
Abstract
There appears to be a mismatch between the assumed therapeutic equivalence of generic drugs, their interchangeability, and reported clinical discomfort following generic drug use and drug switches. In this article, we describe why we are of the opinion that the current regulatory approach to the evaluation of generic drugs based on average bioequivalence is sufficient to expect therapeutic equivalence in the clinical setting. This has often been debated, specifically as adverse drug reactions related to generic drug switches are regularly reported. We agree that clinical discomfort during a bioequivalent drug switch may indeed be caused by different exposures to the active substance. However, this difference in exposure is not a result of the characteristics or quality of generic drugs; it is caused by the pharmacokinetic within-subject variability of the active substance, i.e., the variability on the bioavailability of the active substance, when comparing two occasions of administration of the same drug product, to the same patient. Therefore, reported clinical discomfort following generic drug use and drug switches does not warrant a change in the regulatory approach to the evaluation of the bioequivalence of generic drugs. Switching from a brand-name drug to currently approved generic drugs, or between different generic drugs, will in principle result in comparable exposure, within boundaries determined by the within-subject variability of the pharmacokinetics of the active substance involved.
Similar content being viewed by others
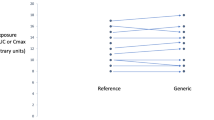

Individual differences in exposure upon switching to a bioequivalent drug, even outside the acceptance range for average bioequivalence, are known to occur. However, these are not because of differences in the characteristics of the drugs, but rather the intra-individual variability of the active substance involved. |
In the case of an approved generic drug with comparable quality, given the identical intra-subject variability, the variation in exposure following a switch from a brand-name drug to a generic drug is comparable to that observed upon repeated administration of either the brand-name drug or the generic drug. |
The current regulatory approach to the evaluation of bioequivalence appears to be sufficient. For approved generic drugs, comparable exposure between drugs are expected, within boundaries in individual blood drug concentrations determined by the within-subject variability of the active substance. |
A demonstration of bioequivalence is the cornerstone of generic drug approval. Two pharmaceutically comparable drug products of high quality are bioequivalent if there is statistical proof of comparable bioavailability. For immediate-release drugs with systemic action, this comparable bioavailability is usually investigated in a single-dose crossover study, in which both drug products are administered in two separate periods, in a randomized sequence. The evaluation is predominantly based on the area under the plasma concentration–time curve (AUC) and the maximum plasma concentration (Cmax), which reflect the extent of exposure and peak exposure, respectively, and therefore allow for a comparison of exposure over time. Already in the early 1970s, the Canadian authorities used the arbitrary cut-off of at least 80% bioavailability to compare different drug products [1], and the use of a confidence interval for the statistical test was proposed [2]. Current guidance dictates that, generally, the 90% confidence intervals for the geometric mean ratio of Cmax and AUC between the two products should be within the acceptance interval of 80.00–125.00% [3].
For active substances with a high pharmacokinetic variability, of over 30%, the US Food and Drug Administration allows a widening of the acceptance limits for AUC and Cmax to a range depending on the extent of the within-subject variability, i.e., the variability on the bioavailability of an active substance, when comparing two occasions of administration of the same drug product, to the same patient [4, 5]. In the European Union, widening of the acceptance limits is similarly allowed but can only be applied to Cmax, which may be increased in a scaled approach, to a maximum range of 69.84–143.19%, which is the limit when a within-subject variability of 50% is determined. In addition, for active substances with a narrow therapeutic index, acceptance limits for both AUC and Cmax can be scaled to the variability (USA), or tightened to 90.00–111.11% for AUC only or for both, AUC and Cmax (European Union) [6, 7].
Several aspects of the regulatory approach to the concept of bioequivalence have been debated over the years. Ever since the first proposal of the confidence interval limits, the width of this acceptance range has been challenged [8], mainly raising the question as to why the same arbitrary limits are applied to all active substances. Partly because of those discussions, in current regulation, a narrowing or widening of the confidence interval limits is allowed. Independent of the confidence interval limits applied, it is known that the ratio of the exposure to the active substance from the generic and brand-name drug is within a much smaller range than allowed with the above-mentioned criteria, as indicated by an analysis of 2070 bioequivalence studies by Davit et al. in 2009 [9]. In that analysis, they demonstrated that the mean estimated difference between approved generic and brand-name drugs was only 4.35% for Cmax and 3.56% for AUC. For the European situation, based on a smaller subset of 120 bioequivalence studies, comparable figures were obtained [10].
Another point of debate has been that bioequivalence studies are usually conducted with healthy subjects and not with the intended patient population. It has often been questioned whether the bioequivalence conclusion, as drawn in a healthy subject population, can be extrapolated to patients. Indeed, absolute exposure may be different for patients compared to healthy subjects. However, this difference is active substance related and similarly influenced by, for example, patient comorbidities, for both brand-name drugs and generic drugs. As discussed by Versantvoort et al., the relative exposure between the brand-name and generic drugs will be the same in healthy subjects and patients [11]. One further reason for assessing bioequivalence using healthy subjects is that in these subjects, with generally less comorbidities than patients, it is expected that less variability of exposure is observed that is unrelated to the differences between the tested drug products [3]. This is relevant, as the primary goal of a bioequivalence study is to investigate formulation-related differences between the tested drug products, and to measure them in the most sensitive manner, other variability should be minimal. Healthy subjects are the most suitable model for that purpose. Likewise, the pharmacokinetic behavior of drugs can be markedly different in men vs women [12] or in patients with different degrees of, for instance, renal function; but this also applies to an identical extent for brand-name drugs and generic drugs. Therefore, demonstrating bioequivalence for both sexes does not seem to be necessary; bioequivalence demonstrated in male individuals can safely be extrapolated to female individuals and vice versa, as discussed by Gonzalez-Rojano et al. [13].
Nevertheless, it is useful to empirically test the assumption of comparable relative exposure in patients vs healthy subjects. For this purpose, a well-controlled comparative bioavailability study was recently performed with kidney and liver transplant patients, and similar tacrolimus exposure for brand-name and generic drugs was concluded, in line with the expectation based on studies in healthy subjects [14]. Likewise, bioequivalence as determined in healthy subjects between different generic lamotrigine drugs (generic–generic switch) was confirmed in patients with epilepsy [15].
These findings support the current approval pathway for generic drugs and the notion that bioequivalent drugs are expected to be therapeutically equivalent. However, this assumption is based on bioequivalence: comparable average exposure in the whole population. A treating physician or prescriber would predominantly be interested in therapeutic equivalence for their individual patient. This presumed therapeutic equivalence is often debated, for instance for cardiovascular drugs [16, 17] and drugs for psychiatric illness [18]. Nonetheless, no substantial differences in clinical outcomes were observed between patients who started treatment with either generic or brand-name warfarin [19]. Further, in a clinical crossover study evaluating bupropion exposure in patients with major depression, the bioequivalence of three generic drugs to the brand-name drug and between the three generic drugs was confirmed, and no differences in depressive symptoms or side effects were noted [20]. However, clinical evidence from unbiased randomized controlled trials, for the assumed absence of clinically relevant differences (in exposure) between generic and brand-name drugs, is limited [21].
Indeed, if bioequivalent drugs are therapeutically equivalent, then patients would not be expected to experience clinical discomfort related to generic drug switches. Adverse drug reactions (ADRs) related to generic drug switches are reported regularly, as evident from case reports [22, 23] and observational studies [24, 25]. The following questions then arise: What causes this apparent discrepancy between theory (assumed interchangeability of generic drugs) and real-world observations (reported clinical discomfort upon switching to a generic drug), and could the explanation still be found in pharmacokinetic aspects underlying such a generic switch?
For example, a recent article by Concordet et al. describes issues related to a levothyroxine drug switch in France affecting more than 2.5 million patients in 2017 [24]. Even though bioequivalence was demonstrated between the old and new formulation of levothyroxine used in the switch, applying the tightened 90.00–111.11% acceptance range [26], more than 30,000 ADRs were reported following this massive drug switch. Based on this levothyroxine case, Concordet et al. argue that a demonstration of average bioequivalence is not sufficient to ensure drug switchability. They also argue that tightening the acceptance limits for the average bioequivalence 90% confidence intervals is insufficient, as the width of the 90% confidence interval is inversely related to the number of studied subjects and therefore still may allow marked differences in exposure upon switching [27]. Moreover, according to the authors, as more than 50% of the subjects in the supporting levothyroxine bioequivalence study [26] demonstrated an individual pharmacokinetic exposure ratio outside the 90.00–111.11% confidence interval, acceptance criteria should have been a warning signal of a possible lack of individual bioequivalence. Therefore, the authors argue that a priori characterization of the within-subject variability and the extent to which the pharmacokinetic exposure ratios differ between individuals (subject-by-formulation interaction), as in the concept of individual bioequivalence, would have identified switchability issues and would have allowed for a better regulatory assessment of the new levothyroxine formulation.
The Concordet et al. articles have been extensively commented upon, with discussions mainly related to difficulties and shortcomings of the individual bioequivalence approach and of the calculation and interpretation of the number of subjects in a bioequivalence study with an individual exposure ratio outside the bioequivalence acceptance criteria [28,29,30,31,32,33,34,35]. The debate clearly indicates that a consensus has not been reached on the issue of the interchangeability of drugs based on demonstrated average bioequivalence.
In our opinion, within-subject pharmacokinetic variability is the most important factor in this debate. As demonstrated by Yu et al., there are clear indications that within-subject variability has far more influence than subject-by-formulation interaction for drugs for which bioequivalence has been demonstrated [36]. In Yu et al.’s article, it was demonstrated for a selection of drugs that the variability of exposure following a switch from a brand-name drug to a generic drug is comparable to that observed upon repeated administration of either the brand-name drug or the generic drug. It is indeed acknowledged that in bioequivalence studies, subjects will often have an individual exposure ratio outside the acceptance criteria [37], a situation we also observe in results from bioequivalence studies filed for regulatory approval of generic drugs. However, it is crucial to realise that the same phenomenon occurs when comparing the exposure of two repeated administrations of the same brand-name or generic drug; therefore, this finding is merely a reflection of the daily clinical situation upon treating a patient, with his or her inherent within-subject variability leading to different exposures per occasion, than a reflection of differences in the quality and/or exposure of a generic drug. Experimental imprecision adds to the observation of different individual exposures in studies. Indeed, Munafo et al. [33] describe simulations in which individual exposures outside the acceptance range are observed, with a bioanalytical error of 10% as the only source of variability.
Thus, when a patient experiences clinical discomfort during a bioequivalent drug switch, it may indeed be caused by a different exposure to the active substance. However, this different exposure does not seem to be the result of different characteristics of the bioequivalent drug, but is highly likely because of pharmacokinetic within-subject variability of the active substance, unrelated to the different drug products. When we assume that the difference in exposure is related to the perceived clinical discomfort in a patient, this clinical discomfort is not attributable to differences between the original and switched drug, but merely to pharmacokinetic within-subject variability.
The question then remains as to why larger numbers of ADRs are reported in the case of drug switches than in the case of drug continuation. In line with the above reasoning, a potential explanation could be that patients and prescribers are more prone to report ADRs, as negative perceptions about generic drug switching and forced drug switches persist. This would imply that drug switching does not increase the actual number of ADRs, but merely increases the reporting rate of ADRs. In relation to another relatively large-scale levothyroxine drug switch, in 2007 and 2008 in New Zealand, the increased number of reported ADRs in that setting has been postulated to be caused by an increased reporting rate because of inaccurate guidance information and media attention [38]. Another potential explanation for an increased number of reported ADRs related to drug switches is postulated to be a nocebo effect, particularly owing to the negative perceptions of generic drugs [39, 40]. However, the psychological aspects potentially affecting increased ADR reporting are outside the scope of this opinion paper, and any such hypothesis would warrant further investigation. Regardless of the true cause of the increased number of reported ADRs, clinical discomfort for an individual patient should never be underestimated and should always undergo thorough causal and clinical review, both on an individual level (physician) and on a population level (pharmacovigilance). With regard to the individual patient specifically, it is important to stress that we do not want to disregard any clinical discomfort that a patient may experience. The discomfort exists, and it can have a significant impact on the quality of life. In highly exceptional cases, this could be a result of an allergic response to excipients used in the new drug product.
Furthermore, as a regulatory agency, we see it as our responsibility to be critical of our own rules and regulations. For this reason, we investigated bioequivalence and the underlying assumptions of generic interchangeability, for instance by performing a bioequivalence study between approved gabapentin generics [41]; a retrospective analysis of bioequivalence studies submitted for regulatory evaluation [36], modeling and simulation efforts towards generic drugs [42]; and an analysis of 1348 reported ADRs and 23.8 million drug switches [25], to investigate the potential consequences of generic drug switching.
So far, these and other data have not provided evidence to support a change in our current regulatory approach to the evaluation of bioequivalence. The data remain in support of the assumption that switching from a brand-name drug to currently approved generic drugs, or between different generic drugs, will result in comparable exposure within boundaries determined by the within-subject variability of the pharmacokinetics of the active substance involved.
References
Davies RO, Zarowny DP, Robin HR, Ruedy J. Bioavailability. Can Med Assoc J. 1972;107(3):183–4.
Westlake WJ. Use of confidence intervals in analysis of comparative bioavailability trials. J Pharm Sci. 1972;61(8):1340–1.
EMA. Guideline on the investigation of bioequivalence. Ref.: CPMP/EWP/QWP/1401/98 Rev. 1/Corr **. 2010. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf. Accessed 24 Jan 2020.
Davit BM, Chen ML, Conner DP, Haidar SH, Kim S, Lee CH, et al. Implementation of a reference-scaled average bioequivalence approach for highly variable generic drug products by the US Food and Drug Administration. AAPS J. 2012;14(4):915–24.
US FDA. Bioavailability studies submitted in NDAs or INDs: general considerations. Guidance for industry. 2019. https://www.fda.gov/media/121311/download. Accessed 12 May 2020.
European Medicines Agency. Questions and answers: positions on specific questions addressed to the Pharmacokinetics Working Party (PKWP). 2015. https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/clinical-pharmacology-pharmacokinetics/clinical-pharmacology-pharmacokinetics-questions-answers. Accessed 12 May 2020.
US FDA. Draft guidance on warfarin sodium. 2012. https://www.accessdata.fda.gov/drugsatfda_docs/psg/Warfarin_Sodium_tab_09218_RC12-12.pdf. Accessed 12 May 2020.
Levy G. Bioavailability limits. Can Med Assoc J. 1972;107(8):722.
Davit B. Comparing generic and innovator drugs: a review of 12 years of bioequivalence data from the United States Food and Drug Administration. Ann Pharmacother. 2009;43(10):1583–97.
Yu Y, Teerenstra S, Neef C, Burger D, Maliepaard M. Investigation into the interchangeability of generic formulations using immunosuppressants and a broad selection of medicines. Eur J Clin Pharmacol. 2015;71(8):979–90.
Versantvoort C, Maliepaard M, Lekkerkerker F. Generics: what is the role of registration authorities. Neth J Med. 2008;66(2):62–6.
Soldin OP, Mattison DR. Sex differences in pharmacokinetics and pharmacodynamics. Clin Pharmacokinet. 2009;48(3):143–57.
Gonzalez-Rojano E, Marcotegui J, Ochoa D, Roman M, Alvarez C, Gordon J, et al. Investigation on the existence of sex-by-formulation interaction in bioequivalence trials. Clin Pharmacol Ther. 2019;106(5):1099–112.
Alloway RR, Vinks AA, Fukuda T, Mizuno T, King EC, Zou Y, et al. Bioequivalence between innovator and generic tacrolimus in liver and kidney transplant recipients: a randomized, crossover clinical trial. PLoS Med. 2017;14(11):e1002428.
Privitera MD, Welty TE, Gidal BE, Diaz FJ, Krebill R, Szaflarski JP, et al. Generic-to-generic lamotrigine switches in people with epilepsy: the randomised controlled EQUIGEN trial. Lancet Neurol. 2016;15(4):365–72.
Kesselheim AS, Misono AS, Lee JL, Stedman MR, Brookhart MA, Choudhry NK, et al. Clinical equivalence of generic and brand-name drugs used in cardiovascular disease: a systematic review and meta-analysis. JAMA. 2008;300(21):2514–26.
Leclerc J, Thibault M, Midiani Gonella J, Beaudoin C, Sampalis J. Are generic drugs used in cardiology as effective and safe as their brand-name counterparts? A systematic review and meta-analysis. Drugs. 2020;80(7):697–710.
Blier P, Margolese HC, Wilson EA, Boucher M. Switching medication products during the treatment of psychiatric illness. Int J Psychiatry Clin Pract. 2019;23(1):2–13.
Desai RJ, Gopalakrishnan C, Dejene S, Sarpatwari AS, Levin R, Dutcher SK, et al. Comparative outcomes of treatment initiation with brand versus generic warfarin in older patients. Clin Pharmacol Ther. 2019;107(6):1334–40.
Kharasch ED, Neiner A, Kraus K, Blood J, Stevens A, Schweiger J, et al. Bioequivalence and therapeutic equivalence of generic and brand bupropion in adults with major depression: a randomized clinical trial. Clin Pharmacol Ther. 2019;105(5):1164–74.
Flacco ME, Manzoli L, Boccia S, Puggina A, Rosso A, Marzuillo C, et al. Registered randomized trials comparing generic and brand-name drugs: a survey. Mayo Clin Proc. 2016;91(8):1021–34.
Goldberg JF. A case of akathisia after switching from branded to generic high-dose olanzapine. J Clin Psychiatry. 2012;73(4):497.
Gallelli LGG, Codamo G, Argentieri A, Michniewicz A, Siniscalchi A, et al. Recognizing severe adverse drug reactions: two case reports after switching therapies to the same generic company. Curr Drug Saf. 2015;11:104–8.
Concordet D, Gandia P, Montastruc JL, Bousquet-Melou A, Lees P, Ferran A, et al. Levothyrox® new and old formulations: are they switchable for millions of patients? Clin Pharmacokinet. 2019;58(7):827–33.
Glerum PJ, Maliepaard M, de Valk V, Scholl JHG, van Hunsel F, van Puijenbroek EP, et al. Quantification of adverse drug reactions related to drug switches in the Netherlands. Clin Transl Sci. 2020;13(3):599–607.
Gottwald-Hostalek U, Uhl W, Wolna P, Kahaly GJ. New levothyroxine formulation meeting 95-105% specification over the whole shelf-life: results from two pharmacokinetic trials. Curr Med Res Opin. 2017;33(2):169–74.
Concordet D, Gandia P, Montastruc JL, Bousquet-Melou A, Lees P, Ferran AA, et al. Why were more than 200 subjects required to demonstrate the bioequivalence of a new formulation of levothyroxine with an old one? Clin Pharmacokinet. 2019;59(1):1–5.
Coste J, Bertagna X, Zureik M. Comment on: “Levothyrox® new and old formulations: are they switchable for millions of patients?”. Clin Pharmacokinet. 2019;58(7):965–6.
Trechot P. Comment on: Levothyrox® new and old formulations: are they switchable for millions of patients? Clin Pharmacokinet. 2019;58(7):977–8.
Nicolas P. Comment on: “Levothyrox® new and old formulations: are they switchable for millions of patients?”. Clin Pharmacokinet. 2019;58(7):959–60.
Lechat P, Ropers J, Barre J, Mouly S. Comment on: “Levothyrox® new and old formulations: are they switchable for millions of patients?”. Clin Pharmacokinet. 2019;58(10):1351–2.
Yu Y, Maliepaard M. Comment on “Levothyrox® new and old formulations: are they switchable for millions of patients?”. Clin Pharmacokinet. 2019;59(2):281–2.
Munafo A, Krebs-Brown A, Gaikwad S, Urgatz B, Castello-Bridoux C. Comment on “Levothyrox® new and old formulations: are they switchable for millions of patients?”. Clin Pharmacokinet. 2019;58(7):969–71.
Nicolas P. Comment on: “Why were more than 200 subjects required to demonstrate the bioequivalence of a new formulation of levothyroxine with an old one?”. Clin Pharmacokinet. 2019;59(2):273–5.
Krebs-Brown A, Munafo A, Gaikwad S, Urgatz B, Castello-Bridoux C. Comment on: “Why were more than 200 subjects required to demonstrate the bioequivalence of a new formulation of levothyroxine with an old one?”. Clin Pharmacokinet. 2019;59(2):265–7.
Yu Y, Teerenstra S, Neef C, Burger D, Maliepaard M. A comparison of the intrasubject variation in drug exposure between generic and brand-name drugs: a retrospective analysis of replicate design trials. Br J Clin Pharmacol. 2016;81(4):667–78.
Hammami MM, De Padua SJS, Hussein R, Al Gaai E, Khodr NA, Al-Swayeh R, et al. Generic-reference and generic-generic bioequivalence of forty-two, randomly-selected, on-market generic products of fourteen immediate-release oral drugs. BMC Pharmacol Toxicol. 2017;18(1):78.
Faasse K, Cundy T, Petrie KJ. Medicine and the media. Thyroxine: anatomy of a health scare. BMJ. 2009;339:b5613.
Faasse K, Martin LR. The power of labeling in nocebo effects. Int Rev Neurobiol. 2018;139:379–406.
Weissenfeld J, Stock S, Lungen M, Gerber A. The nocebo effect: a reason for patients’ non-adherence to generic substitution? Pharmazie. 2010;65(7):451–6.
Yu Y, Teerenstra S, Vanmolkot F, Neef C, Burger D, Maliepaard M. Interchangeability of gabapentin generic formulations in the Netherlands: a comparative bioavailability study. Clin Pharmacol Ther. 2013;94(4):519–24.
Glerum PJ, Yu Y, Yamada WM, Neely MN, Maliepaard M, Burger DM, et al. Interchangeability of generic drugs: a nonparametric pharmacokinetic model of gabapentin generic drugs. Clin Pharmacol Ther. 2018;104(5):966–73.
Author information
Authors and Affiliations
Contributions
PG, YY, CN, DB, and MM wrote the article.
Corresponding author
Ethics declarations
Conflict of interest
Pieter J. Glerum, Yang Yu, and Marc Maliepaard are full-time employees of the Medicines Evaluation Board in the Netherlands. Cees Neef and David M. Burger have no conflicts of interest that are directly relevant to the content of this article.
Funding
No sources of funding were used to assist with the preparation of this article.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Glerum, P.J., Neef, C., Burger, D.M. et al. Pharmacokinetics and Generic Drug Switching: A Regulator’s View. Clin Pharmacokinet 59, 1065–1069 (2020). https://doi.org/10.1007/s40262-020-00909-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-020-00909-8