
Abstract
Conventional vaccine adjuvants enhance peptide-specific T-cell and B-cell responses by modifying peptide stability or uptake or by binding to pattern-recognition receptors on antigen-presenting cells (APCs). This article discusses the application of a distinct mechanism of adjuvant activity: the activation of type I, or invariant, natural killer T (iNKT) cells to drive cellular and humoral immune responses. Using a semi-invariant T-cell receptor (TCR), iNKT cells recognize glycolipid antigens presented on cluster of differentiation (CD)-1d molecules. When their ligands are presented in concert with peptides, iNKT cells can provide T-cell help, ‘licensing’ APCs to augment peptide-specific T-cell and antibody responses. We discuss the potential benefits and limitations of exploiting iNKT cells as ‘universal helpers’ to enhance vaccine responses for the treatment and prevention of cancer and infectious diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Natural killer T (NKT) cells are a subset of ‘innate-like’ T lymphocytes that respond to glycolipid, rather than peptide, antigens. |
NKT cell-activating glycolipids can act as adjuvants, enhancing immune responses against co-administered peptides and proteins. |
Vaccine strategies for co-delivering glycolipids and peptide antigens, including nanoparticles and synthetic conjugates, are effective in vivo and are entering clinical development. |
1 Harnessing the Immune System Through Vaccination
The contribution of vaccination to human health is considered second only to that provided by the supply of clean drinking water [1]. For decades, vaccines have successfully been used to elicit protective antibodies against a broad range of infectious diseases. However, traditional vaccination strategies have proven less successful for the efficient induction of cytotoxic cluster of differentiation (CD)-8+ T-cell responses. As these T cells recognize peptides derived from intracellular antigens, they can respond to antigens that antibodies often cannot reach, including novel intracellular proteins (neoantigens) expressed by transformed or infected cells [2]. Accordingly, considerable effort is being invested in developing adjuvants that help CD8+ T-cell-recruiting vaccines to target and eradicate malignant or virus-infected cells [3–7].
Vaccine adjuvants are broadly defined as substances capable of enhancing and/or shaping antigen-specific immune responses [2]. The addition of an adjuvant to a vaccine can enhance the functionality, magnitude, or breadth of an immune response and enable dose sparing [2]. To date, clinically approved adjuvants fall broadly into two categories: (1) delivery systems that improve antigen stability, release, or cellular uptake, e.g., liposomes, virosomes, or immune-stimulating complexes (ISCOMs), or (2) immunostimulants that engage pattern recognition receptors (PRRs) to drive cytokine and chemokine release by innate immune cells leading to antigen-presenting cell (APC) activation, e.g., Toll-like receptor (TLR) ligands [8, 9]. Adjuvants currently approved for human use include aluminum salt (alum), oil-in-water emulsions (MF59 and AS03), the TLR4 ligand monophosphoryl lipid A (MPL), the TLR2 ligand Pam3Cys, and the imidazoquinoline resiquimod (R-848), and alum-absorbed MPL (AS04) [10–12], all of which can enhance antibody-mediated immune responses. In contrast, the licensed vaccines best able to generate CD8+ T-cell-mediated immunity are live attenuated pathogens, such as the yellow fever and varicella zoster virus vaccines [13–15], which also contain intrinsic adjuvants such as unmethylated CpG sequences.
Although few therapeutic cancer vaccines have been approved for clinical use, the success of checkpoint blockade monotherapy has reinvigorated interest in this approach by providing ‘proof of principal’ that T cells can act against solid cancers [16]. While some therapeutic cancer vaccines attempt to induce antibodies against growth factors, e.g., CIMAvax-EGF, most current investigational approaches focus on generating cellular, principally CD8+ T-cell, responses targeted directly against malignant cells. For example, the licensed prostate cancer vaccine sipuleucel-T (Provenge®, Dendreon Corporation), which comprises autologous mononuclear cells pulsed with prostatic acid phosphatase (PAP) linked directly to the cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF) [3], induces modest survival benefits and elicits T-cell responses against PAP [17]. A second prostate cancer vaccine, PROSTVAC® (Bavarian Nordic), which utilizes recombinant poxviruses to express prostate-specific antigen (PSA) along with three co-stimulatory molecules (lymphocyte function-associated antigen [LFA]-3, intercellular adhesion molecule [ICAM]-1, and B7.1), has generated T-cell responses in in vivo mouse models and in early phase clinical trials [18, 19]; the results of a phase III study are awaited.
Despite the relative success of viral vectors as vaccines, they continue to be associated with safety and manufacturing difficulties [20]. Thus, an important goal is to develop synthetic adjuvants capable of driving potent CD8+ T-cell responses. While some conventional vaccine adjuvants can help induce CD8+ T-cell responses in humans, including proprietary oil-in-water emulsions [21], liposomes [22, 23], and TLR agonists [24], only 3-O-desacyl-4′-MPL has been licensed for clinical use in the prophylactic human papilloma virus (HPV) vaccine Cervarix® (GlaxoSmithKline).
In this article, we discuss the ability of glycolipid ligands of type I, or invariant, natural killer T (iNKT) cells to function as vaccine adjuvants. Rather than just eliciting an innate immune response in which iNKT cells and trans-activated natural killer (NK) cells directly lyse cancer cells, these adjuvants instead act via a unique mechanism, recruiting iNKT cells to act as ‘universal helper’ T cells to enhance cellular and humoral immune responses against co-administered peptide antigens. We emphasize the mechanistic and functional differences between the adjuvant activity of iNKT cell ligands, which act via a third-party mechanism akin to that of conventional CD4+ T-cell help, and the activity of conventional adjuvants. Finally, we review methods of exploiting iNKT cell ligands for therapeutic vaccination and their preclinical and clinical results to date.
2 Adaptive Immune Responses
Dendritic cells (DCs) are professional APCs that take up microbial products and/or infected or dying cells from peripheral tissues or the circulation and present the acquired material to T cells in the form of antigenic peptide bound to major histocompatibility complex (MHC) molecules. Given the host’s diverse repertoire of T cells, each bearing a uniquely recombined T-cell receptor (TCR), there is a high likelihood of peptide recognition for a given antigen. The ability of DCs to stimulate proliferation and differentiation of peptide-specific T cells into effector cells, which takes several days, is central to the specific and targeted eradication of pathogens and malignant cells by the adaptive immune system.
Exogenous antigens, such as those delivered in the form of a vaccine, can be processed in vacuolar compartments within DCs, with some of the peptides generated then loaded onto MHC class II molecules and presented on the cell surface to CD4+ T cells. Pathways involving either vacuolar presentation or proteasomal cleavage in the cytosol have been described for the ‘cross-presentation’ of exogenous antigens via MHC class I molecules to CD8+ T cells, which otherwise recognise only endogenous antigens. In both humans and mice, where conventional DCs have been classified into two subtypes based on expression of XCR1, evidence that XCR1+ DCs have a heightened capacity for cross-presentation [25] is accumulating, and these cells are therefore likely to be critical in the initiation of vaccine-induced CD8+ T-cell responses.
Pathogens or dead cells taken up by DCs not only contain potentially antigenic peptides but also typically contain pathogen-associated or danger-associated molecular patterns (PAMPs and DAMPs). These molecules engage cellular PRRs expressed by DCs, such as TLRs, nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), and cytoplasmic DNA sensors. In response, DCs up-regulate peptide presentation on MHC and express co-stimulatory molecules, enhancing the effector T-cell response against the presented peptide. Many current and proposed adjuvants exploit the activity of PRRs by serving as PAMPs or DAMPs to mimic the effect of an immunogenic infection and enhance T-cell priming by DCs. For example, R-848 activates immune cells via TLR7/8 signaling and is approved for use in topical anti-viral creams [26, 27]. Similarly, co-administration of the TLR4 ligand MPL with alum has been shown to enhance differentiation of activated T cells into cytotoxic CD8+ T cells in clinical trials [28, 29].
Once activated, CD8+ T cells move into infected or distressed tissues, where they can recognize and eliminate affected cells through recognition of antigenic peptides presented on the MHC class I molecules of target cells. Cytotoxicity is mediated by a variety of possible mechanisms, including the secretion of cytokines, targeted secretion of cytotoxins, such as perforin or granzymes [30], or Fas/Fas ligand (FasL) interactions. Activated CD4+ T cells exert their effector functions through interactions with MHC class II+ APCs in infected or malignant tissues, releasing cytokines into the local region. Alternatively, it has recently been shown that they too can have direct cytotoxic activity against MHC class II+ targets [31].
The expression of MHC class II is generally limited to APCs. This expression profile is fundamental to another important role of CD4+ T cells: that of ‘helper’ cells. In this capacity, interactions with CD4+ T cells can enhance the function of APCs, such as antibody production by B cells, antimicrobial activity by macrophages, and—importantly for this article—the stimulation of CD8+ T cells by DCs.
2.1 Help for CD8+ T-Cell Responses
Peptide-specific CD8+ T-cell responses can be enhanced by either CD4+ T-cell help or strong PRR stimulation. These mechanisms appear to be distinct but can cooperate. In non-inflammatory conditions, CD4+ T-cell help is essential for the generation of long-lived functional memory CD8+ T cells [32, 33]. In contrast, CD8+ T-cell responses can be generated against some viral infections in the absence of CD4+ T-cell help through sufficient early type I interferons (IFNs) invoked by PRR stimulation [34]. These systems may cooperate: APCs acquiring antigens in conjunction with sub-maximal danger signals may seek ‘validation’ from CD4+ T cells to become fully activated [35]. Other factors contributing to memory CD8+ T-cell responses include TCR avidity, the rate of antigen clearance, and the magnitude of the effector CD8+ T-cell burst [36].
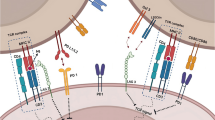
Upon recognition of peptides presented by DCs on MHC class II, CD4+ helper T cells can ‘license’ DCs to augment CD8+ T-cell responses against a co-presented CD8+ T-cell epitope [37–39]. Unlike the enhancement of CD8+ T-cell responses induced by PRRs, this CD4+ T-cell help depends upon an interaction between CD40 on the APC and CD40 ligand (CD154) on the helper T cell [40–43] (Fig. 1). A similar CD40-dependent mechanism allows CD4+ T cells to facilitate antibody production by B cells [44, 45].

Classical cluster of differentiation (CD)-4+ T-cell help. CD4+ helper T cells recognize long peptides presented by dendritic cells on major histocompatibility complex class II molecules leading to the release of pro-inflammatory cytokines, such as interferon-γ, and the CD40/CD40L-dependent ‘licensing’ of dendritic cells that together augment cytotoxic CD8+ T-cell responses against co-presented CD8+ T-cell epitopes. A similar CD40-dependent mechanism also allows CD4+ T cells to facilitate antibody production by B cells (not shown). CCL chemokine ligand, CCR chemokine receptor, IFN interferon, IL interleukin, MHC major histocompatibility complex, TCR T-cell receptor, TNF tumor necrosis factor
In the case of DCs, CD4+ helper and CD8+ cytotoxic T cells must recognize their cognate peptides presented on the same cell [37, 41, 42]. This requirement for dual recognition of two peptides by discrete TCRs on the same APC may have evolved as a mechanism to avoid undesirable autoimmunity. Viral infection models in mice indicate that, while the initial activation of CD4+ and CD8+ T cells can involve antigen presentation by spatially and temporally separate DC subsets, the cross-presenting XCR1+ DC population ultimately provides the critical platform for both CD4+ T-cell help [46, 47] and generation of cytotoxic effector CD8+ T cells [48].
2.2 Challenges to Providing CD4+ T-Cell Help
While many vaccine adjuvants are able to activate PRRs within DCs, the reliable generation of CD4+ T-cell help in humans is more problematic. Full-length proteins can be expected to include some CD4+ T-cell epitopes that can attract help for CD8+ T cells. However, the cross-presentation of intact proteins is generally not efficient enough to generate strong CD8+ T-cell responses [49]. The use of peptide vaccines to circumvent this problem is hampered by the high degree of inter-individual MHC polymorphism, which narrows the range of individuals capable of responding to a given binding peptide. Moreover, an individual MHC class II binding site can accept peptides of variable lengths, complicating prediction of class II epitopes [50]. Furthermore, naïve CD4+ T cells specific for a given peptide are rare [51], and T-cell receptor diversity declines with age [52, 53], potentially leading to gaps in the T-cell repertoire [54, 55]. Together, this may limit opportunities to engage CD4+ T-cell help even if an individual’s MHC class II status is known. While ‘universal’ CD4+ T-cell epitopes capable of binding multiple MHC class II molecules have been identified, they have not yet shown unequivocal clinical utility [56, 57].
Synthetic long peptides (SLPs) can be designed to contain both CD8+ and CD4+ T-cell epitopes and lead to stronger CD8+ T-cell responses than both intact proteins and minimal MHC class I-binding short peptides [49, 58]. Unlike short peptides, which can load directly onto MHC class I, SLPs require processing by DCs. This avoids peptide presentation by non-professional APCs lacking co-stimulatory molecules, which may promote tolerance. Peptide-specific T-cell responses and regression of vulvar intraepithelial neoplasia in situ has been reported in women vaccinated with human papilloma virus (HPV)-derived SLPs [59].
Other methods have been employed to overcome the difficulties in providing CD4+ T-cell help, including the administration of autologous DCs loaded ex vivo with peptides and the administration of agonistic anti-CD40 monoclonal antibodies to license DCs without the requirement for CD4+ help. However, each of these approaches has practical limitations, and vaccines that can stimulate peptide-specific CD8+ T-cell responses independently of classical CD4+ T-cell help are desirable.
2.3 Innate-Like T Cells
In addition to traditional peptide-reactive T cells, some T-cell populations bear TCRs capable of recognizing non-peptide antigens presented by molecules other than classical MHC. These include glycolipid-responsive NKT cells, phosphoantigen-responsive Vγ9Vδ2 T cells, and riboflavin-derivative-responsive mucosal-associated invariant T cells (MAITs). Unlike conventional T cells, which are restricted by highly polymorphic classical MHC molecules, these ‘innate-like’ T cells express stereotyped TCRs that respond to defined non-peptide ligands presented by non-polymorphic molecules. As a result, defined ligands can activate innate-like T cells across entire populations, irrespective of tissue type. Indeed, the TCRs of CD1d-restricted NKT cells are so highly conserved that mouse CD1d molecules can present glycolipid antigens to human NKT cells and vice versa [60], greatly facilitating pre-clinical studies in animal models.
3 Type I or Invariant Natural Killer T (iNKT) Cells
NKT cells have been categorized into two principal subsets: type I and type II. The focus of this article, type I or so-called ‘invariant’, NKT cells, bear a semi-invariant TCR α chain (Vα24-Jα18 in humans, Vα14-Jα18 in mice) with a limited but not invariant TCR β-chain repertoire. These cells recognize self and foreign glycolipids presented by the non-polymorphic MHC class I-like CD1 family member, CD1d, as cognate antigens. The modified marine-sponge-derived glycolipid, α-galactosylceramide (α-GalCer), is the prototypical ligand for iNKT cell activation [61]. A second population of NKT cells, type II, are also CD1d-restricted but do not recognize α-GalCer and lack the semi-invariant TCR α-chain characteristic of iNKT cells [62, 63]. Their antigen specificity is poorly understood but includes sulfatide [64], lysophosphatidylcholine [65], lysophosphatidylethanolamine [66], phosphatidylglycerol [67, 68], and small aromatic (non-lipid) molecules [69]. Finally, some NKT cells recognize α-GalCer via a range of TCR α and β chains using alternative docking topologies and have been termed ‘atypical’ NKT cells [70, 71].
As their name suggests, iNKT cells express surface receptors traditionally associated with NK cells, including CD161 in humans and NK1.1 and NKG2D in mice. There is evidence that these NK receptors are functional [72–74]. Most circulating human iNKT cells have an effector memory phenotype (CD45RO+, CCR7–, CD62L–) [75], which is linked with the production of cytotoxic molecules [76, 77]. Existing in a semi-activated state [78], iNKT cells are able to respond rapidly to self or bacterial glycolipids presented by CD1d or by pathogen-induced cytokines in the absence of direct antigenic stimulation [79].
In humans, iNKT cells account for a median 0.05% of T cells in the peripheral blood. However, this frequency varies widely between individuals, ranging from undetectable to over 1% [80–82], with older patients tending to have fewer circulating iNKT cells [83]. Although these numbers are considerably lower than in mice [84], iNKT cells nevertheless comprise a high proportion of T cells relative to antigen-specific CD4+ T cells, which can represent as few as 0.005% of the total CD4+ T-cell pool [85]. Moreover, circulating iNKT cell frequencies may not accurately reflect systemic numbers [84], with iNKT cells being enriched in the human liver, where they account for a median of 0.5% of T cells [86]. In outbred swine, which have a variety of iNKT cell frequencies similar to those seen in humans, the frequency of iNKT cells in blood correlates poorly with frequency in tissues/nodes and with the immune response to α-GalCer stimulation [87].
In mice, NKT cells are known to contribute to the immune response against a variety of bacterial, fungal, protozoan, and viral pathogens [88], and several microbial glycolipids that bind to CD1d have been described [89–92]. In humans, a reduction in iNKT cells in the peripheral blood is correlated with several autoimmune and inflammatory disorders [93–95], as well as susceptibility to graft-versus-host disease (GVHD) [96]. A protective role for iNKT cells in surveillance against malignancy has been reported for spontaneous tumors initiated by the chemical carcinogen methylcholanthrene (MCA) [97] and in p53-deficient mice [98]. It has been proposed that iNKT-cell activity is modulated by changes in lipid metabolism brought about by endoplasmic reticulum (ER) stress in cancer cells [99], which may account for this protective role. The frequency of iNKT cells is reduced in patients with many cancers compared with that in healthy controls, and low iNKT cell numbers are associated with poor clinical outcomes in head and neck cancer [100], colorectal cancer [101], and neuroblastoma [102].
Despite bearing nearly identical TCR α-chain sequences, iNKT cells are made up of functionally distinct CD4+ and CD4– subsets. In humans, both CD4+ and CD4– iNKT cells are potent producers of IFN-γ, but CD4+ iNKT cells also produce T helper (TH)-2-type cytokines, such as interleukin (IL)-4 and IL-13 [81, 103]. In addition to CD4 status, the balance of TH1 and TH2 cytokines released upon antigen recognition by iNKT cells can be influenced by (1) the lipid tail composition of CD1d ligands [104], for example, a truncated OCH ligand induces IL-4 production and a TH2 response [105, 106]; (2) the presence of the TH1-biasing cytokine IL-12 [107]; (3) the degree and nature of co-stimulation received from the APC [108, 109]; and (4) the presence or absence of an immunosuppressive milieu [110].
Cytokine release by iNKT cells influences other effectors, including antigen-specific CD8+ T cells, helper CD4+ T cells, and NK cells. For example, systemic administration of α-GalCer in mice leads to cytokine-mediated NK cell expansion and increased cytotoxicity [111, 112] that is impaired by blocking both IFN-γ and IL-12 [113]. In addition, iNKT cells can exhibit direct cytotoxicity against target cells presenting a potent ligand, such as α-GalCer, on CD1d. As well as expressing granzyme-B and perforin, iNKT cells can up-regulate expression of FasL, tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL), and TNF. There is evidence for involvement of each of these cytotoxicity pathways in the lysis of CD1d-expressing target cells by iNKT cells [81, 114–117]. The degree of cytotoxicity exhibited by iNKT cells correlates with both target-cell CD1d expression and the potency of the iNKT cell agonist [114]. However, direct cytotoxicity towards tumor cells is often hampered by a lack of CD1d expression on these cells [114].
The localization of iNKT cells in the spleen enables them to respond to blood-borne antigens and to shape the adaptive immune response by sensitizing lymphoid-resident cross-presenting DCs to PRR signaling [118]. In particular, the marginal zone (MZ) of the spleen contains a wide vascular bed, the marginal sinus, where the arterial blood supply terminates [119]. In mice, iNKT cells are widely distributed throughout the spleen during steady-state conditions and rapidly consolidate in the MZ following delivery of self or foreign glycolipid antigens [120]. Here, their activation by DCs is amplified by other bystander cells, including MZ B cells and macrophages [121, 122]. In lymph nodes, glycolipid antigens that enter the subcapsular sinus (SCS) are retained and presented to iNKT cells by CD169+ macrophages, leading to iNKT cell arrest within the SCS, cytokine production, and expansion [123]. Thus, the rapid activation of iNKT cells is a natural phenomenon that can shape immune outcomes by modulating the responses of many different cell types, including DCs [124], CD4+ and CD8+ T cells [125], and B cells [126, 127].
4 Using iNKT Ligands as Universal Helper Epitopes
Like CD4+ T cells, iNKT cells are capable of providing cognate help to APCs. DC-mediated presentation of α-GalCer on CD1d leads to a reciprocal interaction between iNKT cells and the DC, in which iNKT cells up-regulate CD40L and release cytokines, including GM-CSF [107, 128, 129]. This promotes DC licensing and maturation, leading to up-regulation of co-stimulatory markers, such as CD80 and CD86, and production of IL-12, which further activates iNKT cells as well as CD8+ T cells [130]. The help provided by iNKT cells can enhance CD8+ T-cell responses against co-presented peptides [131]. In a manner similar to conventional CD4+ T-cell help, iNKT-cell help depends critically on peptide and glycolipid presentation by the same APC and on the CD40/CD40L interaction [37, 124, 125]. Thus, through the recruitment of third-party iNKT cells, the glycolipid α-GalCer can act as a ‘universal helper’ epitope for vaccination strategies (Fig. 2).

Invariant natural killer T (iNKT) cell help. Similar to cluster of differentiation (CD)-4+ T cells, iNKT cells can provide ‘help’ to antigen-presenting cells. Presentation of α-GalCer on CD1d by dendritic cells leads to up-regulation of CD40L on iNKT cells and release of pro-inflammatory cytokines, such as interferon-γ and granulocyte-macrophage colony-stimulating factor, which promote dendritic cell licensing. Interleukin-12 production further activates iNKT cells as well as CD8+ T cells. The help provided by iNKT cells can enhance CD8+ T-cell responses against co-presented peptides. In a manner similar to that of conventional CD4+ T-cell help, iNKT-cell help depends critically on peptide and glycolipid presentation by the same antigen-presenting cell and on the CD40/CD40L interaction. CD cluster of differentiation, CCL chemokine ligand, CCR chemokine receptor, GM-CSF granulocyte-macrophage colony-stimulating factor, IFN interferon, IL interleukin, iNKT invariant natural killer T cell, MHC major histocompatibility complex, TCR T-cell receptor, TNF tumor necrosis factor
Although the mechanisms mirror those of classical CD4+ T-cell help, the nature of the help provided by iNKT cells differs qualitatively. While DCs licensed by CD4+ T cells recruit CD8+ T cells using CCR5 ligands [132], licensing of DCs by iNKT cells results in production of the chemokine CCL17, which attracts naïve CD8+ T cells expressing the chemokine receptor CCR4 and results in improved migration to CCL17-expressing DCs and increased DC-CD8+ T-cell contact time [133]. The relevance of this in terms of function and homing of the peptide-specific CD8+ T cells is yet to be defined. In addition, iNKT cells may promote cross-talk between XCR1+ DCs and plasmacytoid DCs, which is reportedly important for efficient CD8+ memory T-cell induction [134].
By providing a universal form of T-cell help, ligands for iNKT cells fulfill the broad definition of an adjuvant: “a substance that enhance[s] the immunogenicity of an antigen” [135]. The adjuvant activity of iNKT ligands can be combined with that of conventional adjuvants that activate PRRs. Administration of TLR agonists in vitro strongly enhances the production of IFN-γ in mouse splenocytes pre-treated with α-GalCer [136]. In mice, the proliferation and effector function of antigen-specific CD8+ T cells is significantly improved by administration of the TLR4 ligand MPL in conjunction with α-GalCer [137]. Similarly, in human DCs, exposure to a TLR ligand and α-GalCer-activated iNKT cells leads to an increase in expression of maturation markers [137]. Notably, an initial α-GalCer-mediated activation of iNKT cells can condition multiple DC subsets to respond more effectively to TLR ligands [118], shaping the downstream CD8+ T-cell response.
5 Pre-Clinical and Clinical Evidence of iNKT Cell Help
While several studies report that systemic administration of IL-12 or α-GalCer activates iNKT cells and leads to tumor regression in vivo [138, 139], others report only a brief inhibition of tumor growth and a limited impact on overall survival [140, 141]. It is likely that this approach invokes lysis of cancer cells by activated iNKT cells or via the downstream activation of NK cells. Moreover, sequential systematic α-GalCer administration leads to TH2 biasing of iNKT cells [142] and, ultimately, a hypo-responsive state similar to anergy that may be associated with the development of an IL-10-secreting NKT cell subset (NKT10) [143, 144]. This outcome may be related to α-GalCer presentation by non-professional APCs.
5.1 Co-Administration
Co-administration of α-GalCer with soluble antigens [124, 125], DNA vaccines [145], or recombinant viruses expressing a malarial antigen [146] significantly enhances peptide-specific CD8+ and CD4+ T-cell responses as well as antibody-mediated responses. The drawback of this approach is that co-administration does not necessarily ensure uptake of both antigen and adjuvant by the same APC, especially if the individual vaccine components have different pharmacokinetics. Indeed, a study in macaques demonstrated no enhancement of influenza-specific T- or B-cell immunity following intravenous co-administration of α-GalCer with live attenuated influenza virus [147]. The presentation of some of the α-GalCer by non-professional APCs may limit immune responses and provoke iNKT cell anergy. To fully exploit iNKT cell help and induce robust CD8+ T-cell responses, it is critical that the iNKT ligand and antigenic peptide are targeted to the same professional APC; this has been the goal of recent approaches [125, 133].
5.2 α-GalCer-Loaded Antigen-Expressing Dendritic Cells (DCs)
Administration of DCs loaded ex vivo with α-GalCer promotes prolonged IFN-γ production and prevents iNKT cell anergy, resulting in more potent anti-tumor effects [148–152]. By loading DCs with both α-GalCer and peptide, the co-presentation of a peptide CD8+ epitope and iNKT cell ligand is ensured, improving the activity of the DC vaccine. An in vitro study of human monocyte-derived DCs (moDCs) found that pulsing these cells with α-GalCer and a tumor-associated antigen (TAA) induced more robust expansion of TAA-specific CD8+ T cells than did non-α-GalCer-pulsed moDCs, suggesting that the α-GalCer adjuvant effect might be applicable to humans [153]. A limitation of this approach in clinical practice is the need for a large number of peripheral blood mononuclear cells (PBMCs) and Good Manufacturing Practice (GMP)-compliant facilities to generate the patient-specific cellular vaccines.
A related approach is to generate artificial APCs, such as α-GalCer-pulsed, CD1d-transfected embryonic kidney cells transfected with messenger RNA (mRNA) encoding the target antigen [154]. This entirely avoids the need to generate patient-specific cellular vaccines, although it does not overcome the need for GMP-compliant cell culture.
The observation that exogenous α-GalCer-pulsed DC vaccines made from CD1d–/– APCs still activate iNKT cells in vivo [155] suggests that DC vaccines can simply function as a vehicle for the co-delivery of antigen and glycolipid adjuvant to an endogenous APC population. In mice, CD8+ T-cell responses could be generated to protein-pulsed CD1d–/– DCs, with the critical endogenous DC subset involved being splenic langerin+ CD8α+ DCs, a subset of cross-presenting XCR1+ DCs [155]. If such transfer of antigen and adjuvant to resident APCs occurs in humans, the ex vivo generation of DCs may be unnecessary.
5.3 α-GalCer-Pulsed Whole Tumor Cell Vaccines
For the treatment of cancer, vaccines comprising irradiated tumor cells treated with α-GalCer elicit stronger protective immunity than do unpulsed tumor cells in vivo. In this model, resident APCs take up α-GalCer-loaded tumor cells and present TAAs alongside α-GalCer, resulting in iNKT cell-mediated DC licensing and enhanced CD4+ and CD8+ T-cell-dependent anti-tumor immunity [156]. An advantage of using autologous whole tumor vaccines is that they provide a broad range of patient-specific TAAs.
In animal studies, vaccines comprising whole tumor cells pulsed with α-GalCer led to effective tumor protection in various malignancies, including myeloma [157], melanoma [156, 158], murine B-cell lymphomas [159, 160], and myeloid leukemias [160, 161]. Not all of these tumors expressed CD1d, emphasizing the role of resident APCs in these responses. Nonetheless, for tumors that express low levels of CD1d, the efficacy of an α-GalCer-pulsed whole tumor vaccine could be enhanced by transfection of the tumor cell with CD1d [158], making the injected cells susceptible to iNKT cell-mediated lysis. The enhanced memory responses observed were attributed to tumor cell remnants being taken up and cross-presented on DCs [156, 159]; this promoted DC maturation and IL-12 production, enabling fully active DCs to recruit peptide-specific CD8+ and CD4+ T cells. In a model of intracranial glioma, the therapeutic impact of administration of irradiated glioma cells pulsed with α-GalCer was most significant when regulatory T cells were first eradicated [162].
The practical use of α-GalCer-loaded tumor cells is complicated by the need to harvest tumor cells for the generation of patient-specific vaccines, the robust precautions necessary to minimize the risk of inoculating a patient with viable tumor cells, the need for GMP-compliant facilities to make the personalised vaccine, and the potential to induce autoimmunity. This may favor synthetic methods of delivering TAA and iNKT cell ligands.
5.4 Microorganisms/Micro-Particles
Nano-particulate delivery systems, such as lipo-particles or microorganisms, offer an alternative to patient-specific cellular vaccines. Co-delivery of a TAA with α-GalCer within cationic liposomes enhanced anti-tumor cytotoxic T-cell responses in vivo [163]. Vaccination with virus-like particles (VLPs) bound to α-GalCer in a composite particle activated splenic iNKT cells, generated antigen-specific CD8+ T cells, and promoted tumor rejection in vivo [164]. A recent study found that co-delivery of α-GalCer with ovalbumin (OVA) in poly(lactic-co-glycolic) acid (PLGA) nanoparticles elicited more powerful antigen-specific CD8+ T-cell responses and improved overall survival in a B16.OVA tumor model than did PLGA nanoparticles containing the TLR ligands R848 and poly I:C with the OVA antigen [165].
Exploiting intact microorganisms as a delivery vehicle may carry the additional benefit of delivering a TLR signal alongside iNKT ligand and antigenic peptide. Therapeutic vaccination with a Listeria monocytogenes-based vaccine expressing a TAA in complex with α-GalCer prevented liver metastases in a model of metastatic breast cancer [166]. This was associated with a significant increase in the percentage of active iNKT cells in the spleen as well as an increase in CD8+ T-cell responses. Similarly, incorporation of α-GalCer into a recombinant Mycobacterium bovis bacillus Calmette–Guèrin (rBCG) strain presenting a simian immunodeficiency viral (SIV) epitope significantly enhanced antigen-specific CD8+ T-cell responses following SIV re-challenge in mice [167].
Thus, nano-particulate delivery systems can be successfully employed to co-deliver antigenic peptides and iNKT cell ligands to APCs. Microorganism-based delivery vehicles may carry the additional advantage of activating PRRs within APCs. Disadvantages of the latter approach include the possibility of forming neutralizing antibody responses against the microorganism or eliciting off-target immunodominant CD8+ T-cell responses against microbial peptides.
5.5 Synthetic iNKT-Cell Activating Vaccine Conjugates
Given the difficulty of manufacturing autologous cellular vaccines in humans [168], more efficient and effective strategies to co-deliver iNKT cell-activating glycolipids and peptide antigens to the same APC are needed. Recent studies have demonstrated the efficacy of so-called ‘conjugate vaccines’, in which an antigen is conjugated, via an enzymatically cleavable linker, to a glycolipid iNKT cell agonist, such as α-GalCer [169] (Fig. 3). Our center has undertaken in vivo studies of synthetic vaccines comprising short peptides chemically conjugated to an α-GalCer ‘pro-drug’ via cleavable linkers. Following uptake by an APC, the conjugated components of the vaccine are cleaved enzymatically within the endosomal compartment. Incorporation of additional amino acids at the N-terminus of the peptide helps to promote proteolytic generation of the respective epitopes within APCs [170]. The antigenic peptide component of the vaccine is then cross-presented on MHC class I molecules to activate CD8+ T cells, while the α-GalCer prodrug spontaneously converts to active α-GalCer and is presented on CD1d molecules on the same APC to stimulate iNKT cells. In this way, activated iNKT cells provide targeted T-cell help to license DCs engaged in stimulating CD8+ T-cell responses against the peptide. Indeed, a conjugate incorporating the immunodominant OVA peptide elicited a strong cytotoxic CD8+ T-cell response that suppressed OVA-induced allergic airway inflammation by killing allergen-bearing APCs [171]. A similar glycolipid–peptide conjugate vaccine, this time using a cathepsin B-cleavable linker, promoted a robust CD8+ T-cell-dependent immune response against established OVA-expressing B16 melanoma tumors in mice [172]. The activity was CD1d dependent and associated with cytotoxicity mediated by activated CD8+ T cells. Importantly, in both studies, the conjugate vaccine demonstrated biological activity superior to that of unconjugated admixed peptide and glycolipid, highlighting the importance of targeting antigen and iNKT cell ligand to the same APC [172, 173].

Structure of a glycolipid–peptide conjugate vaccine. A prodrug form of α-GalCer is linked via a cathepsin-B-cleavable linker to a major histocompatibility complex class I-restricted peptide sequence. Combining both peptide and glycolipid in one structure ensures uptake by the same antigen-presenting cell, facilitating co-presentation of both vaccine epitopes, which enhances the CD8+ T-cell response
The clinical potential of these α-GalCer conjugate vaccines is supported by data demonstrating their capacity to activate both iNKT cells and peptide-specific CD8+ T cells in human cells in vitro [172]. Advantageously, synthetic vaccines based on established antigenic peptides are relatively easy and cost effective to manufacture [172]. This approach may therefore have utility in the treatment of cancer as well as infectious or allergic disease, although further research will be needed to investigate conjugate vaccines containing antigens associated with specific disease states. Many tumor-specific antigens have already been cataloged [174], and high-throughput gene sequencing technologies will enable identification of additional targets, the importance of cancer-specific neoantigens in tumor control being increasingly recognized [175]. To exploit this therapeutically, a highly flexible platform would be needed to manufacture such personalized vaccines.
6 Induction of Humoral Immunity by iNKT Cell Adjuvants
While this article focuses primarily on CD8+ T-cell responses, α-GalCer also promotes humoral immune responses [176]. Indeed, in the presence of α-GalCer, robust antibody responses can be elicited even in MHC class II knockout animals, which have no potential for classical CD4+ T-cell help [176, 177]. A cognate interaction between iNKT cells and CD1d on B cells can promote differentiation of iNKT cells into CXCR5+ PD-1hi follicular helper NKT (NKTFH) cells [178]. Co-administration of α-GalCer in conjunction with a protein vaccine results in rapid germinal center (GC) formation and class-switched antigen-specific antibody production [127, 179, 180] that is dependent on NKTFH cells, CD40L [126], and the expression of CD1d on B cells [181].
Some studies have indicated that, unlike classical TFH cells, NKTFH-cell help does not promote long-term B-cell memory formation [178, 182, 183]. However, two independent studies have demonstrated that inoculation with a polysaccharide–glycolipid vaccine can promote iNKT-dependent long-lived memory B-cell formation. Liposomal nanoparticles displaying a synthetic glycolipid and Streptococcus pneumoniae polysaccharide antigens elicited a direct iNKT–B-cell interaction in vivo, despite the absence of NKTFH-cell differentiation [184]. The iNKT cells were first activated by CD1d-expressing APCs before providing cognate help to B cells that promoted prolonged antibody responses with isotype switching, affinity maturation, and long-lasting B-cell memory. In an alternative approach similar to the conjugate vaccines above, direct conjugation of an S. pneumoniae capsule protein to α-GalCer stimulated NKTFH cells to promote generation of antigen-specific, long-lived memory B cells and plasmablasts in mice [173], leading to the development of potent anti-polysaccharide immunity and protection from pneumonia following re-challenge. Thus, analogous to the enhancement of cytotoxic CD8+ T-cell responses, vaccines that ensure co-delivery of a glycolipid–antigen complex have the potential to stimulate robust and long-lasting humoral immune responses.
Activated iNKT cells can also provide help to B cells indirectly by boosting conventional T-cell help: DCs activated through an interaction with iNKT cells via CD1d have an enhanced ability to induce classical TFH cells, which in turn interact with B cells [185, 186]. Co-delivery of α-GalCer and protein antigens from a number of infectious pathogens activates this ‘non-cognate’ B-cell help by iNKT cells to effect protective humoral responses and long-lasting B-cell memory [120, 187–190].
A clinical trial has demonstrated the adjuvant effect of the potent synthetic iNKT-activating glycolipid, ABX196, in humans [191]. A phase I/II dose-escalation study in healthy volunteers showed that co-administration of ABX196 with the poorly immunogenic Hepatitis B (HBs) antigen resulted in activated IFN-γ-producing iNKT cells in the peripheral blood as well as protective anti-HBs antibodies in 79% of the patient cohort, suggesting that ABX196 is an effective adjuvant in humans. Transient hepatotoxicity was observed in some patients who received ABX196, although most cases were mild to moderate. This might be ameliorated by using a less potent activating ligand or through changes to the mode of delivery.
7 Challenges to the Clinical Application of iNKT Help
A number of barriers to the exploitation of iNKT cell help as adjuvants may exist. In mice, strong TCR-mediated activation, coupled with co-stimulation, can induce profound iNKT-cell anergy that renders these cells resistant to subsequent stimulation [143, 192]. However, anergy has most frequently been observed when iNKT cell ligands are administered systemically, whereas the targeting of α-GalCer to professional APCs appears to prevent or reduce this phenomenon in mice [193]. Similarly, in humans, repeated systemic injections of a glycolipid–liposomal vaccine have been reported to reduce circulating iNKT cell numbers as well as serum IFN-γ [191], whereas a second administration of α-GalCer-pulsed DCs led to an increase in serum IFN-γ compared with the first administration [150].
In some circumstances, iNKT cells also have an immunosuppressive role [94, 194]. They can protect against the development of certain autoimmune diseases, such as type I diabetes mellitus [195, 196] and experimental autoimmune encephalitis [197, 198], and can impede CD8+ T cell responses to viral proteins in the skin [199]. In the skin, an immunosuppressive population of iNKT cells plays a role in a number of pathologies [200], including oncogene-driven skin hyperplasia [201]. iNKT cells are also vital for the establishment of tolerance to antigens introduced into an immunologically privileged site [202]. Although questions remain surrounding how iNKT cells are programmed to promote different immune responses, it does appear that potent activating ligands, like α-GalCer, generate predominantly pro-inflammatory TH1-biased responses as opposed to suppression [203, 204]. Nonetheless, a recent report did show that iNKT cells stimulated with α-GalCer in vivo can develop characteristics of regulatory cells, including production of the immunomodulatory cytokine IL-10 [144]. Thus, further investigation of this phenomenon is warranted.
The liver is rich in iNKT cells, and—in mice—hepatotoxicity has been observed in response to systemically administered α-GalCer, although not when administered in the context of a bacterial delivery system [166]. Hepatotoxicity has not been reported in phase I/II clinical trials of systemically administered α-GalCer or α-GalCer-pulsed APCs [140, 205–207]; however, a phase I clinical trial using a more potent iNKT-cell agonist (ABX196) indicated that stronger ligands can lead to hepatotoxicity in humans [191]. This, in conjunction with the potential to stimulate an immunosuppressive iNKT cell population detailed above, underscores the importance of careful selection of glycolipid and mode of delivery for vaccine development.
The lower, and highly variable, frequency of iNKT cells in humans compared with mice could impact the effectiveness of iNKT cell-based immunotherapies. However, even in humans, iNKT cells are enriched in tissues such as the liver [86], and the frequency of iNKT cells is likely to be far greater than that of a CD4+ T cell with a given peptide specificity. Inter-individual variations in iNKT frequency may not be critical: α-GalCer was an effective adjuvant when administered with ultraviolet (UV)-killed swine influenza (SI) virus in outbred swine, which have similarly disparate iNKT cell frequencies to humans [208], with no reported correlation between iNKT-cell frequencies and the immune response in individual pigs. Finally, the cytokine and antibody responses observed in early clinical trials of iNKT cell ligands suggest that humans have sufficient functional iNKT cells to trigger immunologic responses [140, 150, 191]. Ultimately, only the results of randomized controlled trials of iNKT cell-activating vaccines will establish their efficacy in humans.
8 Concluding Remarks and Perspectives
Optimal cytotoxic CD8+ T-cell responses require cognate T-cell help for the APC and are not optimally induced by the adjuvants traditionally used in the clinic. In practice, issues of peptide selection and specificity complicate the reliable induction of CD4+ T-cell help. In contrast, iNKT cells can be specifically and universally activated by defined glycolipids, irrespective of an individual’s tissue type, and provide a universal form of T-cell help that differs both qualitatively and quantitatively from conventional CD4+ T-cell help. Critically, the effective use of iNKT cell ligands as adjuvants requires co-presentation of peptide and glycolipid antigens on the same APC. A variety of approaches may achieve this, of which the most practical for clinical application may be their synthetic combination, either via liposome/microparticle packaging or chemical conjugation.
Preclinical data suggest that iNKT-cell help can be employed to enhance both cellular and humoral immune responses against malignancies and infections. The experience accumulated from clinical trials to date demonstrates that iNKT cell agonists are immunologically active in humans and have good safety profiles [140, 149, 209]. Nonetheless, informed selection of iNKT cell agonist, target antigen, delivery system, and dosing schedule will be necessary to minimize the risks of toxicity and to maximize the chances of eliciting an effective targeted and sustained immune response.
We conclude that iNKT cell ligands can provide a universal form of T-cell help to enhance peptide-specific CD8+ T-cell and antibody responses. Synthetic approaches to the co-delivery of peptide antigens and innate-like T-cell ligands to DCs represent a promising strategy to improve vaccine responses against cancer and infectious diseases.
References
Andre FE, et al. Vaccination greatly reduces disease, disability, death and inequity worldwide. Bull World Health Organ. 2008;86:140–6.
Reed SG, et al. Key roles of adjuvants in modern vaccines. Nat Med. 2013;19:1597–608.
Kantoff PW, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22.
Jeanbart L, Swartz MA. Engineering opportunities in cancer immunotherapy. Proc Natl Acad Sci USA. 2015;112:14467–72.
Tan HX, Gilbertson BP, Jegaskanda S, et al. Recombinant influenza virus expressing HIV-1 p24 capsid protein induces mucosal HIV-specific CD8 T-cell responses. Vaccine. 2016;34(9):1172–9.
Wilson NS, et al. ISCOMATRIX vaccines mediate CD8+ T-cell cross-priming by a MyD88-dependent signaling pathway. Immunol Cell Biol. 2012;90:540–52.
Leroux-Roels G, et al. Vaccine adjuvant systems containing monophosphoryl lipid A and QS-21 induce strong humoral and cellular immune responses against hepatitis B surface antigen which persist for at least 4 years after vaccination. Vaccine. 2015;33:1084–91.
Lambrecht BN, et al. Mechanism of action of clinically approved adjuvants. Curr Opin Immunol. 2009;21:23–9.
De Gregorio E, et al. Immunology of TLR-independent vaccine adjuvants. Curr Opin Immunol. 2009;21:339–45.
Apostolico Jde S, et al. Adjuvants: classification, modus operandi, and licensing. J Immunol Res. 2016;2016:1459394.
Temizoz B, et al. Vaccine adjuvants as potential cancer immunotherapeutics. Int Immunol. 2016;28:329–38.
Banday AH, et al. Cancer vaccine adjuvants: recent clinical progress and future perspectives. Immunopharmacol Immunotoxicol. 2015;37:1–11.
Akondy RS, et al. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J Immunol. 2009;183:7919–30.
Querec T, et al. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J Exp Med. 2006;203:413–24.
Sei JJ, et al. Effector and central memory poly-functional CD4(+) and CD8(+) T cells are boosted upon ZOSTAVAX (R) vaccination. Front Immunol. 2015;6:553.
Larkin J, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:1270–1.
Small EJ, et al. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol. 2000;18:3894–903.
Gulley JL, et al. Immune impact induced by PROSTVAC (PSA-TRICOM), a therapeutic vaccine for prostate cancer. Cancer Immunol Res. 2014;2:133–41.
Mandl SJ, et al. PROSTVAC, PSA-targeted immunotherapy: new evidence for mechanism of action. J Clin Oncol. 2014;32:5 Suppl (abstract 3080).
Larocca C, Schlom J. Viral vector-based therapeutic cancer vaccines. Cancer J. 2011;17:359–71.
Leroux-Roels I, et al. Adjuvant system AS02 V enhances humoral and cellular immune responses to pneumococcal protein PhtD vaccine in healthy young and older adults: randomised, controlled trials. Vaccine. 2015;33:577–84.
Orr MT, et al. Adjuvant formulation structure and composition are critical for the development of an effective vaccine against tuberculosis. J Control Release. 2013;172:190–200.
van Dissel JT, et al. A novel liposomal adjuvant system, CAF01, promotes long-lived Mycobacterium tuberculosis-specific T-cell responses in human. Vaccine. 2014;32:7098–107.
Navabi H, et al. A clinical grade poly I:C-analogue (Ampligen) promotes optimal DC maturation and Th1-type T cell responses of healthy donors and cancer patients in vitro. Vaccine. 2009;27:107–15.
Joffre OP, et al. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557–69.
Hemmi H, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196–200.
Miller RL, et al. Imiquimod applied topically: a novel immune response modifier and new class of drug. Int J Immunopharmacol. 1999;21:1–14.
MacLeod MK, et al. Vaccine adjuvants aluminum and monophosphoryl lipid A provide distinct signals to generate protective cytotoxic memory CD8 T cells. Proc Natl Acad Sci USA. 2011;108:7914–9.
Cluff CW. Monophosphoryl lipid A (MPL) as an adjuvant for anti-cancer vaccines: clinical results. In: Jeannin J-F, editor. Lipid A in cancer therapy. Advances in experimental medicine and biology, vol. 667. New York: Springer Science + Business Media; 2009. p. 111–23.
Chavez-Galan L, et al. Cell death mechanisms induced by cytotoxic lymphocytes. Cell Mol Immunol. 2009;6:15–25.
Quezada SA, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207:637–50.
Janssen EM, et al. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–6.
Bourgeois C, et al. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science. 2002;297:2060–3.
Wiesel M, et al. Type I IFN substitutes for T cell help during viral infections. J Immunol. 2011;186:754–63.
Greyer M, et al. T cell help amplifies innate signals in CD8(+) DCs for optimal CD8(+) T cell priming. Cell Rep. 2016;14:586–97.
Kim J, et al. Memory programming in CD8(+) T-cell differentiation is intrinsic and is not determined by CD4 help. Nat Commun. 2015;6:7994.
Ridge JP, et al. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–8.
Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–9.
Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–42.
Lipsky PE, et al. Analysis of CD40–CD40 ligand interactions in the regulation of human B cell function. Ann NY Acad Sci. 1997;815:372–83.
Bennett SR, et al. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–80.
Schoenberger SP, et al. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature. 1998;393:480–3.
Smith CM, et al. Cognate CD4(+) T cell licensing of dendritic cells in CD8(+) T cell immunity. Nat Immunol. 2004;5:1143–8.
Scherle PA, Gerhard W. Functional analysis of influenza-specific helper T cell clones in vivo. T cells specific for internal viral proteins provide cognate help for B cell responses to hemagglutinin. J Exp Med. 1986;164:1114–28.
Russell AS, et al. Antibody dependent cell-mediated cytotoxicity to herpes simplex virus in man: the influence of drugs on polymorphonuclear leucocyte and mononuclear effector cells. Prostaglandins Med. 1979;3:147–58.
Eickhoff S, et al. Robust anti-viral immunity requires multiple distinct T cell–dendritic cell interactions. Cell. 2015;162:1322–37.
Hor JL, et al. Spatiotemporally distinct interactions with dendritic cell subsets facilitates CD4+ and CD8+ T cell activation to localized viral infection. Immunity. 2015;43:554–65.
Dorner BG, et al. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity. 2009;31:823–33.
Zhang H, et al. Comparing pooled peptides with intact protein for accessing cross-presentation pathways for protective CD8+ and CD4+ T cells. J Biol Chem. 2009;284:9184–91.
Wang P, et al. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008;4:e1000048.
Moon JJ, et al. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity. 2007;27:203–13.
Britanova OV, et al. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J Immunol. 2014;192:2689–98.
Haynes BF, et al. The human thymus during aging. Immunol Res. 2000;22:253–61.
Yager EJ, et al. Age-associated decline in T cell repertoire diversity leads to holes in the repertoire and impaired immunity to influenza virus. J Exp Med. 2008;205:711–23.
Lee JB, et al. Decline of influenza-specific CD8+ T cell repertoire in healthy geriatric donors. Immun Ageing. 2011;8:6.
Fraser CC, et al. Generation of a universal CD4 memory T cell recall peptide effective in humans, mice and non-human primates. Vaccine. 2014;32:2896–903.
Wen X, et al. Inclusion of a universal tetanus toxoid CD4(+) T cell epitope P2 significantly enhanced the immunogenicity of recombinant rotavirus DeltaVP8* subunit parenteral vaccines. Vaccine. 2014;32:4420–7.
Bijker MS, et al. CD8+ CTL priming by exact peptide epitopes in incomplete Freund’s adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. J Immunol. 2007;179:5033–40.
Kenter GG, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009;361:1838–47.
Brossay L, et al. CD1d-mediated recognition of an alpha-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J Exp Med. 1998;188:1521–8.
Kawano T, et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science. 1997;278:1626–9.
Rhost S, et al. Immunomodulatory type II natural killer T lymphocytes in health and disease. Scand J Immunol. 2012;76:246–55.
Godfrey DI, et al. NKT cells: what’s in a name? Nat Rev Immunol. 2004;4:231–7.
Jahng A, et al. Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J Exp Med. 2004;199:947–57.
Chang DH, et al. Inflammation-associated lysophospholipids as ligands for CD1d-restricted T cells in human cancer. Blood. 2008;112:1308–16.
Zeissig S, et al. Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity. Nat Med. 2012;18:1060–8.
Leon L, et al. Saposins utilize two strategies for lipid transfer and CD1 antigen presentation. Proc Natl Acad Sci USA. 2012;109:4357–64.
Wolf BJ, et al. Identification of a potent microbial lipid antigen for diverse NKT cells. J Immunol. 2015;195:2540–51.
Van Rhijn I, et al. CD1d-restricted T cell activation by nonlipidic small molecules. Proc Natl Acad Sci USA. 2004;101:13578–83.
Le Nours J, et al. Atypical natural killer T-cell receptor recognition of CD1d-lipid antigens. Nat Commun. 2016;7:10570.
Uldrich AP, et al. A semi-invariant Valpha10+ T cell antigen receptor defines a population of natural killer T cells with distinct glycolipid antigen-recognition properties. Nat Immunol. 2011;12:616–23.
Exley M, et al. CD161 (NKR-P1A) costimulation of CD1d-dependent activation of human T cells expressing invariant Vα24JαQ T cell receptor alpha chains. J Exp Med. 1998;188:867–76.
Vilarinho S, et al. Blockade of NKG2D on NKT cells prevents hepatitis and the acute immune response to hepatitis B virus. Proc Natl Acad Sci USA. 2007;104:18187–92.
Kawamura T, et al. NKG2A inhibits invariant NKT cell activation in hepatic injury. J Immunol. 2009;182:250–8.
Sandberg JK, et al. Dominant effector memory characteristics, capacity for dynamic adaptive expansion, and sex bias in the innate Valpha24 NKT cell compartment. Eur J Immunol. 2003;33:588–96.
Yang S, et al. The shedding of CD62L (L-selectin) regulates the acquisition of lytic activity in human tumor reactive T lymphocytes. PLoS One. 2011;6:e22560.
Sallusto F, et al. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–63.
Stetson DB, et al. Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J Exp Med. 2003;198:1069–76.
Holzapfel KL, et al. Antigen-dependent versus -independent activation of invariant NKT cells during infection. J Immunol. 2014;192:5490–8.
Montoya CJ, et al. Characterization of human invariant natural killer T subsets in health and disease using a novel invariant natural killer T cell-clonotypic monoclonal antibody, 6B11. Immunology. 2007;122:1–14.
Gumperz JE, et al. Functionally distinct subsets of CD1d-restricted natural killer T cells revealed by CD1d tetramer staining. J Exp Med. 2002;195:625–36.
Chan AC, et al. Immune characterization of an individual with an exceptionally high natural killer T cell frequency and her immediate family. Clin Exp Immunol. 2009;156:238–45.
Jing Y, et al. Aging is associated with a rapid decline in frequency, alterations in subset composition, and enhanced Th2 response in CD1d-restricted NKT cells from human peripheral blood. Exp Gerontol. 2007;42:719–32.
Berzins SP, et al. Systemic NKT cell deficiency in NOD mice is not detected in peripheral blood: implications for human studies. Immunol Cell Biol. 2004;82:247–52.
Long HM, et al. MHC II tetramers visualize human CD4+ T cell responses to Epstein–Barr virus infection and demonstrate atypical kinetics of the nuclear antigen EBNA1 response. J Exp Med. 2013;210:933–49.
Kenna T, et al. NKT cells from normal and tumor-bearing human livers are phenotypically and functionally distinct from murine NKT cells. J Immunol. 2003;171:1775–9.
Artiaga BL, et al. Adjuvant effects of therapeutic glycolipids administered to a cohort of NKT cell-diverse pigs. Vet Immunol Immunopathol. 2014;162:1–13.
Kinjo Y, et al. The role of invariant natural killer T cells in microbial immunity. J Infect Chemother. 2013;19:560–70.
Kinjo Y, et al. Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat Immunol. 2006;7:978–86.
Kinjo Y, et al. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature. 2005;434:520–5.
Mattner J, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature. 2005;434:525–9.
Sriram V, et al. Cell wall glycosphingolipids of Sphingomonas paucimobilis are CD1d-specific ligands for NKT cells. Eur J Immunol. 2005;35:1692–701.
Berzins SP, et al. Presumed guilty: natural killer T cell defects and human disease. Nat Rev Immunol. 2011;11:131–42.
van der Vliet HJ, et al. The immunoregulatory role of CD1d-restricted natural killer T cells in disease. Clin Immunol. 2004;112:8–23.
Weinkove R, et al. Functional invariant natural killer T-cell and CD1d axis in chronic lymphocytic leukemia: implications for immunotherapy. Haematologica. 2013;98:376–84.
Rubio MT, et al. Early posttransplantation donor-derived invariant natural killer T-cell recovery predicts the occurrence of acute graft-versus-host disease and overall survival. Blood. 2012;120:2144–54.
Smyth MJ, et al. Differential tumor surveillance by natural killer (NK) and NKT cells. J Exp Med. 2000;191:661–8.
Swann JB, et al. Type I natural killer T cells suppress tumors caused by p53 loss in mice. Blood. 2009;113:6382–5.
McEwen-Smith RM, et al. The regulatory role of invariant NKT cells in tumor immunity. Cancer Immunol Res. 2015;3:425–35.
Schneiders FL, et al. Circulating invariant natural killer T-cell numbers predict outcome in head and neck squamous cell carcinoma: updated analysis with 10-year follow-up. J Clin Oncol. 2012;30:567–70.
Tachibana T, et al. Increased intratumor Valpha24-positive natural killer T cells: a prognostic factor for primary colorectal carcinomas. Clin Cancer Res. 2005;11:7322–7.
Metelitsa LS, et al. Natural killer T cells infiltrate neuroblastomas expressing the chemokine CCL2. J Exp Med. 2004;199:1213–21.
Lee PT, et al. Distinct functional lineages of human V(alpha)24 natural killer T cells. J Exp Med. 2002;195:637–41.
Lawson V. Turned on by danger: activation of CD1d-restricted invariant natural killer T cells. Immunology. 2012;137:20–7.
Stanic AK, et al. Another view of T cell antigen recognition: cooperative engagement of glycolipid antigens by Va14Ja18 natural T(iNKT) cell receptor [corrected]. J Immunol. 2003;171:4539–51.
Oki S, et al. The clinical implication and molecular mechanism of preferential IL-4 production by modified glycolipid-stimulated NKT cells. J Clin Invest. 2004;113:1631–40.
Kitamura H, et al. The natural killer T (NKT) cell ligand alpha-galactosylceramide demonstrates its immunopotentiating effect by inducing interleukin (IL)-12 production by dendritic cells and IL-12 receptor expression on NKT cells. J Exp Med. 1999;189:1121–8.
Hayakawa Y, et al. Differential regulation of Th1 and Th2 functions of NKT cells by CD28 and CD40 costimulatory pathways. J Immunol. 2001;166:6012–8.
Arora P, et al. A single subset of dendritic cells controls the cytokine bias of natural killer T cell responses to diverse glycolipid antigens. Immunity. 2014;40:105–16.
Molano A, et al. Modulation of invariant natural killer T cell cytokine responses by indoleamine 2,3-dioxygenase. Immunol Lett. 2008;117:81–90.
Smyth MJ, et al. Sequential activation of NKT cells and NK cells provides effective innate immunotherapy of cancer. J Exp Med. 2005;201:1973–85.
Smyth MJ, et al. Sequential production of interferon-gamma by NK1.1(+) T cells and natural killer cells is essential for the antimetastatic effect of alpha-galactosylceramide. Blood. 2002;99:1259–66.
Eberl G, MacDonald HR. Selective induction of NK cell proliferation and cytotoxicity by activated NKT cells. Eur J Immunol. 2000;30:985–92.
Wingender G, et al. Antigen-specific cytotoxicity by invariant NKT cells in vivo is CD95/CD178-dependent and is correlated with antigenic potency. J Immunol. 2010;185:2721–9.
Nakagawa R, et al. Mechanisms of the antimetastatic effect in the liver and of the hepatocyte injury induced by alpha-galactosylceramide in mice. J Immunol. 2001;166:6578–84.
Nieda M, et al. TRAIL expression by activated human CD4(+)V alpha 24NKT cells induces in vitro and in vivo apoptosis of human acute myeloid leukemia cells. Blood. 2001;97:2067–74.
Metelitsa LS, et al. Expression of CD1d by myelomonocytic leukemias provides a target for cytotoxic NKT cells. Leukemia. 2003;17:1068–77.
Osmond TL, et al. Activated NKT cells can condition different splenic dendritic cell subsets to respond more effectively to TLR engagement and enhance cross-priming. J Immunol. 2015;195:821–31.
Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol. 2005;5:606–16.
King IL, et al. The mechanism of splenic invariant NKT cell activation dictates localization in vivo. J Immunol. 2013;191:572–82.
Barral P, et al. The location of splenic NKT cells favours their rapid activation by blood-borne antigen. EMBO J. 2012;31:2378–90.
Bialecki E, et al. Role of marginal zone B lymphocytes in invariant NKT cell activation. J Immunol. 2009;182:6105–13.
Barral P, et al. CD169(+) macrophages present lipid antigens to mediate early activation of iNKT cells in lymph nodes. Nat Immunol. 2010;11:303–12.
Fujii S, et al. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med. 2003;198:267–79.
Hermans IF, et al. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol. 2003;171:5140–7.
Galli G, et al. Invariant NKT cells sustain specific B cell responses and memory. Proc Natl Acad Sci USA. 2007;104:3984–9.
Leadbetter EA, et al. NK T cells provide lipid antigen-specific cognate help for B cells. Proc Natl Acad Sci USA. 2008;105:8339–44.
Tomura M, et al. A novel function of Valpha14+ CD4+ NKT cells: stimulation of IL-12 production by antigen-presenting cells in the innate immune system. J Immunol. 1999;163:93–101.
Vincent MS, et al. CD1-dependent dendritic cell instruction. Nat Immunol. 2002;3:1163–8.
Brigl M, et al. Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat Immunol. 2003;4:1230–7.
Gottschalk C, et al. The role of invariant natural killer T cells in dendritic cell licensing, cross-priming, and memory CD8(+) T cell generation. Front Immunol. 2015;6:379.
Castellino F, et al. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature. 2006;440:890–5.
Semmling V, et al. Alternative cross-priming through CCL17-CCR4-mediated attraction of CTLs toward NKT cell-licensed DCs. Nat Immunol. 2010;11:313–20.
Shimizu K, et al. Invariant NKT cells induce plasmacytoid dendritic cell (DC) cross-talk with conventional DCs for efficient memory CD8+ T cell induction. J Immunol. 2013;190:5609–19.
Murphy K, Travers P, Walport M, Janeway C. Janeway’s immunobiology. 8th ed. New York: Garland Science; 2012.
Ando T, et al. Toll-like receptor agonists and alpha-galactosylceramide synergistically enhance the production of interferon-gamma in murine splenocytes. Sci Rep. 2013;3:2559.
Hermans IF, et al. Dendritic cell function can be modulated through cooperative actions of TLR ligands and invariant NKT cells. J Immunol. 2007;178:2721–9.
Cui J, et al. Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science. 1997;278:1623–6.
Nakagawa R, et al. Treatment of hepatic metastasis of the colon26 adenocarcinoma with an alpha-galactosylceramide, KRN7000. Cancer Res. 1998;58:1202–7.
Giaccone G, et al. A phase I study of the natural killer T-cell ligand alpha-galactosylceramide (KRN7000) in patients with solid tumors. Clin Cancer Res. 2002;8:3702–9.
Schneiders FL, et al. Clinical experience with alpha-galactosylceramide (KRN7000) in patients with advanced cancer and chronic hepatitis B/C infection. Clin Immunol. 2011;140:130–41.
Burdin N, et al. Immunization with alpha-galactosylceramide polarizes CD1-reactive NK T cells towards Th2 cytokine synthesis. Eur J Immunol. 1999;29:2014–25.
Parekh VV, et al. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J Clin Invest. 2005;115:2572–83.
Sag D, et al. IL-10-producing NKT10 cells are a distinct regulatory invariant NKT cell subset. J Clin Invest. 2014;124:3725–40.
Huang Y, et al. Enhancement of HIV DNA vaccine immunogenicity by the NKT cell ligand, alpha-galactosylceramide. Vaccine. 2008;26:1807–16.
Gonzalez-Aseguinolaza G, et al. Natural killer T cell ligand alpha-galactosylceramide enhances protective immunity induced by malaria vaccines. J Exp Med. 2002;195:617–24.
Fernandez CS, et al. In-vivo stimulation of macaque natural killer T cells with alpha-galactosylceramide. Clin Exp Immunol. 2013;173:480–92.
Chang DH, et al. Sustained expansion of NKT cells and antigen-specific T cells after injection of alpha-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J Exp Med. 2005;201:1503–17.
Ishikawa A, et al. A phase I study of alpha-galactosylceramide (KRN7000)-pulsed dendritic cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res. 2005;11:1910–7.
Nieda M, et al. Therapeutic activation of Valpha24+ Vbeta11+ NKT cells in human subjects results in highly coordinated secondary activation of acquired and innate immunity. Blood. 2004;103:383–9.
Fujii S, et al. Prolonged IFN-gamma-producing NKT response induced with alpha-galactosylceramide-loaded DCs. Nat Immunol. 2002;3:867–74.
Toura I, et al. Cutting edge: inhibition of experimental tumor metastasis by dendritic cells pulsed with alpha-galactosylceramide. J Immunol. 1999;163:2387–91.
Moreno M, et al. IFN-gamma-producing human invariant NKT cells promote tumor-associated antigen-specific cytotoxic T cell responses. J Immunol. 2008;181:2446–54.
Shimizu K, et al. Vaccination with antigen-transfected, NKT cell ligand-loaded, human cells elicits robust in situ immune responses by dendritic cells. Cancer Res. 2013;73:62–73.
Petersen TR, et al. Exploiting the role of endogenous lymphoid-resident dendritic cells in the priming of NKT cells and CD8+ T cells to dendritic cell-based vaccines. PLoS One. 2011;6:e17657.
Shimizu K, et al. Cross-presentation of glycolipid from tumor cells loaded with alpha-galactosylceramide leads to potent and long-lived T cell mediated immunity via dendritic cells. J Exp Med. 2007;204:2641–53.
Liu K, et al. Innate NKT lymphocytes confer superior adaptive immunity via tumor-capturing dendritic cells. J Exp Med. 2005;202:1507–16.
Shimizu K, et al. Tumor cells loaded with alpha-galactosylceramide induce innate NKT and NK cell-dependent resistance to tumor implantation in mice. J Immunol. 2007;178:2853–61.
Chung Y, et al. An NKT-mediated autologous vaccine generates CD4 T-cell dependent potent antilymphoma immunity. Blood. 2007;110:2013–9.
Mattarollo SR, et al. NKT cell adjuvant-based tumor vaccine for treatment of myc oncogene-driven mouse B-cell lymphoma. Blood. 2012;120:3019–29.
Gibbins JD, et al. An autologous leukemia cell vaccine prevents murine acute leukemia relapse after cytarabine treatment. Blood. 2014;124:2953–63.
Hunn MK, et al. Vaccination with irradiated tumor cells pulsed with an adjuvant that stimulates NKT cells is an effective treatment for glioma. Clin Cancer Res. 2012;18:6446–59.
Neumann S, et al. Synthetic TRP2 long-peptide and alpha-galactosylceramide formulated into cationic liposomes elicit CD8(+) T-cell responses and prevent tumour progression. Vaccine. 2015;33:5838–44.
McKee SJ, et al. Virus-like particles and alpha-galactosylceramide form a self-adjuvanting composite particle that elicits anti-tumor responses. J Control Release. 2012;159:338–45.
Dolen Y, et al. Co-delivery of PLGA encapsulated invariant NKT cell agonist with antigenic protein induce strong T cell-mediated antitumor immune responses. Oncoimmunology. 2016;5:e1068493.
Singh M, et al. Direct incorporation of the NKT-cell activator alpha-galactosylceramide into a recombinant Listeria monocytogenes improves breast cancer vaccine efficacy. Br J Cancer. 2014;111:1945–54.
Venkataswamy MM, et al. Improving Mycobacterium bovis bacillus Calmette–Guerin as a vaccine delivery vector for viral antigens by incorporation of glycolipid activators of NKT cells. PLoS One. 2014;9:e108383.
Hailemichael Y, et al. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med. 2013;19:465–72.
Savage PB. Vaccine development: NKT-cell adjuvants in conjugate. Nat Chem Biol. 2014;10:882–3.
Flechtner JB, et al. High-affinity interactions between peptides and heat shock protein 70 augment CD8+ T lymphocyte immune responses. J Immunol. 2006;177:1017–27.
Anderson RJ, et al. A self-adjuvanting vaccine induces cytotoxic T lymphocytes that suppress allergy. Nat Chem Biol. 2014;10:943–9.
Anderson RJ, et al. NKT cell-dependent glycolipid-peptide vaccines with potent anti-tumour activity. Chem Sci. 2015;6:5120–7.
Cavallari M, et al. A semisynthetic carbohydrate-lipid vaccine that protects against S. pneumoniae in mice. Nat Chem Biol. 2014;10:950–6.
Vigneron N, et al. Database of T cell-defined human tumor antigens: the 2013 update. Cancer Immun. 2013;13:15.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74.
Galli G, et al. CD1d-restricted help to B cells by human invariant natural killer T lymphocytes. J Exp Med. 2003;197:1051–7.
Dellabona P, et al. iNKT-cell help to B cells: a cooperative job between innate and adaptive immune responses. Eur J Immunol. 2014;44:2230–7.
Chang PP, et al. Identification of Bcl-6-dependent follicular helper NKT cells that provide cognate help for B cell responses. Nat Immunol. 2012;13:35–43.
Barral P, et al. B cell receptor-mediated uptake of CD1d-restricted antigen augments antibody responses by recruiting invariant NKT cell help in vivo. Proc Natl Acad Sci USA. 2008;105:8345–50.
Ko SY, et al. alpha-Galactosylceramide can act as a nasal vaccine adjuvant inducing protective immune responses against viral infection and tumor. J Immunol. 2005;175:3309–17.
Lang GA, et al. Requirement for CD1d expression by B cells to stimulate NKT cell-enhanced antibody production. Blood. 2008;111:2158–62.
King IL, et al. Invariant natural killer T cells direct B cell responses to cognate lipid antigen in an IL-21-dependent manner. Nat Immunol. 2012;13:44–50.
Tonti E, et al. Follicular helper NKT cells induce limited B cell responses and germinal center formation in the absence of CD4(+) T cell help. J Immunol. 2012;188:3217–22.
Bai L, et al. Natural killer T (NKT)-B-cell interactions promote prolonged antibody responses and long-term memory to pneumococcal capsular polysaccharides. Proc Natl Acad Sci USA. 2013;110:16097–102.
Tonti E, et al. NKT-cell help to B lymphocytes can occur independently of cognate interaction. Blood. 2009;113:370–6.
Scanlon ST, et al. Airborne lipid antigens mobilize resident intravascular NKT cells to induce allergic airway inflammation. J Exp Med. 2011;208:2113–24.
Youn HJ, et al. A single intranasal immunization with inactivated influenza virus and alpha-galactosylceramide induces long-term protective immunity without redirecting antigen to the central nervous system. Vaccine. 2007;25:5189–98.
Kopecky-Bromberg SA, et al. Alpha-C-galactosylceramide as an adjuvant for a live attenuated influenza virus vaccine. Vaccine. 2009;27:3766–74.
Devera TS, et al. CD1d-dependent B-cell help by NK-like T cells leads to enhanced and sustained production of Bacillus anthracis lethal toxin-neutralizing antibodies. Infect Immun. 2010;78:1610–7.
Kamijuku H, et al. Mechanism of NKT cell activation by intranasal coadministration of alpha-galactosylceramide, which can induce cross-protection against influenza viruses. Mucosal Immunol. 2008;1:208–18.
Tefit JN, et al. Efficacy of ABX196, a new NKT agonist, in prophylactic human vaccination. Vaccine. 2014;32:6138–45.
Iyoda T, et al. Invariant NKT cell anergy is induced by a strong TCR-mediated signal plus co-stimulation. Int Immunol. 2010;22:905–13.
Wang J, et al. Cutting edge: CD28 engagement releases antigen-activated invariant NKT cells from the inhibitory effects of PD-1. J Immunol. 2009;182:6644–7.
Godfrey DI, Kronenberg M. Going both ways: immune regulation via CD1d-dependent NKT cells. J Clin Invest. 2004;114:1379–88.
Lehuen A, et al. Overexpression of natural killer T cells protects Valpha14- Jalpha281 transgenic nonobese diabetic mice against diabetes. J Exp Med. 1998;188:1831–9.
Hammond KJ, et al. alpha/beta-T cell receptor (TCR) + CD4-CD8- (NKT) thymocytes prevent insulin-dependent diabetes mellitus in nonobese diabetic (NOD)/Lt mice by the influence of interleukin (IL)-4 and/or IL-10. J Exp Med. 1998;187:1047–56.
Jahng AW, et al. Activation of natural killer T cells potentiates or prevents experimental autoimmune encephalomyelitis. J Exp Med. 2001;194:1789–99.
Mars LT, et al. Cutting edge: V alpha 14-J alpha 281 NKT cells naturally regulate experimental autoimmune encephalomyelitis in nonobese diabetic mice. J Immunol. 2002;168:6007–11.
Matarollo SR, et al. NKT cells inhibit antigen-specific effector CD8 T cell induction to skin viral proteins. J Immunol. 2011;187:1601–8.
McKee SJ, et al. Immunosuppressive roles of natural killer T (NKT) cells in the skin. J Leukoc Biol. 2014;96:49–54.
Mattarollo SR, et al. Invariant NKT cells in hyperplastic skin induce a local immune suppressive environment by IFN-gamma production. J Immunol. 2010;184:1242–50.
Sonoda KH, et al. CD1-reactive natural killer T cells are required for development of systemic tolerance through an immune-privileged site. J Exp Med. 1999;190:1215–26.
Araki M, et al. Synthetic glycolipid ligands for human iNKT cells as potential therapeutic agents for immunotherapy. Curr Med Chem. 2008;15:2337–45.
Guillaume J, et al. Synthesis of C-5″ and C-6″-modified alpha-GalCer analogues as iNKT-cell agonists. Bioorg Med Chem. 2015;23:3175–82.
Motohashi S, et al. A phase I-II study of alpha-galactosylceramide-pulsed IL-2/GM-CSF-cultured peripheral blood mononuclear cells in patients with advanced and recurrent non-small cell lung cancer. J Immunol. 2009;182:2492–501.
Nicol AJ, et al. Comparison of clinical and immunological effects of intravenous and intradermal administration of alpha-galactosylceramide (KRN7000)-pulsed dendritic cells. Clin Cancer Res. 2011;17:5140–51.
Yamasaki K, et al. Induction of NKT cell-specific immune responses in cancer tissues after NKT cell-targeted adoptive immunotherapy. Clin Immunol. 2011;138:255–65.
Artiaga BL, et al. alpha-Galactosylceramide protects swine against influenza infection when administered as a vaccine adjuvant. Sci Rep. 2016;6:23593.
Uchida T, et al. Phase I study of alpha-galactosylceramide-pulsed antigen presenting cells administration to the nasal submucosa in unresectable or recurrent head and neck cancer. Cancer Immunol Immunother. 2008;57:337–45.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
The writing of this manuscript was supported by Genesis Oncology Trust (NZ) (grant number GOT-1352-RPG to RW).
Conflict of interest
IH: Proprietary interest and member of management team of Avalia Immunotherapies. RW: Scientific Advisory Board member for Avalia Immunotherapies; part-time employee of the Malaghan Institute of Medical Research, which has an interest in Avalia Immunotherapies. MS: Employed by the Malaghan Institute of Medical Research, which has an interest in Avalia Immunotherapies.
Rights and permissions
About this article

Cite this article
Speir, M., Hermans, I.F. & Weinkove, R. Engaging Natural Killer T Cells as ‘Universal Helpers’ for Vaccination. Drugs 77, 1–15 (2017). https://doi.org/10.1007/s40265-016-0675-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-016-0675-z