
Abstract
Disturbed proteostasis as reflected by a massive accumulation of misfolded protein aggregates is a central feature in Alzheimer’s disease. Proteostatic disturbances may be caused by a shift in protein production and clearance. Whereas rare genetic causes of the disease affect the production side, sporadic cases appear to be directed by dysfunction in protein clearance. This review focusses on the involvement of lysosome-mediated clearance. Autophagy is a degradational system where intracellular components are degraded by lysosomal organelles. In addition, “outside-to-inside” trafficking through the endosomes converges with the autolysosomal pathway, thereby bringing together intracellular and extracellular components. Recent findings demonstrate that disturbance in the endo- and autolysosomal pathway induces “inside-to-outside” communication via induction of unconventional secretion, which may bear relevance to the spreading of disease pathology through the brain. The involvement of these pathways in the pathogenesis of the disease is discussed with an outlook to the opportunities it provides for diagnostics as well as therapeutic interventions.
Similar content being viewed by others

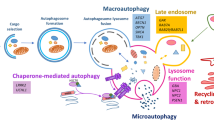
Endo- and autolysosomal impairment contributes to Alzheimer’s disease pathogenesis. |
Endo- and autolysosomal impairment affects the intracellular clearance of proteins as well as unconventional secretion to the extracellular space. |
Endo- and autolysosomal dysfunction provides opportunities for diagnostics and therapeutic intervention. |
1 Introduction
The key hallmark of Alzheimer’s disease (AD) and other neurodegenerative diseases is a severe disturbance in the protein homeostasis or proteostasis. This is reflected by the accumulation of misfolded proteins that are ultimately deposited in aggregates. In AD, two distinct types of inclusion bodies are found: extracellular plaques mainly comprising amyloid β (Aβ) aggregates, and intracellular tangles, of which the microtubule binding protein tau is the main constituent. Aβ and tau pathology together determine the pathological definition of AD, but how the two pathologies are connected is still largely unknown. The currently leading hypothesis in the field states that Aβ is the initiator of the AD pathogenic process and tau the executor [1, 2]. In strong support of the latter, the cognitive deficit of patients displays a strong correlation with the tangle load, originally extrapolated from post-mortem studies [3] and more recently confirmed by positron emission tomography (PET) studies in living patients [4]. The spatio–temporal distribution of the plaques and tangles follows a stereotypical pattern in the majority of patients with AD that differs for the two pathologies [3]. This spatial paradox illustrates that there is no linear mechanistic link between Aβ and tau pathology. Recently, accumulating data support a prion-like propagation and cell-to-cell spreading of tau pathology, in both cell and animal models (reviewed in Goedert and Spillantini [5]), indicating that tau pathology can self-propagate independent of Aβ. Indeed, tau aggregation by itself is sufficient to induce neurodegeneration in primary tauopathies, including familial frontotemporal dementias (FTD) that are directly associated with mutations in tau [6].
Familial variants of AD (FAD) typically enhance the formation of Aβ or its aggregation properties, supporting a role for Aβ early in the pathological cascade. FAD mutations are found in the amyloid precursor protein (APP) or the catalytic subunit of one of the APP-processing enzymes, presenilin (PSEN) 1 or 2 [7]. In contrast to these rare familial cases, impaired clearance of misfolded proteins and aggregates is considered to underlie the pathological protein accumulation of both Aβ and tau in sporadic AD, involving the large majority of AD cases [8]. In AD, in particular the endolysosomal and autolysosomal systems are impaired, which are involved in the degradation of both intracellular and extracellular material. In this concise review, we discuss the role of autolysosomal dysfunction in AD pathogenesis and how this can be exploited for diagnostic and therapeutic purposes.
2 Lysosomal Organelles: Where Intracellular Meets Extracellular: “Outside-In” and “Inside-Out”
Lysosomal degradation of proteins is a key aspect of maintaining proteostasis in the cell, which occurs in a heterogeneous group of hybrid organelles termed autolysosomes [9] or endolysosomes [10]. These organelles are morphologically similar and share molecular markers and thus represent a major crossroad of the endolysosomal and autolysosomal pathways in the cell (Fig. 1). The endolysosomal system regulates extracellular and organelle-contained cargo where extracellular components enter the cell by endo- or phagocytosis and are delivered to the early endosome. Early endosomes are also the major entry point of membrane-bound and luminal cargo of organelles that are delivered by membrane transport vesicles. These early endosomes mature to late endosomes, concentrating the cargo targeted for degradation in their lumen. This maturation coincides with several sorting and recycling events and acidification of the endosome lumen. Late endosomes fuse with lysosomes, which triggers the degradation of all luminal content by activated acid hydrolases. Cytosolic content can also be degraded by lysosomal organelles through different types of autophagy, the “self-eating” of the cell [11].

Convergence of endolysosomal and autolysosomal pathways. Molecules can enter the cell by endo- or phagocytosis and are delivered to the early endosome. Intracellular organelle-contained cargo is delivered to the early endosome though transport vesicles. On the maturing endosome membrane, vesicles bud inward to form intraluminal vesicles (ILVs), thereby sorting both membrane-bound endosome cargo and cytosolic microautophagy cargo for lysosomal degradation. Alternatively, cytosolic unfolded proteins can directly be delivered to the endosome/lysosome lumen by chaperone-mediated autophagy (CMA) translocation complexes. The late endosome or multivesicular body (MVB) will fuse with the lysosome to degrade all content. The macroautophagy pathway captures cytosolic proteins and organelles by the phagophore that closes to form an autophagosome. These autophagosomes also fuse with lysosomes, thus at the lysosome intracellular and extracellular protein degradation converge. Furthermore, MVBs, autolysosomes and other intermediates of these pathways can fuse with the plasma membrane to release their content, including ILVs that are then named exosomes (exo), to the extracellular space. Thereby these organelles mediate both “outside-to-inside” and “inside-to-outside” signaling
Autophagy comprises macroautophagy, chaperone-mediated autophagy (CMA) and microautophagy (Fig. 1). Macroautophagy is a multi-step process (see, for example, Bento et al. [12] for details on molecular steps) starting with the enclosure of substrate by a double lipid membrane. After formation of these autophagosomes, fusion with lysosomes results in generation of autolysosomes that degrade the autophagic substrates. Macroautophagy can be induced by inhibition of the mammalian target of rapamycin (mTOR; a serine/threonine kinase), for instance through amino acid starvation or rapamycin, but also by mTOR-independent activators. The entire autolysosomal pathway is affected by activation of the transcription factor EB (TFEB), a broad transcriptional master regulator of autophagy and autophagosome biogenesis (reviewed in Bajaj et al. [13] and Cortes and La Spada [14]). The cytosolic content enters autophagosomes largely on a stochastic basis, but increasing numbers of specific autophagy receptors are being identified as involved in selective autophagy and hence enrich for specific substrates [15]. Microautophagy involves the invagination of the endosomal membrane by a series of ESCRT complexes to generate intraluminal vesicles (ILVs; Fig. 1), thereby capturing cytosolic and membrane-bound molecules [16]. As ILVs accumulate in the maturing endosome, these organelles are also called multivesicular bodies (MVBs) due to their distinct morphology [17]. The biogenesis of ILVs comprises another crossroad of the endo- and autolysosomal pathway where intracellular cytosolic content and membrane-bound cargo from organelles and the plasma membrane are sorted together to lysosomal degradation (Fig. 1). CMA involves a sequence-directed targeting of unfolded cytosolic proteins to the lysosome by the chaperone HSC70. These proteins do not require membrane traffic but can be directly delivered to the lumen of autolysosomal organelles via a translocation complex that also comprises LAMP2 [18]. There is considerable crosstalk between CMA and microautophagy; for example, HSC70 can also target components to microautophagy that depends on ESCRT complexes I and III [19, 20]. The different intermediates in the endo- and autolysosomal pathways provide an “outside-to-inside” connection where intracellular and extracellular proteins meet (Fig. 1). Hence the collective endocytic and autophagic pathways can mediate the degradation of internalized soluble molecules, membrane components and soluble cytosolic content by convergence on lysosomal degradation.
Apart from the established role of endo- and autolysosomal compartments in degradation, an exciting new field of research focusses on their role in secretion. In the conventional secretory pathway, cargo produced in the endoplasmic reticulum (ER) passes through the Golgi before trafficking to the plasma membrane for release. Unconventional secretory pathways bypass the Golgi and typically employ the autophagic and endolysosomal compartments. Fusion of vesicles derived from the endo-autolysosomal system with the plasma membrane was first described for secretory lysosomes, where lysosomal catabolic enzymes can be released into the extracellular space. More recently it has been demonstrated that cytosolic cargo can also be released via unconventional secretion [21], indicating that endo-autolysosomal organelles are not only important for “outside-to-inside” transport but also mediate “inside-to-outside” signaling. When MVBs fuse with the plasma membrane, the ILVs are also released to the extracellular space and are then called “exosomes”. Exosomes are a subclass of the wider family of extracellular vesicles that also comprise plasma membrane-formed microvesicles and larger membrane structures [22]. Exosomes carry a wide variety of cytosolic proteins and microRNAs (miRNAs) and can be purified from many tissues, including blood, urine, and cerebrospinal fluid. As their content is protected in the extracellular milieu by the exosomal membrane, these vesicles can facilitate spreading of pathological proteins and RNAs to neighboring cells. In addition, the improved exosome-purification protocols provide a powerful tool for biomarker detection from fluid biopsies.
3 Dysfunctional Endo-Autolysosomal System in Alzheimer’s Disease—Cause and Effect
The extreme polarity and limited regenerative capacity of neurons demand strong functional autophagy to clear long-lived proteins, cell organelles, and aggregates to ensure cell survival. The unique spatial architecture of neurons results in cell type-specific aspects regarding biogenesis, fusion, and maturation of the different intermediates in the pathway [23, 24]. Impairment of autophagy has been observed in several neurodegenerative diseases. In AD and in primary tauopathies, the extensive accumulation of endo- and auto-lysosomal structures is a pronounced feature [25, 26]. This involves accumulation of aberrant intermediates both in dystrophic neurites that surround plaques [27] and in the somata, where it presents as granulo-vacuolar-degeneration bodies (GVBs) [25]. The latter carry signatures of cellular stress [28]. Recently, it was shown that the removal of defective mitochondria via a type of selective autophagy (“mitophagy”) is also impaired in the AD brain and neurons derived from patient induced pluripotent stem cells (iPSCs) [29]. The strong association between disturbed autophagy and AD may indicate that it contributes to the accumulation of Aβ and tau.
Indeed, mice with a brain-specific deletion of general autophagy factors such as ATG7 or TFEB accumulate phosphorylated tau [30] and secrete and accumulate more Aβ [31, 32]. Defective lysosomal clearance hence contributes to accumulation of pathological Aβ and tau species and thereby to plaque and tangle formation in AD. BACE1, which together with PSEN is responsible for the formation of Aβ, is degraded by autophagy which presents another way in which autophagic dysfunction will result in higher Aβ levels [33, 34]. APP is chaperoned by SORL1 through the endolysosomal system and recycles back to the plasma membrane [35]. Prolonged retention of APP in BACE1-positive endosomes results in increased Aβ production, whereas stimulation of APP exit from the endolysosomal system decreases Aβ levels, indicating that endolysosomal structures are a key location for Aβ generation [36, 37]. Furthermore, APP binds to HSC70 and contains an ESCRT-recognition motif, linking its processing directly to CMA and microautophagy [38]. Interestingly, apart from effects on the formation of Aβ, FAD mutations in PSEN1 are associated with impairment of lysosomal function for which effects on the pH, calcium homeostasis, and regulation of auto-lysosomal signaling have been reported [32, 39,40,41]. In addition, both disturbed Aβ metabolism and tau pathology are observed in different lysosomal storage diseases (LSDs), indicating another direct connection between the lysosome dysfunction and AD pathology [42,43,44,45].
In turn, both Aβ peptide [46,47,48] and pathogenic tau [49], have been demonstrated to induce lysosomal dysfunction. In brains of patients with FAD or primary tauopathies, cathepsin D is observed localized in the cytosol, indicating that the lysosomal integrity is impaired [26]. Tau was also shown to directly impair mitophagy by sequestering parkin, a ubiquitin ligase involved in the initiation of the clearance of mitochondria by autophagy [50].
Interestingly, it has been shown that cells with impaired lysosomal function activate unconventional secretory routes to remove misfolded proteins, including tau and Aβ [51,52,53,54]. This is, for instance, observed in Neu1 null mutant mice, which show enhanced Aβ secretion through lysosomal exocytosis [55]. This may relieve the protein load inside the cell while contributing to the cell-to-cell transmission of pathology, which is the prevailing hypothesis underlying AD progression.
The connection between AD pathology and endo-autolysosomal dysfunction therefore appears to comprise a vicious cycle: endo-autolysosomal dysfunction contributes to the accumulation of aberrant Aβ and tau species, which in turn contribute to dysfunction of auto- and endolysosome pathways.
4 Possibilities for Diagnosis
Diagnostic tools for neurodegenerative disease such as AD are limited by the long presymptomatic development of the disease and the fact that the central nervous system is poorly accessible for taking samples for diagnostic testing. Noninvasive imaging of Aβ and tau pathology in living patients has been considerably improved over recent years by the development of new tracers [56]. Although these tools render detailed images of specific protein accumulations, they do not provide information on the cause of the proteostatic dysregulation that underlies the pathology, which may differ between patients with AD. Such information is essential for effective therapeutic intervention.
The inside-to-outside signaling of endo-autolysosomal organelles can be utilized to obtain information on intracellular protein homeostasis. Exosomes contain to a large extent a protein and miRNA footprint of the cell from which they were released [22]. Small fractions of the secreted Aβ and phosphorylated tau are carried by exosomes [57,58,59]. Different miRNA levels have been detected in exosome samples of patients with AD from blood [60, 61] and cerebrospinal fluid (CSF) [62]. Furthermore, miRNA profiles from CSF exosome fractions differentiated patients with AD and those with Parkinson’s disease, indicating they could be used for diagnostic purposes [63]. L1CAM-immunopurified exosome preparations from patient serum samples indicated different protein profiles between AD and FTD [64], suggesting that exosome profiling could assist in differential diagnosis. Interestingly, patients with mild cognitive impairment that remained stable versus patients who developed AD showed distinct exosome profiles [65] indicating potential to serve as a prognostic marker. Furthermore, differences in synaptic proteins and autolysosomal markers (including cathepsin D and HSC70) have been demonstrated in these blood-derived exosome fractions of patients with AD and are already present in individuals before the symptomatic onset of AD [66, 67]. The extent to which these markers can be utilized as diagnostics tools of presymptomatic AD warrants further replication and exploration. If confirmed, such biomarkers will be of major importance for the inclusion of patients in clinical trials for drugs that target disease mechanisms involving the endo-autolysosomal system as well as efficacy monitoring of the treatment.
5 Therapeutic Opportunities
To date, no therapy is available to halt the progression of AD, and therapeutic options are limited to the treatment of behavioral and cognitive symptoms. Given the impairment of autophagy in AD and its intricate connection to the pathogenesis, stimulation of the pathway it is an attractive target that may ameliorate pathological effects [68].
Here, a great deal can be learned from studies on LSDs, neurological diseases where the lysosomal defect is the direct cause of the disease. Specific mutations in lysosomal genes provide an obvious target for restoration of the defect, and enzyme-replacement therapies are actively pursued for many of these disorders (see Marques and Saftig [69] and Beck [70] for details). Approaches using direct replacement with recombinant protein are faced with hurdles that are also relevant for AD: Targeting across the blood–brain barrier and side effects caused by the extracellular presence of recombinant proteins. Alternatively, viral delivery using specific adeno-associated virus (AAV0 serotypes is employed to deliver a copy of the intact gene to brain cells [71]. In this case, the mutant gene product is still present. Current developments in gene-editing technology, particularly in increasing the safety and efficiency of Crispr/Cas-mediated methods [72, 73] are a promising future direction in combination with appropriate viral delivery methods. For AD, the connection with lysosomal dysfunction cannot be directly connected to a single gene defect, and the single enzyme-replacement approach does not address the problem. Nevertheless, in preclinical studies, proof of concept has been obtained that modulation of the autolysosomal pathway ameliorates the pathology as well as the downstream neurodegeneration. Intervention at different steps of the pathway has been studied, from the initiation of phagophore formation to increasing the proteolytic capacity of the autolysosomes. Stimulation of autophagy by genetic reduction of mTOR [74], as well as by treatment with rapamycin [75], can reduce Aβ plaque load as well as behavioral deficits in mouse models. Also, sarkosyl-insoluble tau and behavioral deficits are reduced by rapamycin treatment in tauopathy mouse models [75, 76]. Rapamycin affects the earliest steps of autophagy via phosphoinositide 3 kinase (PI3K)/mTOR and has pleiotropic effects because of the broad spectrum of downstream effectors of these kinases. A cell-permeable peptide consisting of a fragment of Beclin 1 induces autophagy and clears protein aggregates downstream of mTOR [77]. This is a very interesting and specific approach to activate autophagy independent of mTOR but has currently only been demonstrated in cultured cells. Recently, inhibition of the small GTPase Rhes was shown to reduce tau pathology and neurodegeneration in a mouse model by inducing autophagy [78]. Inhibition of Rhes was accomplished using a farnesyltransferase inhibitor, lonafarnib, which will also affect the function of other farnesylated proteins. In addition, lonafarnib was only protective in the early stages of tau pathology, which limits its clinical application. Targeting the autophagy pathway in an mTOR-independent manner is potentially interesting, but more study is required to enable this approach for drug development for AD. Overexpression of TFEB reduces pathology and degeneration in models for tau as well as Aβ pathology [49, 79, 80]. Studies into the role of TFEB to date have been mainly limited to disease models, although a recent transcriptomics study indicated increased expression of TFEB and TFEB targets in the AD hippocampus [81]. The natural compound trehalose can be used to induce autophagy in an mTOR-independent manner via activation of TFEB. Trehalose treatment indeed reduced tau pathology in a mouse model [82, 83]. Trehalose has also been protective in amyloid mouse models, although this could not always be attributed to effects on autophagy [84,85,86].
This may relate to the mechanism by which trehalose leads to TFEB induction, which has only recently been discovered. Trehalose treatment was shown to cause a slight impairment of lysosomal function that induced TFEB [87]. This may strongly limit its potential for clinical use. It will be interesting to investigate other candidate drugs that enhance TFEB signaling. One of these is the commonly used over-the-counter drug aspirin, which reduced Aβ plaque load in a mouse model in a TFEB/peroxisome proliferator-activated receptor α (PPARα)-dependent manner [88].
The more global lysosomal targeting by stimulation of the TFEB response is also being investigated for the treatment of LSDs and has been shown to reduce the accumulation of undegraded cargo and ameliorate the clinical phenotype in a mouse model for Pompe disease [89, 90]. The removal of intracellular undegraded material is (partly) attributed to secretion of lysosomal content to the extracellular space [89, 91]. Indeed, TFEB has been shown to induce lysosomal secretion [92]. This is extremely relevant in view of employing TFEB as a target in AD, because the secreted material may further exacerbate the neuroinflammatory response, which is a major contributor to neurodegeneration. In addition, the potential release of pathological Aβ and tau species may further ameliorate the neurodegeneration by direct extracellular toxicity of oligomeric species. Moreover, the released Aβ and tau species could serve as “seeds” that facilitate extracellular Aβ intracellular tau pathology.
It is good to note that pleiotropic effects may also provide additional benefit. Recently, deletion of the broad effector kinase p38α mitogen-activated protein kinase (MAPK) was used to enhance autophagy and lysosomal degradation in an Aβ mouse model and shown to reduce plaque pathology by reducing the levels of BACE1 [34]. In addition to this effect on neuronal autophagy, MAPK inhibitors also inhibit the pro-inflammatory microglial response, which is a secondary pathogenic contributor [93]. It is important to note that the positive effects of compounds on lysosomal clearance may be partly mediated by glia cells via uptake of extracellular pathological proteins [94, 95]. Microglia have been implicated in the spreading of tau pathology [96] and as such have been termed “tau distributors” [97]. Inhibition of exosome release from microglia inhibits spreading of neuronal tau pathology in a mouse model, directly pointing to the involvement of the glial endolysosomal system in the spreading of disease and hence a target to halt progression of the disease.
In addition to these “general” activators of the pathway, more selective molecular targeting of autophagy induction is being investigated (e.g., via Beclin1 [88]). Alternatively, more specifically, increasing mitophagy by urolithin A or actinonin was shown to also ameliorate both Aβ and tau burden in worms and mice [29, 98]. However, for therapeutic targeting in AD, the initiation of autophagy may not be the target of choice. The accumulation of different autophagy intermediates in the AD brain indicates that the induction of autophagy is not the primary problem but rather a disturbance in the flux in the pathway. In addition, treatments such as rapamycin are not effective against preexisting plaques and tangles [99]. Intervention further downstream in the pathway at the level of lysosomal proteolytic capacity can be achieved by increasing the levels of lysosomal enzymes. Overexpression of glucocerebrosidase was demonstrated to ameliorate the toxicity of Aβ oligomers [100]. Another way to increase lysosomal proteolysis is by reducing the levels of cystatins, the endogenous inhibitors of the lysosomal cathepsins. This has been demonstrated to decrease the Aβ levels in wild-type APP mice [101] as well as the pathology and neurodegeneration in a mouse model for Aβ plaque pathology [47].
The primary cause of impairment in the autophagy or endolysosomal pathway is an important unresolved issue that is essential for effective targeting. It is possible that the overload of aberrant proteins overwhelms the degradational capacity. This seems unlikely, as lysosomal accumulation of tau and Aβ is not an outstanding feature of the AD brain. In fact, cathepsin D levels are increased selectively in correlation with tau pathology in the AD brain, suggestive of a response to increased lysosomal degradative capacity to the accumulating pathology [102]. The lack of effectiveness of this response, despite the higher proteolytic enzyme levels, may indicate that the delivery of specific cargo is hampered in AD, thereby decreasing the degradation of tau and Aβ. Therefore, the study of specific autophagy receptors may identify a more selective target. In fact, although still in its infancy, specificity of cargo receptors for different tau species has been reported: NDP52 targets phosphorylated tau [103], OPTN physiological tau, whereas SQSTM1 selectively targets aggregated tau [104]. Alternatively, the lysosomal dysfunction could point to a problem with the trafficking and/or fusion of the autophagy organelles. This is supported by the accumulation of GVBs with increasing tau pathology [105]. These relatively enigmatic organelles are immune-positive for endo- and autolysosomal markers and are therefore considered as accumulated endo-autolysosomal intermediates [106]. More knowledge on the precise origin of GVBs will provide crucial clues on what is key to the deficit in the pathway in AD.
6 Concluding Remarks
Dysfunction of endo-autolysosome systems result in accumulation of pathological Aβ and tau and vice versa, comprising a vicious cycle in AD pathogenesis. In particular, the flux in the pathways appears severely disturbed. Enhancing the flux through the lysosomal pathway can directly reduce Aβ and tau levels, but also potentially reduces the release of pathological Aβ and tau species via unconventional secretion. According to the spreading hypothesis, reducing the amount of available seeds would inhibit the progression of pathology and neurodegeneration. This concept is employed in the various vaccination strategies for Aβ and tau where proof of concept has been obtained in preclinical studies and are currently in several clinical trials. The poor blood–brain barrier passage of antibodies is a major obstacle in such approaches. Alternatively, inhibiting seed release by stimulation of autophagy provides a potentially more druggable target as it is more readily targeted using small molecules. Importantly, when targeting Aß or tau by immunotherapy, it is good to be aware that some antibody approaches may require an intact endo-autolysosomal system [107,108,109].
In conclusion, stimulation of endo-autolysosome systems may be beneficial for AD, not only in the clearance of both intracellular and extracellular aggregates but also in the prevention of seed secretion. The unique position of these organelles at the intersection of intra- and extracellular proteostatic pathways therefore opens an increasingly attractive avenue for therapeutic intervention.
References
Scheltens P, Blennow K, Breteler MMB, de Strooper B, Frisoni GB, Salloway S, et al. Alzheimer’s disease. Lancet (Elsevier Ltd.). 2016;388:505–17.
Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and “wingmen”. Nat Neurosci. 2015;18:800–6.
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59.
Jack CR, Wiste HJ, Schwarz CG, Lowe VJ, Senjem ML, Vemuri P, et al. Longitudinal tau PET in ageing and Alzheimer’s disease. Brain. 2018;141:1517–28.
Goedert M, Spillantini MG. Propagation of tau aggregates. Mol Brain. 2017;10:18.
Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12:609–22.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med (Wiley-Blackwell). 2016;8:595–608.
Baranello RJ, Bharani KL, Padmaraju V, Chopra N, Lahiri DK, Greig NH, et al. Amyloid-beta protein clearance and degradation (ABCD) pathways and their role in Alzheimer’s disease. Curr Alzheimer Res. 2015;12:32–46.
Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811–36.
Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30:3481–500.
Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol. 1992;119:301–11.
Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, Vicinanza M, et al. Mammalian autophagy: how does it work? Annu Rev Biochem. 2016;85:685–713.
Bajaj L, Lotfi P, Pal R, di Ronza A, Sharma J, Sardiello M. Lysosome biogenesis in health and disease. J Neurochem. 2019;148:573–89.
Cortes CJ, La Spada AR. TFEB dysregulation as a driver of autophagy dysfunction in neurodegenerative disease: molecular mechanisms, cellular processes, and emerging therapeutic opportunities. Neurobiol Dis. 2019;122:83–93.
Rogov V, Dötsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014;53:167–78.
Hurley JH, Emr SD. The ESCRT complexes: structure and mechanism of a membrane-trafficking network. Annu Rev Biophys Biomol Struct. 2006;35:277–98.
Von Bartheld CS, Altick AL. Multivesicular bodies in neurons: distribution, protein content, and trafficking functions. Prog Neurobiol. 2011;93:313–40.
Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–3.
Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. 2011;20:131–9.
Uytterhoeven V, Lauwers E, Maes I, Miskiewicz K, Melo MN, Swerts J, et al. Hsc70-4 deforms membranes to promote synaptic protein turnover by endosomal microautophagy. Neuron. 2015;88:735–48.
Kim J, Gee HY, Lee MG. Unconventional protein secretion—new insights into the pathogenesis and therapeutic targets of human diseases. J Cell Sci. 2018;131:213686.
van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213–28.
Ferguson SM. Neuronal lysosomes. Neurosci Lett. 2018;697:1–9.
Ferguson SM. Axonal transport and maturation of lysosomes. Curr Opin Neurobiol. 2018;51:45–51.
Köhler C. Granulovacuolar degeneration: a neurodegenerative change that accompanies tau pathology. Acta Neuropathol. 2016;132:339–59.
Piras A, Collin L, Grüninger F, Graff C, Rönnbäck A. Autophagic and lysosomal defects in human tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol Commun. 2016;4:22.
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22.
Scheper W, Hoozemans JJM. The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol. 2015;130:315–31.
Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019;22:401–12.
Norambuena A, Wallrabe H, Cao R, Wang DB, Silva A, Svindrych Z, et al. A novel lysosome-to-mitochondria signaling pathway disrupted by amyloid-β oligomers. EMBO J. 2018;37:e100241.
Nilsson P, Sekiguchi M, Akagi T, Izumi S, Komori T, Hui K, et al. Autophagy-related protein 7 deficiency in amyloid β (Aβ) precursor protein transgenic mice decreases Aβ in the multivesicular bodies and induces Aβ accumulation in the golgi. Am J Pathol. 2014;185:305–13.
Reddy K, Cusack CL, Nnah IC, Khayati K, Saqcena C, Huynh TB, et al. Dysregulation of nutrient sensing and CLEARance in presenilin deficiency. Cell Rep. 2016;14:2166–79.
Saido TC, Taniguchi N, Manya H, Saito T, Kizuka Y, Murayama S, et al. An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer’s disease. EMBO Mol Med. 2015;7:175–89.
Schnöder L, Hao W, Qin Y, Liu S, Tomic I, Liu X, et al. Deficiency of neuronal p38α MAPK attenuates amyloid pathology in alzheimer disease mouse and cell models through facilitating lysosomal degradation of BACE1. J Biol Chem. 2016;291:2067–79.
Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–77.
Bhalla A, Vetanovetz CP, Morel E, Chamoun Z, Di Paolo G, Small SA. The location and trafficking routes of the neuronal retromer and its role in amyloid precursor protein transport. Neurobiol Dis. 2012;47:126–34.
Mecozzi VJ, Berman DE, Simoes S, Vetanovetz C, Awal MR, Patel VM, et al. Pharmacological chaperones stabilize retromer to limit APP processing. Nat Chem Biol. 2014;10:443–9.
Park JS, Kim DH, Yoon SY. Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep. 2016;49:337–42.
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–58.
Coen K, Flannagan RS, Baron S, Carraro-Lacroix LR, Wang D, Vermeire W, et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J Cell Biol. 2012;198:23–35.
Lee J-H, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y, et al. Presenilin 1 maintains lysosomal Ca2 + homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep. 2015;12:1430–44.
Keilani S, Lun Y, Stevens AC, Williams HN, Sjoberg ER, Khanna R, et al. Lysosomal dysfunction in a mouse model of sandhoff disease leads to accumulation of ganglioside-bound amyloid- peptide. J Neurosci. 2012;32:5223–36.
Ginsberg SD, Galvin JE, Lee VM, Rorke LB, Dickson DW, Wolfe JH, et al. Accumulation of intracellular amyloid-beta peptide (A beta 1-40) in mucopolysaccharidosis brains. J Neuropathol Exp Neurol. 1999;58:815–24.
Ohmi K, Kudo LC, Ryazantsev S, Zhao H-Z, Karsten SL, Neufeld EF. Sanfilippo syndrome type B, a lysosomal storage disease, is also a tauopathy. Proc Natl Acad Sci. 2009;106:8332–7.
Malnar M, Hecimovic S, Mattsson N, Zetterberg H. Bidirectional links between Alzheimer’s disease and Niemann–Pick type C disease. Neurobiol Dis. 2014;72:37–47.
Kaminari A, Hoozemans JJM, Rutjes H, Scheper W, De Kimpe L, Zwart R, et al. Intracellular accumulation of aggregated pyroglutamate amyloid beta: convergence of aging and Aβ pathology at the lysosome. Age. 2012;35:673–87.
Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–77.
Lafay-Chebassier C, Paccalin M, Page G, Barc-Pain S, Perault-Pochat MC, Gil R, et al. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J Neurochem. 2005;94:215–25.
Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM, et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009;18:4153–70.
Cummins N, Tweedie A, Zuryn S, Bertran-Gonzalez J, Götz J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019;38:e99360.
Mohamed N-V, Plouffe V, Rémillard-Labrosse G, Planel E, Leclerc N. Starvation and inhibition of lysosomal function increased tau secretion by primary cortical neurons. Sci Rep. 2015;4:5715.
Lee J-G, Takahama S, Zhang G, Tomarev SI, Ye Y. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat Cell Biol. 2016;18:765–76.
Fontaine SN, Zheng D, Sabbagh JJ, Martin MD, Chaput D, Darling A, et al. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 2016;35:1537–49.
Nilsson P, Iwata N, Tsubuki S, Sekiguchi M, Matsuba Y, Saito T, et al. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013;5:61–9.
Annunziata I, Patterson A, Helton D, Hu H, Moshiach S, Gomero E, et al. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-β secretion via deregulated lysosomal exocytosis. Nat Commun. 2013;4:2734.
Ossenkoppele R, Smith R, Ohlsson T, Strandberg O, Mattsson N, Insel PS, et al. Associations between tau, Aβ, and cortical thickness with cognition in Alzheimer disease. Neurology. 2019;92:e601–12.
Chiasserini D, Van Weering JRT, Piersma SR, Pham TV, Malekzadeh A, Teunissen CE, et al. Proteomic analysis of cerebrospinal fluid extracellular vesicles: a comprehensive dataset. J Proteom. 2014;106:191–204.
Saman S, Kim WH, Raya M, Visnick Y, Miro S, Saman S, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. 2012;287:3842–9.
Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, et al. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci. 2006;103:11172–7.
Lugli G, Cohen AM, Bennett DA, Shah RC, Fields CJ, Hernandez AG, et al. Plasma exosomal miRNAs in persons with and without alzheimer disease: altered expression and prospects for biomarkers. PLoS One. 2015;10:e0139233.
Cheng L, Doecke JD, Sharples RA, Villemagne VL, Fowler CJ, Rembach A, et al. Prognostic serum miRNA biomarkers associated with Alzheimer’s disease shows concordance with neuropsychological and neuroimaging assessment. Mol Psychiatry. 2015;20:1188–96. https://doi.org/10.1038/mp.2014.127.
Boxer AL, Karydas A, Tartaglia MC, Weichert A, Schneider R, McKeever PM, et al. microRNA expression levels are altered in the cerebrospinal fluid of patients with young-onset Alzheimer’s disease. Mol Neurobiol. 2018;55:8826–41.
Gui Y, Liu H, Zhang L, Lv W, Hu X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget. 2015;6:37043–53.
Fiandaca MS, Kapogiannis D, Mapstone M, Boxer A, Eitan E, Schwartz JB, et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: a case–control study. Alzheimer’s Dement. 2015;11(600–607):e1.
Winston CN, Goetzl EJ, Akers JC, Carter BS, Rockenstein EM, Galasko D, et al. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimer’s Dement. 2016;3:63–72.
Goetzl EJ, Kapogiannis D, Schwartz JB, Lobach IV, Goetzl L, Abner EL, et al. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 2016;30:4141–8.
Goetzl EJ, Boxer A, Schwartz JB, Abner EL, Petersen RC, Miller BL, et al. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology. 2015;85:40–7.
Boland B, Yu WH, Corti O, Mollereau B, Henriques A, Bezard E, et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2018;17:660–88.
Marques ARA, Saftig P. Lysosomal storage disorders—challenges, concepts and avenues for therapy: beyond rare diseases. J Cell Sci. 2019;132:221–739.
Beck M. Treatment strategies for lysosomal storage disorders. Dev Med Child Neurol. 2018;60:13–8.
Tordo J, O’Leary C, Antunes ASLM, Palomar N, Aldrin-Kirk P, Basche M, et al. A novel adeno-associated virus capsid with enhanced neurotropism corrects a lysosomal transmembrane enzyme deficiency. Brain. 2018;141:2014–31.
Schaefer KA, Wu W-H, Colgan DF, Tsang SH, Bassuk AG, Mahajan VB. Unexpected mutations after CRISPR–Cas9 editing in vivo. Nat Methods. 2017;14:547–8.
Schneller JL, Lee CM, Bao G, Venditti CP. Genome editing for inborn errors of metabolism: advancing towards the clinic. BMC Med. 2017;15:43.
Caccamo A, De Pinto V, Messina A, Branca C, Oddo S. Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J Neurosci. 2014;34:7988–98.
Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One. 2010;5:e9979.
Ozcelik S, Fraser G, Castets P, Schaeffer V, Skachokova Z, Breu K, et al. Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PLoS One. 2013;8:2–8.
Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494:201–6.
Hernandez I, Luna G, Rauch JN, Reis SA, Giroux M, Karch CM, et al. A farnesyltransferase inhibitor activates lysosomes and reduces tau pathology in mice with tauopathy. Sci Transl Med. 2019;11:eaat3005.
Polito VA, Li H, Martini-Stoica H, Wang B, Yang L, Xu Y, et al. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med. 2014;6:1142–60.
Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, et al. Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. J Neurosci. 2014;34:9607–20.
Bordi M, Berg MJ, Mohan PS, Peterhoff CM, Alldred MJ, Che S, et al. Autophagy flux in CA1 neurons of Alzheimer hippocampus: increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy. 2016;12:2467–83.
Schaeffer V, Lavenir I, Ozcelik S, Tolnay M, Winkler DT, Goedert M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain. 2012;135:2169–77.
Krüger U, Wang Y, Kumar S, Mandelkow E-M. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging. 2012;33:2291–305.
Tien NT, Karaca I, Tamboli IY, Walter J. Trehalose alters subcellular trafficking and the metabolism of the Alzheimer-associated amyloid precursor protein. J Biol Chem. 2016;291:10528–40.
Portbury SD, Hare DJ, Sgambelloni C, Perronnes K, Portbury AJ, Finkelstein DI, et al. Trehalose improves cognition in the transgenic Tg2576 mouse model of alzheimer’s disease. J Alzheimers Dis. 2017;60:549–60.
Du J, Liang Y, Xu F, Sun B, Wang Z. Trehalose rescues Alzheimer’s disease phenotypes in APP/PS1 transgenic mice. J Pharm Pharmacol. 2013;65:1753–6.
Rusmini P, Cortese K, Crippa V, Cristofani R, Cicardi ME, Ferrari V, et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy. 2018;15:1–21.
Chandra S, Jana M, Pahan K. Aspirin induces lysosomal biogenesis and attenuates amyloid plaque pathology in a mouse model of Alzheimer’s disease via PPARα. J Neurosci. 2018;38:6682–99.
Spampanato C, Feeney E, Li L, Cardone M, Lim J-A, Annunziata F, et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med. 2013;5:691–706.
Gatto F, Rossi B, Tarallo A, Polishchuk E, Polishchuk R, Carrella A, et al. AAV-mediated transcription factor EB (TFEB) gene delivery ameliorates muscle pathology and function in the murine model of Pompe disease. Sci Rep. 2017;7:15089.
Feeney EJ, Spampanato C, Puertollano R, Ballabio A, Parenti G, Raben N. What else is in store for autophagy? Exocytosis of autolysosomes as a mechanism of TFEB-mediated cellular clearance in Pompe disease. Autophagy. 2013;9:1117–8.
Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011;21:421–30.
Alam J, Scheper W. Targeting neuronal MAPK14/p38α activity to modulate autophagy in the Alzheimer disease brain. Autophagy. 2016;12:2516–20.
Pomilio C, Pavia P, Gorojod RM, Vinuesa A, Alaimo A, Galvan V, et al. Glial alterations from early to late stages in a model of Alzheimer’s disease: evidence of autophagy involvement in Aβ internalization. Hippocampus. 2016;26:194–210.
Shibuya Y, Chang CCY, Huang L-H, Bryleva EY, Chang T-Y. Inhibiting ACAT1/SOAT1 in microglia stimulates autophagy-mediated lysosomal proteolysis and increases Aβ1-42 clearance. J Neurosci. 2014;34:14484–501.
Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 2015;138:1738–55.
Microglia Carr F. Tau distributors. Nat Rev Neurosci. 2015;16:702.
Fatouros C, Pir GJ, Biernat J, Koushika SP, Mandelkow E, Mandelkow E-M, et al. Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Hum Mol Genet. 2012;21:3587–603.
Majumder S, Richardson A, Strong R, Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One. 2011;6:e25416.
Choi S, Kim D, Kam T-I, Yun S, Kim S, Park H, et al. Lysosomal enzyme glucocerebrosidase protects against Aβ1-42 oligomer-induced neurotoxicity. PLoS One. 2015;10:e0143854.
Wang C, Sun B, Zhou Y, Grubb A, Gan L. Cathepsin B degrades amyloid-β in mice expressing wild-type human amyloid precursor protein. J Biol Chem. 2012;287:39834–41.
Chai YL, Chong JR, Weng J, Howlett D, Halsey A, Lee JH, et al. Lysosomal cathepsin D is upregulated in Alzheimer’s disease neocortex and may be a marker for neurofibrillary degeneration. Brain Pathol. 2019;29:63–74.
Jo C, Gundemir S, Pritchard S, Jin YN, Rahman I, Johnson GVW. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun. 2014;5:3496.
Xu Y, Zhang S, Zheng H. The cargo receptor SQSTM1 ameliorates neurofibrillary tangle pathology and spreading through selective targeting of pathological MAPT (microtubule associated protein tau). Autophagy. 2018;15:1–16.
Thal DR, Del Tredici K, Ludolph AC, Hoozemans JJM, Rozemuller AJ, Braak H, et al. Stages of granulovacuolar degeneration: their relation to Alzheimer’s disease and chronic stress response. Acta Neuropathol. 2011;122:577–89.
Funk KE, Mrak RE, Kuret J. Granulovacuolar degeneration (GVD) bodies of Alzheimer’s disease (AD) resemble late-stage autophagic organelles. Neuropathol Appl Neurobiol. 2011;37:295–306.
Collin L, Bohrmann B, Göpfert U, Oroszlan-Szovik K, Ozmen L, Grüninger F. Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer‘s disease. Brain. 2014;137:2834–46.
Krishnamurthy PK, Deng Y, Sigurdsson EM. Mechanistic studies of antibody-mediated clearance of tau aggregates using an ex vivo brain slice model. Front Psychiatry. 2011;2:59.
Wang H-C, Zhang T, Kuerban B, Jin Y-L, Le W, Hara H, et al. Autophagy is involved in oral rAAV/Aβ vaccine-induced Aβ clearance in APP/PS1 transgenic mice. Neurosci Bull. 2015;31:491–504.
Acknowledgements
The authors are grateful to all the scientists whose excellent work contributes to the broader landscape of the topic of this review, and we apologize that we were unable to cite all contributions due to space restrictions. We thank the members of the Van Weering and Scheper labs for stimulating discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Opens access was paid by Vrije Universiteit Amsterdam, The Netherlands. JvW has received research funding from Alzheimer Nederland (WE.03-2016-05), Alzheimer Association (AARG-17-498856) with instrument support of ZonMW (91111009). WS has received research funding from ZonMW Memorabel/Alzheimer Nederland (Deltaplan Dementie) 733050101, ZonMW/Stichting Proefdiervrij MKMD 114022506, Health Holland (Stichting LSH-TKI) LSHM17104 & LSHM18024, Weston Brain Institute NR160014, Alzheimer Nederland WE.03-2017-10, and Joint Programme-Neurodegenerative Disease Research (NAB3) JPCOFUND_FP-829-031.
Conflict of interest
JvW and WS have no conflicts of interest that are directly relevant to the content of this article.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
van Weering, J.R.T., Scheper, W. Endolysosome and Autolysosome Dysfunction in Alzheimer’s Disease: Where Intracellular and Extracellular Meet. CNS Drugs 33, 639–648 (2019). https://doi.org/10.1007/s40263-019-00643-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-019-00643-1