological chaperone, is currently approved in several countries for the treatment of patients [aged ≥ 18 years in the USA and Canada; ≥ 16 years of age in other countries] who have a confirmed diagnosis of Fabry disease and a migalastat-amenable GLA mutation, based on in vitro assay data (Sect. 6). Recommendations for the use of oral migalastat therapy in Fabry disease management has yet to be included in international guidelines [1].
In the pivotal phase 3 FACETS and ATTRACT trials in patients with Fabry disease (encompassing a broad spectrum of disease manifestations), oral migalastat was efficacious in ERT-naive (Sect. 4.1) and -experienced (Sect. 4.2) patients with migalastat-amenable GLA-mutations. Migalastat treatment resulted in increased and sustained endogenous α-Gal A activity levels, with consequent reductions in substrate accumulation in renal tissue, urine and plasma. During long-term therapy, these beneficial effects on endogenous α-Gal A activity levels were sustained, renal function was maintained across a wide spectrum of baseline function, and cardiac mass was reduced.
Although there was no significant difference between the migalastat and placebo group in the percentage of patients with ≥ 50% reduction from baseline in the number of GL-3 inclusions/KIC at 6 months in the primary ITT analysis in the overall FACETS population, migalastat recipients with amenable GLA mutations had significantly greater improvements in other surrogate endpoints (mITT analyses; Sect. 4.1.1). Efficacy outcomes appeared to be driven by gender, potentially reflecting differences in disease burden/severity between male and female patients, with males tending to have more severe disease at baseline and therefore, experiencing a much greater improvement in surrogate endpoints [36]. The greater reductions in the number of GL-3 inclusions/KIC in males than females may, at least in part, also reflect that Fabry cells co-exist with normal cells in female patients (Sect. 1); hence, the proportion of Fabry versus normal cells may vary from biopsy to biopsy in the same patient [49]. The majority of participants in FACETS were females (Sect. 4.1.1).
In the ATTRACT trial in ERT-experienced patients with migalastat-amenable GLA mutations (Sect. 4.2), three patients had p.A143T mutations, with the pathogenic role of this mutation in Fabry disease equivocal [50, 51], and ten had amenable p.N215S mutations, which are primarily associated with cardiac manifestations of the disease and less associated with the progressive loss of renal function, particularly in female Fabry patients [52]. The inclusion and/or lack of differentiation of 25% (13 out of 52) of patients with p.A143T or p.N215S mutations in the mITT analyses could potentially have impacted the findings of this study with regards to renal function. In addition, interpretation of data from this trial may potentially be impacted by the fact that patients treated with agalsidase alfa and agalsidase beta were pooled into one treatment group [1].
Migalastat was generally well tolerated in patients with Fabry disease, where headache and nasopharyngitis were the most commonly reported adverse reactions in migalastat and placebo recipients in phase 3 trials (Sect. 5).
There are several attributes of migalastat that make it an attractive alternative option to ERT for treating Fabry disease. Firstly, migalastat has a convenient oral regimen, thereby eliminating the requirement for lifelong IV infusions and complications that have been associated with IV ERT (agalsidase alfa and agalsidase beta), such as infusion-related reactions (e.g. fever, chills, flushing, headache, pruritus, and nausea) and hypersensitivity reactions (e.g. allergic or anaphylactic-type reactions) [5, 14, 53,54,55]. The non-immunogenic nature of migalastat means that such antibody-related tolerability issues, which have been described for several ERTs, are not expected [14]. Secondly, as a small molecule, migalastat is likely to have enhanced cellular and tissue distribution and the potential to cross the blood-brain barrier [as evidenced in Fabry transgenic mice (Sect. 2)], which may assist in the treatment of disease symptoms originating in the central nervous system [53]. The improved efficacy of migalastat compared with ERT in reducing cardiac mass (Sect. 4.2) suggests that migalastat may be more effective than ERT at penetrating cardiac tissue [56]. As an orally administered therapy, migalastat may also facilitate earlier intervention than ERT in patients with Fabry disease.
A general advantage of pharmacological chaperone therapy over ERT is that, given the appropriate dosing regimen, it allows for sustained and stable enzyme levels that more closely mimic those of endogenous wild-type enzymes, whereas ERT leads to fluctuating and intermittent enzymatic activity [14]. In order to maximise the in situ activity of α–galactosidase A and GL-3 substrate reduction with migalastat therapy (i.e. the maximal pharmacological chaperone effect; Sect. 2), the recommended dosage regimen of migalastat (Sect. 6) is based on a balance between migalastat target organ concentration and clearance (as assessed in preclinical and clinical studies; Sect. 2) [57].
Patients should be made aware of the importance of compliance with migalastat therapy in order to optimise its efficacy [58]. Of note, a prospective, observational cohort study is currently recruiting patients with genetically confirmed Fabry disease who are taking migalastat, in order to assess patient adherence [59].
Monitoring of migalastat therapy is advised in several countries. In the EU and Australia, it is recommended that the response to migalastat therapy is monitored in patients every 6 months (e.g. renal function, echocardiographic parameters), and if meaningful clinical deterioration occurs, further clinical evaluation and/or discontinuation of migalastat treatment should be considered [3, 48]. Similarly, in Canada, patients should be assessed for treatment response or failure following the initiation of migalastat therapy, and monitored every ≤ 6 months over the course of treatment; if meaningful clinical deterioration occurs, migalastat therapy should be stopped, further clinical evaluation initiated and other treatment options should be considered [46]. Additionally, according to the 2017 Canadian Fabry disease guidelines [58], it is recommended that patients who switch from ERT to migalastat are followed up every 6 months for the first 3–5 years of therapy to ensure clinically stable parameters. No recommendations for the monitoring of migalastat therapy have been included in the prescribing information for the USA [2] or Japan [47]. In the USA, the manufacturer assists patients in accessing migalastat by liaising with health insurers (via the patients access scheme), who have specific requirements for treatment (e.g. completion of renal function assessments prior to the initiation of migalastat therapy, with continued monitoring during treatment as clinically appropriate [60]).
Migalastat therapy is approved for use in patients with Fabry disease who have migalastat-amenable GLA mutations (Sect. 6). The future identification of pharmacological agents with chaperoning profiles that target a larger number of GLA mutations (e.g. mutations that are non-amenable to migalastat) would be of high interest, and would broaden the potential for the use of pharmacological chaperone therapy in Fabry disease [14].
There is a paucity of data pertaining to the effects of migalastat therapy on severe clinical events (e.g. major kidney events) and in special populations of patients with Fabry disease, such as female patients who are pregnant or breast-feeding, patients aged ≥ 65 or < 16 years and/or patients with severe renal impairment (eGFR < 30 mL/min/1.73 m2/year) [2]. Three pregnant women with Fabry disease have been exposed to migalastat in clinical trials [2]. No adverse developmental effects have been observed in animal studies with exposure to migalastat; however, current data is not sufficient to draw conclusions about the risks of adverse maternal/foetal outcomes (e.g. major birth defects or miscarriage) associated with migalastat therapy [2]. The effects of migalastat on fertility have not been studied in humans; in animal studies, migalastat therapy was associated with transient and fully reversible infertility (after 4 weeks without migalastat) in male rats, and had no effects on fertility in female rats [2]. These factors highlight the need for ongoing studies and clinical data [including long-term (i.e. ≥ 5 year) data on clinical outcomes], which will ultimately help to establish a more informed approach to the treatment of Fabry disease with migalastat. As part of the post-marketing commitment, there is a planned trial to evaluate the appropriate dosing regimen of migalastat in patients with Fabry disease who have migalastat-amenable GLA mutations and have severe renal impairment or who are on kidney dialysis [61]. A post-marketing, worldwide, prospective, single-arm, observational study in women exposed to migalastat during pregnancy and lactation is also required to assess the risks of pregnancy complications, adverse effects on the developing foetus and neonate, and adverse effects on lactation and the breastfed infant [61].
In conclusion, given its efficacy, convenient oral regimen, extensive tissue penetration and the limited therapeutic options available, migalastat is an important option for the treatment of patients with Fabry disease and migalastat-amenable GLA mutations.
Data Selection Migalastat: 207 records identified
Duplicates removed | 27 |
Excluded during initial screening (e.g. press releases; news reports; not relevant drug/indication; preclinical study; reviews; case reports; not randomized trial) | 39 |
Excluded during writing (e.g. reviews; duplicate data; small patient number; nonrandomized/phase I/II trials) | 80 |
Cited efficacy/tolerability articles | 14 |
Cited articles not efficacy/tolerability | 47 |
Search Strategy: EMBASE, MEDLINE and PubMed from 1946 to present. Clinical trial registries/databases and websites were also searched for relevant data. Key words were migalastat, Galafold, Amigal, GR181413A, HGT3310, Fabry disease. Records were limited to those in English language. Searches last updated 26 February 2019 | |
Change history
10 July 2019
The article Migalastat: A Review in Fabry Disease, written by Emma H. McCafferty, Lesley J. Scott, was originally published Online First without open access. After publication in volume 79, issue 5, pages 543–554 [funder] requested that the article be Open Choice to make the article an open access publication. Post-publication open access was funded by [funder]. The article is forthwith distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123(4):416–27.
Amicus Therapeutics. Galafold™ (migalastat) capsules, for oral use: US prescribing information. 2018. https://www.fda.gov/. Accessed 18 Oct 2018.
European Medicines Agency. Migalastat (Galafold): EU summary of product characteristics. 2018. https://www.ema.europa.eu/. Accessed 18 Oct 2018.
Brady RO, Gal AE, Bradley RM, et al. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med. 1967;276(21):1163–7.
National Institute for Health and Care Excellence. Migalastat for treating Fabry disease. 2017. https://www.nice.org.uk/guidance/hst4. Accessed 26 Oct 2018.
Arends M, Wanner C, Hughes D, et al. Characterization of classical and nonclassical Fabry disease: a multicenter study. J Am Soc Nephrol. 2017;28(5):1631–41.
Whybra C, Kampmann C, Willers I, et al. Anderson-Fabry disease: clinical manifestations of disease in female heterozygotes. J Inherit Metab Dis. 2001;24(7):715–24.
Kampmann C, Baehner F, Whybra C, et al. Cardiac manifestations of Anderson–Fabry disease in heterozygous females. J Am Coll Cardiol. 2002;40(9):1668–74.
Hauth L, Kerstens J, Yperzeele L, et al. Galactosidase alpha p.A143T variant of Fabry disease may result in a phenotype with multifocal microvascular cerebral involvement at a young age. Front Neurol. 2018;9:336.
Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30.
Lee K, Jin X, Zhang K, et al. A biochemical and pharmacological comparison of enzyme replacement therapies for the glycolipid storage disorder Fabry disease. Glycobiology. 2003;13(4):305–13.
Lidove O, West ML, Pintos-Morell G, et al. Effects of enzyme replacement therapy in Fabry disease—a comprehensive review of the medical literature. Genet Med. 2010;12(11):668–79.
Schiffmann R, Hughes DA, Linthorst GE, et al. Screening, diagnosis, and management of patients with Fabry disease: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) controversies conference. Kidney Int. 2017;91(2):284–93.
Parenti G, Andria G, Valenzano KJ. Pharmacological chaperone therapy: preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol Ther. 2015;23(7):1138–48.
Benjamin ER, Flanagan JJ, Schilling A, et al. The pharmacological chaperone 1-deoxygalactonojirimycin increases alpha-galactosidase A levels in Fabry patient cell lines. J Inherit Metab Dis. 2009;32(3):424–40.
Fan J-Q, Ishii S, Asano N, et al. Accelerated transport and maturation of lysosomal α-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat Med. 1999;5(1):112–5.
Yam GH, Zuber C, Roth J. A synthetic chaperone corrects the trafficking defect and disease phenotype in a protein misfolding disorder. Faseb J. 2005;19(1):12–8.
Asano N, Ishii S, Kizu H, et al. In vitro inhibition and intracellular enhancement of lysosomal alpha-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. Eur J Biochem. 2000;267(13):4179–86.
Yam GH, Bosshard N, Zuber C, et al. Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-incompetent variants. Am J Physiol Cell Physiol. 2006;290(4):C1076–82.
Khanna R, Soska R, Lun Y, et al. The pharmacological chaperone 1-deoxygalactonojirimycin reduces tissue globotriaosylceramide levels in a mouse model of Fabry disease. Mol Ther. 2010;18(1):23–33.
Ishii S, Chang H-H, Yoshioka H, et al. Preclinical efficacy and safety of 1-deoxygalactonojirimycin in mice for Fabry disease. J Pharmacol Exp Ther. 2009;328(3):723–31.
Young-Gqamana B, Brignol N, Chang HH, et al. Migalastat HCl reduces globotriaosylsphingosine (lyso-Gb3) in Fabry transgenic mice and in the plasma of Fabry patients. PLoS ONE. 2013;8(3):e57631.
Johnson FK, Mudd PN Jr, Bragat A, et al. Pharmacokinetics and safety of migalastat HCl and effects on agalsidase activity in healthy volunteers. Clin Pharmacol Drug Dev. 2013;2(2):120–32.
Germain DP, Giugliani R, Hughes DA, et al. Safety and pharmacodynamic effects of a pharmacological chaperone on alpha-galactosidase A activity and globotriaosylceramide clearance in Fabry disease: report from two phase 2 clinical studies. Orphanet J Rare Dis. 2012;7(91):1–11.
Giugliani R, Waldek S, Germain DP, et al. A phase 2 study of migalastat hydrochloride in females with Fabry disease: selection of population, safety and pharmacodynamic effects. Mol Genet Metab. 2013;109(1):86–92.
Tuttolomondo A, Simonetta I, Duro G, et al. Inter-familial and intra-familial phenotypic variability in three Sicilian families with Anderson–Fabry disease. OncoTarget. 2017;8(37):61415–24.
Hughes DA, Nicholls K, Shankar SP, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet. 2017;54(4):288–96.
Benjamin ER, Della Valle MC, Wu X, et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med. 2017;19(4):430–8.
Germain DP, Hughes DA, Nicholls K, et al. Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N Engl J Med. 2016;375(6):545–55.
Wu X, Katz E, Della Valle MC, et al. A pharmacogenetic approach to identify mutant forms of alpha-galactosidase A that respond to a pharmacological chaperone for Fabry disease. Hum Mutat. 2011;32(8):965–77.
Johnson FK, Mudd PN Jr, DiMino T, et al. An open-label study to determine the pharmacokinetics and safety of migalastat HCl in subjects with impaired renal function and healthy subjects with normal renal function. Clin Pharmacol Drug Dev. 2015;4(4):256–61.
Ino H, Takahashi N, Terao T, et al. Pharmacokinetics, safety, and tolerability following single-dose migalastat hydrochloride (GR181413A/AT1001) in healthy male Japanese subjects. J Drug Assess. 2013;2(1):87–93.
Johnson FK, Valenzano KJ, Castelli JP, et al. Pharmacokinetic simulation of a 150 mg QOD dose regimen for the pharmacological chaperone migalastat HCl in Fabry disease [abstract no. 001]. Clin Pharmacol Drug Dev. 2017;6(Suppl. 1):3.
Johnson FK, Mudd PN Jr, Janmohamed SG. Relative bioavailability and the effect of meal type and timing on the pharmacokinetics of migalastat in healthy volunteers. Clin Pharmacol Drug Dev. 2015;4(3):193–202.
Mudd PN, Johnson FK, Churchill A. A phase 1 study to investigate the absorption, metabolism and excretion of [14C] migalastat hydrochloride following a single oral administration in healthy volunteers [abstract no. 1699358]. Clin Pharmacol Drug Dev. 2013;2(Suppl. 1):18–9.
Amicus Therapeutics. NDA multi-disciplinary review and evaluation: Galafold (migalastat). 2017. https://www.fda.gov/. Accessed 6 Nov 2018.
Mauer M, Sokolovskiy A, Barth JA, et al. Reduction of podocyte globotriaosylceramide content in adult male patients with Fabry disease with amenable GLA mutations following 6 months of migalastat treatment. J Med Genet. 2017;54(11):781–6.
Schiffmann R, Bichet DG, Jovanovic A, et al. Migalastat improves diarrhea in patients with Fabry disease: clinical-biomarker correlations from the phase 3 FACETS trial. Orphanet J Rare Dis. 2018;13(68):1–7.
The Renal Association. CKD stages G1 or G2. 2018. https://renal.org/information-resources/the-uk-eckd-guide/stages-1-2-ckd/. Accessed 19 Dec 2018.
National Kidney Foundation. Glomerular filtration rate (GFR). 2017. https://www.kidney.org/atoz/content/gfr. Accessed 19 Dec 2018.
Torra R, Germain D, Bichet D, et al. Clinical outcomes with migalastat in patients with Fabry disease based on degree of renal impairment: results from phase 3 trials [abstract no. SP002 and poster]. Nephrol Dial Transplant. 2018;33(Suppl. 1):i346.
Germain DP, Giugliani R, Bichet DG, et al. Efficacy of migalastat in a cohort of male patients with the classic Fabry phenotype in the FACETS phase 3 study [abstract no. 102]. Mol Genet Metab. 2017;120(1–2):S52.
Schiffmann R, Bichet D, Germain D, et al. Effects of long-term migalastat treatment on renal function by baseline proteinuria in patients (PTS) with Fabry disease [abstract no. SP004 and poster]. Nephrol Dial Transplant. 2018;33(Suppl. 1):i346–7.
Jovanovic A, Schiffmann R, Nicholls K, et al. Improvements in cardiac mass with long-term migalastat treatment in patients with Fabry disease: results from phase 3 trials [abstract no. LBN 02]. J Inborn Errors Metab Screen. 2017;5:1–2.
Muentze J, Gensler D, Salinger T, et al. Treatment of cardiac manifestations in Fabry disease with the oral drug migalastat: first 12 months results from a cohort of amenable all-comers [abstract no. 2355]. Eur Heart J. 2018;39(Suppl. 1):460.
Health Canada. Galafold™ (migalastat) capsules: Canadian product monograph. 2017. https://pdf.hres.ca/dpd_pm/00041104.PDF. Accessed 16 Jan 2019.
Japanese Pharmaceuticals and Medical Devices Agency. Galafold capsule 123 mg: Japanese prescribing information. 2018. http://www.info.pmda.go.jp/go/pack/3999045M1028_1_01/. Accessed 18 Oct 2018.
Therapeutic Goods Administration. Galafold® (migalastat): Australian product information. 2018. https://www.tga.gov.au/sites/default/files/auspar-migalastat-180830-pi.pdf. Accessed 16 Jan 2019.
Mauer M, Glynn E, Svarstad E, et al. Mosaicism of podocyte involvement is related to podocyte injury in females with Fabry disease. PLoS ONE. 2014;9(11):e112188.
Lenders M, Weidemann F, Kurschat C, et al. Alpha-galactosidase A p.A143T, a non-Fabry disease-causing variant. Orphanet J Rare Dis. 2016;11(1):54.
Terryn W, Vanholder R, Hemelsoet D, et al. Questioning the pathogenic role of the GLA p.Ala143Thr “mutation” in Fabry disease: implications for screening studies and ERT. JIMD Rep. 2013;8:101–8.
Germain DP, Brand E, Burlina A, et al. Phenotypic characteristics of the p.Asn215Ser (p.N215S) GLA mutation in male and female patients with Fabry disease: a multicenter Fabry Registry study. Mol Genet Genomic Med. 2018;6(4):492–503.
Parenti G. Treating lysosomal storage diseases with pharmacological chaperones: from concept to clinics. EMBO Mol Med. 2009;1(5):268–79.
European Medicines Agency. Agalsidase beta (Fabrazyme): EU summary of product characteristics. 2018. https://www.ema.europa.eu/. Accessed 26 Oct 2018.
European Medicines Agency. Agalsidase alfa (Replagal): EU summary of product characteristics. 2018. https://www.ema.europa.eu/. Accessed 26 Oct 2018.
Mohamed FE, Al-Gazali L, Al-Jasmi F, et al. Pharmaceutical chaperones and proteostasis regulators in the therapy of lysosomal storage disorders: current perspective and future promises. Front Pharmacol. 2017;8:448.
Therapeutic Goods Administration. Australian public assessment report for migalastat. 2018. https://www.tga.gov.au/sites/default/files/auspar-migalastat-180830.pdf. Accessed 16 Jan 2019.
Sirrs S, Bichet DG, Iwanochko RM, et al. 2017 Canadian Fabry disease guidelines. 2017. http://www.garrod.ca/wp-content/uploads/Canadian-FD-Treatment-Guidelines-2017.pdf. Accessed 16 Jan 2019.
US National Institutes of Health. ClinicalTrials.gov identifier NCT03683966. 2018. https://clinicaltrials.gov/. Accessed 16 Jan 2019.
BlueCross BlueShield of Arizona. Galafold™ (migalastat) oral capsule: pharmacy coverage guidelines. 2018. https://www.azblue.com/~/media/azblue/files/pharmacy-forms-mastery-directory/qualified-health-plans/pharmacy-coverage-guidelines-and-precertification-forms/galafold.pdf. Accessed 17 Jan 2019.
Amicus Therapeutics. Galafold™ (migalastat) capsules: NDA approval letter. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2018/208623Orig1s000ltr.pdf. Accessed 18 Dec 2018.
Acknowledgements
During the peer review process, the manufacturer of migalastat was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflicts of interest
Emma McCafferty and Lesley Scott are salaried employees of Adis/Springer, are responsible for the article content and declare no relevant conflicts of interest.
Additional information
The manuscript was reviewed by:A. Ortiz, Unidad de Dialisis, IIS-Fundacion Jimenez Diaz, School of Medicine, UAM, IRSIN and REDINREN, Madrid, Spain; R. Schiffmann, Institute of Metabolic Disease, Baylor Scott & White Research Institute, Dallas, TX, USA.
The original version of this article was revised due to a retrospective Open Access request.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
About this article

Cite this article
McCafferty, E.H., Scott, L.J. Migalastat: A Review in Fabry Disease. Drugs 79, 543–554 (2019). https://doi.org/10.1007/s40265-019-01090-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-019-01090-4