
Abstract
Objectives
The aim of this study was to assess the pharmacokinetics (PK) and safety/tolerability of siponimod in healthy subjects when coadministered with (1) the moderate cytochrome P450 (CYP) 2C9 and CYP3A inhibitor fluconazole (Study A), and (2) with three different CYP2C9 genotype variants (Study B).
Methods
Study A was an open-label, single-dose study comprising periods 1 (14 days; day 1: siponimod 4 mg) and 2 (20 days; day 1: fluconazole 200 mg twice daily; days 2–19: fluconazole 200 mg once daily; day 3: siponimod 4 mg) in healthy subjects (n = 14) with the wild-type CYP2C9 genotype (CYP2C9*1/*1). Study B was a multicentre, open-label study comprising parts 1 (day 1: siponimod 0.25 mg once daily in the CYP2C9*1/*1, CYP2C9*2/*3 and CYP2C9*3/*3 genotypes) and 2 (days 1–2: 0.25 mg once daily; day 3: 0.5 mg once daily in the CYP2C9*2/*3 and CYP2C9*3/*3 genotypes only) in healthy subjects with polymorphic variants of CYP2C9 (n = 24). Pharmacokinetic parameters were calculated using noncompartmental methods.
Results
In Study A, coadministration with fluconazole produced an approximately twofold increase in mean area under the curve (AUC) versus siponimod alone (from 1110 to 2160 h*ng/mL), and an increase in maximum plasma concentration (Cmax; from 31.2 to 34.0 ng/mL) and elimination half-life (T½; from 40.6 to 61.6 h). In Study B, the AUCs of siponimod were approximately two to fourfold greater in subjects with the CYP2C9*2/*3 and CYP2C9*3/*3 genotypes, with a minor increase in Cmax versus the CYP2C9*1/*1 genotype. The mean T½ was prolonged in the CYP2C9*2/*3 (51 h) and CYP2C9*3/*3 (126 h) genotypes versus the CYP2C9*1/*1 (28 h) genotype. Siponimod did not result in increased adverse events in healthy subjects in both studies.
Conclusions
Changes in siponimod PK, when coadministered with fluconazole at steady-state and in subjects with different CYP2C9 genotypes, indicate that the reduced CYP2C9 enzymatic activity does not affect the absorption phase of siponimod but prolongs the elimination phase. These results confirm the relevance of CYP2C9 activity on siponimod metabolism in humans.
Similar content being viewed by others


Siponimod, a potent, selective sphingosine 1-phosphate receptor subtypes 1 and 5 (S1P1,5) receptor modulator, is eliminated primarily through cytochrome P450 (CYP) 2C9, a polymorphic enzyme. |
We conducted two separate pharmacokinetic studies in healthy subjects to quantitatively describe how siponimod metabolism by CYP2C9 can be modulated by exogenous factors such as the CYP2C9 inhibitor (fluconazole) and the inherited CYP2C9 genotype (CYP2C9*2/*3 and CYP2C9*3/*3 vs. CYP2C9*1/*1). |
We report that when CYP2C9 enzymatic activity is reduced, the systemic clearance of siponimod is significantly decreased. |
1 Introduction
Siponimod (BAF312), a potent, oral, selective modulator of the sphingosine 1-phosphate (S1P) receptor subtypes 1 and 5 (S1P1,5), limits the inflammatory effects mediated by B and T cells [1]. It is currently under advanced phase of clinical development for the treatment of secondary progressive multiple sclerosis (SPMS) [2]. The pharmacokinetic (PK) profile of siponimod in a single-ascending dose range study (0.1–75 mg) in 98 healthy subjects was measurable in the plasma as early as 0.25 h postdose, and the maximum plasma concentration (Cmax) was reached within 3–8 h postdose (minimum–maximum 0.25–24 h). In a multiple-ascending dose range study (0.3–20 mg) in 48 healthy subjects over 28 days, the steady-state of plasma siponimod was reached after approximately 6 days; the mean accumulation ratio was 1.9–2.7 and the effective elimination half-life (T½) was 22–38 h (mean 30 h) [1]. The rate and extent of systemic exposure were increased in a dose-proportional manner after single (0.1–75 mg) and multiple (0.3–20 mg) doses of siponimod [1].
Cytochrome P450 (CYP) 2C9 is the major enzyme responsible for the clearance of siponimod [3, 4]. Siponimod is eliminated from the systemic circulation mainly due to metabolism and subsequent biliary/faecal excretion [4,5,6]. Metabolite M3 is one of the main circulating metabolites of siponimod in humans; it is formed by glucuronidation of the hydroxylated M5 metabolite that results from metabolism primarily via CYP2C9 (79.2%), with a minor contribution from CYP3A4 (18.5%) [3, 6, 7].
A drug–drug interaction (DDI) study was conducted with a CYP2C9 inhibitor to evaluate the effect of CYP2C9 inhibition on siponimod PK. In the absence of any strong CYP2C9 inhibitor, fluconazole was selected as it is one of the most potent CYP2C9 inhibitors used in clinical practice. Fluconazole is a moderate CYP2C9 and CYP3A inhibitor and is recommended in regulatory guidance as a prototype inhibitor to assess potential DDI by CYP2C9 inhibition [8, 9]. This study will therefore support the development of clinical recommendations for siponimod coadministration with CYP2C9/CYP3A inhibitors.
CYP2C9 is a polymorphic enzyme. Of more than 50 single nucleotide polymorphisms (SNPs) described in the regulatory and coding regions of the CYP2C9 gene, only two coding SNPs, namely CYP2C9*2 and CYP2C9*3, have shown clinically relevant reductions in enzyme activity [10]. These two SNPs result in six different genotypes that confer three functionally different phenotypes, namely extensive metabolisers (EMs; CYP2C9*1/*1), intermediate metabolisers (IMs; CYP2C9*1/*2, CYP2C9*1/*3 and CYP2C9*2/*2) and poor metabolisers (PMs; CYP2C9*2/*3 and CYP2C9*3/*3) [10,11,12]. Results from in silico SimCYP simulations [a physiological-based PK (PBPK) model; SimCYP Ltd, Sheffield, UK] suggested an increased plasma exposure of siponimod in subjects with the CYP2C9*2 and CYP2C9*3 genotypes, due to a reduction in the enzyme activity [3]. Exploratory PK/pharmacogenetic (PG) analyses indicated that heterozygous CYP2C9*3 carriers tended to have a higher area under the curve (AUC) of siponimod compared with subjects not carrying the *3 allele (unpublished data). A study was conducted to assess the impact of the reduced CYP2C9 enzyme activity on siponimod PK in CYP2C9 PMs (CYP2C9*2/*3 and CYP2C9*3/*3).
The present article reports on two studies: Study A, in vivo effects of the steady-state CYP2C9 enzyme inhibitor, fluconazole, on the PK and safety/tolerability of a single oral dose of siponimod 4 mg in healthy adult subjects; and Study B, the PK and safety/tolerability of a single dose and 3-day dosing of siponimod in healthy subjects with polymorphic variants of CYP2C9.
2 Methods
2.1 Study A: Siponimod Pharmacokinetics (PK) With and Without Coadministration of Fluconazole in Healthy Subjects
2.1.1 Ethics
The study was conducted at the Clive Berghofer Cancer Research Center, Brisbane, QLD, Australia (trial number CTN 2011/010978). The study protocol and subject consent forms were reviewed and approved by the Queensland Institute of Medical Research Human Research Ethics Committee, and all subjects provided written informed consent. The study was conducted in compliance with the ethical principles of the Declaration of Helsinki and the International Conference on Harmonization (ICH) Good Clinical Practice Guidelines [13] and the US FDA guidelines [14].
2.1.2 Study Population
Healthy males and females aged 18–55 years who had a body mass index (BMI) of 18–30 kg/m2, body weight ≥ 50 kg, sitting vital signs of systolic blood pressure 90–140 mmHg and diastolic blood pressure 50–90 mmHg, pulse rate 50–90 beats/min and the wild-type CYP2C9 genotype (CYP2C9*1 homozygous) were included in the study. Patients were excluded from the study if they had a history of hypersensitivity to study drugs or drugs of similar classes; immunodeficiency; drug abuse or alcohol abuse within the past 12 months; cardiovascular diseases; recent (within the previous 3 years) or recurrent history of fainting or palpitations; acute or chronic bronchospastic disease; clinically significant electrocardiograph (ECG) abnormalities; detection of bradycardia and/or arrhythmic episodes on Holter monitoring; malignancy within the past 5 years; and medical conditions such as inflammatory bowel disease, ulcers, gastrointestinal or rectal bleeding, pancreatic injury or pancreatitis, liver injury or abnormal liver function tests and renal impairment. The study disallowed the use of any prescription drugs, herbal supplements (within 4 weeks prior to initial dosing) and/or over-the-counter (OTC) medication or dietary supplements (within 2 weeks prior to initial dosing).
2.1.3 Study Design
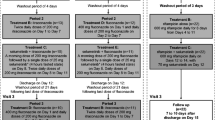
This was an open-label, single-dose, two-period, single-sequence study consisting of a 28-day screening period followed by two treatment periods, each consisting of a 24-h baseline period (day − 1) [Fig. S1, electronic supplementary material (ESM) Online Resource 1].
During period 1 (14 days), subjects received a single oral dose of siponimod 4 mg on day 1. During period 2 (20 days), subjects received fluconazole 200 mg twice on day 1 (as a loading dose), followed by 200 mg administered once daily from days 2–19. A single dose of siponimod 4 mg was coadministered on day 3. There was a washout interval of 14–21 days between the two treatment periods based on the T½ of siponimod as observed in the single-ascending dose study (mean range 27–57 h) (unpublished data) to eliminate any possible carryover effect in the second period of the study.
2.1.4 PK Blood Sampling Schedule and Bioanalysis
At each time point, 2 mL of blood was collected from each subject. During period 1, blood samples were collected predose (0 h), and at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48, 72, 96, 144, 216 and 312 h (day 14) after siponimod administration on day 1. During period 2, blood samples were collected at the same time points as for period 1 and at 408 h (day 18) after siponimod administration on day 3. Further details of the PK blood sampling, as well as the bioanalytical method and assay performance, are provided in ESM Online Resource 2.
2.1.5 PK and Safety Assessments
PK data analyses were conducted with WinNonlin Pro version 5 software (Pharsight Corporation, Mountain View, CA, USA) using standard noncompartmental methods. Plasma concentrations below the lower limit of quantification were treated as zero for all PK calculations. The following plasma siponimod PK parameters were determined from the plasma concentration–time curve: AUC from time zero to the time of the last measurable concentration (AUClast); AUC from time zero extrapolated to infinity (AUC∞); Cmax, time to reach Cmax (Tmax), lag time between drug intake and first quantifiable plasma concentration (Tlag), T½, apparent systemic clearance (CL/F) and apparent volume of distribution (VZ/F).
Safety assessments included recording of all adverse events (AEs) and serious AEs (SAEs), physical examinations, vital sign measurements, and ECG and laboratory evaluations.
2.1.6 Statistical Evaluation
The SimCYP (SimCYP Ltd) modelling and simulation data indicated a two to threefold increase in siponimod AUC in the presence of fluconazole [3]. This increase could have been detected by a sample size of four subjects with power > 90% (significance level 0.05). With a sample size of 10, the 90% confidence intervals (CIs) of the difference between the means (with and without fluconazole) of log-scale, PK exposure would not extend more than 0.156 from the estimated difference, with 90% probability. This estimate was based on the intrasubject coefficient of variation (CV%) of 15% obtained from historical studies (Novartis, data on file). A total of 14 subjects were enrolled to ensure 10 completers.
Only subjects with evaluable PK parameters in both periods were included in the analysis. Log-transformed PK parameters (AUClast, AUC∞ and Cmax) were analysed using a fixed-effects model with treatment and subject as fixed effects. The resulting 90% CIs for the treatment difference between reference (siponimod) and test (siponimod plus fluconazole) groups were back-transformed to obtain point estimates for the ratio of geometric means and the corresponding 90% CIs. The safety data set included all subjects who received at least one dose of siponimod; descriptive statistics were used to analyse the data.
2.2 Study B: Siponimod PK in Healthy Subjects with the Cytochrome P450 (CYP) 2C9 Genetic Polymorphism
2.2.1 Ethics
The study protocol and subject consent forms were reviewed by the Independent Ethics Committee and Institutional Review Board for each centre. All subjects provided written informed consent before study entry. The study was conducted in compliance with the ethical principles of the Declaration of Helsinki and the ICH Good Clinical Practice Guidelines [13].
2.2.2 Study Population
Healthy males and females aged 18–70 years with a BMI between 18 and 30 kg/m2, body weight ≥ 50 kg, sitting vital signs of systolic blood pressure 90–140 mmHg and diastolic blood pressure 50–90 mmHg, pulse rate 50–100 beats/min and the CYP2C9*1/*1 (EM) and CYP2C9*2/*3 and CYP2C9*3/*3 (PM) genotypes were included in the study. Subjects with the CYP2C9 EM phenotype were matched by body weight (± 10%) to those with the CYP2C9 PM phenotype. This multiple matching of PMs to EMs reduced the number of healthy subjects exposed to siponimod. Therefore, an EM subject could serve as a matching partner for up to two PM subjects, provided the two PM subjects had a different CYP2C9 genotype. Key exclusion criteria included clinically significant disease of any major system organ class not resolved within 2 weeks prior to initial dosing; history or presence of any clinically significant ECG abnormalities; adverse cardiovascular findings; surgical or medical conditions other than hepatic impairment that could significantly alter the absorption, distribution, metabolism, or excretion of drugs; and smokers, as defined by use of tobacco products in the previous 3 months. The use of any prescription drugs, herbal supplements (within 4 weeks before initial dosing) and/or OTC medication or dietary supplements (within 2 weeks before initial dosing) was not allowed in the study.
2.2.3 Study Design
This was a two-part, multicentre, open-label study of siponimod in subjects (N = 24) with the CYP2C9*1/*1 (EM), CYP2C9*2/*3 and CYP2C9*3/*3 (PM) genotypes conducted at four centres across Australia (n = 1), France (n = 1), Jordan (n = 1) and the US (n = 1) [Fig. S2, ESM Online Resource 3].
In part 1, following a 41-day screening period and a baseline day (day − 1), a single oral dose of siponimod 0.25 mg was administered to both CYP2C9 EMs and PMs (CYP2C9*1/*1: n = 12; CYP2C9*2/*3: n = 6; CYP2C9*3/*3: n = 6) on day 1, followed by a washout period of 42 days. The washout period between both study parts was decided based on the T½ predicted by SimCYP in subjects with the CYP2C9*3/*3 genotype (a mean T½ of 170 h), thereby setting up a PK sampling period over 840 h (day 36) [3]. PK assessments were conducted throughout the study. EM subjects completed the study after part 1, while PM subjects enrolled in part 1 continued into part 2 of the study.
In part 2, following a 41-day screening period (which overlapped with the washout period of part 1) and a baseline visit (day − 1), multiple oral doses of siponimod were administered over 3 days (days 1 and 2: 0.25 mg; day 3: 0.5 mg; mimicking the first three dosing days of the established dose titration regimen of siponimod used in pivotal studies) to PMs only (CYP2C9*2/*3: n = 6; CYP2C9*3/*3: n = 6), followed by a follow-up period (over 21 days) and a study completion visit. The study did not include CYP2C9*1/*1 subjects in part 2 because historical data were already available for a sufficient comparison (unpublished data). The potential advantage of a within-study comparison was outweighed by the wealth of data that were collected in previous studies favouring the between-study comparison and thereby avoiding an unnecessary additional exposure in CYP2C9*1/*1 subjects in part 2.
2.2.4 PK Blood Sampling Schedule and Bioanalysis
The method of blood sampling was the same as used in Study A, except that 3 mL instead of 2 mL of blood samples were collected at each PK sampling time point. In part 1, blood samples for PK analysis were drawn predose and at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48, 72, 96, 144, 216, 312 (day 14), 408 (day 18), 504 (day 22), 600 (day 26), 720 (day 31), 840 (day 36) and 984 h postdose (day 42). In Part 2, blood samples for PK analysis were drawn predose on days 1, 2, and 3, and postdose on day 3 at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12, 16 and 24 h. Further details on the PK blood sampling, as well as the bioanalytical method and assay performance, are provided in ESM Online Resource 2.
2.2.5 PK and Safety Assessments
The PK parameters of siponimod (Cmax, Tmax, AUClast, AUC∞, AUC24, T½, Tlag, Vz/F and CL/F) and its metabolites (Cmax, Tmax, AUClast, AUC∞, AUC24, T½ and Tlag) were calculated using noncompartmental methods with WinNonlin Pro software version 6.2 (Pharsight Corporation).
Safety assessments comprised collecting all AEs and SAEs and monitoring cardiac function and evaluations of standard clinical laboratory parameters (haematology, blood chemistry and urinalysis).
2.2.6 Statistical Analyses
In total, 36 subjects were planned to be enrolled in the study to obtain 18 completers in part 1. A sample size of six subjects per genotype group was estimated to provide 80% power to detect a 1.5-fold change in PK parameters (Cmax, AUClast and AUC∞) of siponimod at a significance level of 0.05, with an assumption of an intersubject CV of 25% based on historical data. With six completed subjects per group, the 90% CI of the PK parameters ratio was expected to be within 72–138% of the observed mean, with 90% coverage probability. The SimCYP simulations predicted a 4.5-fold higher (based on AUC∞) siponimod exposure in subjects with a CYP2C9*3/*3 versus CYP2C9*1/*1 genotype [3].
The PK analysis set included all subjects who received the study drug and had at least one available valid PK measurement. The safety analysis set included all subjects who received the study drug. Log-transformed PK parameters of Cmax, AUClast and AUC∞ for siponimod were analysed using a fixed-effects model, with genotype as a fixed factor and baseline body weight as a covariate for part 1. The estimated mean and 90% CIs of difference (CYP2C9*2/*3 vs. CYP2C9*1/*1, and CYP2C9*3/*3 vs. CYP2C9*1/*1) were back-transformed to obtain the geometric mean ratio and 90% CI of the ratio.
3 Results
3.1 Study A: Siponimod PK With and Without Coadministration of Fluconazole in Healthy Subjects
3.1.1 Subject Disposition and Baseline Characteristics
Of the 14 subjects enrolled, 11 completed the study, two withdrew consent, and one discontinued due to an upper respiratory tract infection. All subjects were male and predominantly Caucasian (n = 13, 92.9%). The mean (standard deviation [SD]) age and BMI were 24.1 (4.2) years and 23.9 (2.8) kg/m2, respectively (Table S1, ESM Online Resource 4).
3.1.2 PK of Siponimod Alone and When Coadministered with Fluconazole
The mean plasma concentration–time profiles in both study periods are shown in Fig. 1. The mean PK parameters for siponimod administered alone or in combination with fluconazole at steady-state are presented in Table 1. After a single dose of siponimod alone, the geometric mean Cmax was 31.2 ng/mL, and Tmax was approximately 4 h. The AUClast and AUC∞ were 1110 and 1120 h*ng/mL, respectively. Compared with siponimod alone, coadministration with fluconazole resulted in an approximately twofold increase in the geometric mean AUClast from 1110 to 2160 h*ng/mL, and an increase in geometric mean Cmax from 31.2 to 34.0 ng/mL. The mean CL/F of siponimod was reduced in the presence of fluconazole (1.8 L/h) versus siponimod alone (3.6 L/h). The geometric mean T½ increased from 40.6 to 61.6 h when siponimod was coadministered with fluconazole. Fluconazole did not affect the Tmax or Tlag.

Arithmetic mean (SD) plasma concentration–time profiles of siponimod in healthy subjects after administration of siponimod 4 mg alone and in combination with steady-state fluconazole (inset: semi-logarithmic scale) [Study A]. SD standard deviation
Point estimate ratios (90% CI) for AUClast, AUC∞ and Cmax on coadministration of siponimod with fluconazole versus siponimod alone were 1.97 (1.86–2.08), 1.98 (1.87–2.10) and 1.10 (1.04–1.16), respectively (Table 1).
3.1.3 Safety
In period 1 (siponimod alone), 11 subjects (79%) experienced AEs compared with eight subjects (73%) in period 2 (siponimod + fluconazole). The most commonly reported AEs (≥ 10% of subjects) were headache, dizziness, somnolence, bradycardia, fatigue, seasonal allergy, and upper respiratory tract infection. The majority of AEs reported in the study were of mild intensity. The AEs of moderate intensity reported in the treatment groups were bradycardia and abdominal discomfort (siponimod alone) and headache and disturbance in attention (siponimod + fluconazole), all of which were resolved without any treatment. No deaths or SAEs were reported during the study.
No clinically significant changes were observed in vital signs throughout the study. The most common ECG abnormality observed during the study was bradycardia. None of these events were considered clinically significant. The safety and tolerability profile was consistent with the tolerability in a previous siponimod single-ascending dose range study (unpublished data).
3.2 Study B: Siponimod PK in Healthy Subjects with the CYP2C9 Genetic Polymorphism
3.2.1 Subject Disposition and Demographics
A total of 24 subjects with the CYP2C9*1/*1 (n = 12), CYP2C9*2/*3 (n = 6) and CYP2C9*3/*3 (n = 6) genotypes were enrolled and completed part 1 of the study. All 12 subjects with the CYP2C9*2/*3 and CYP2C9*3/*3 genotypes continued and completed part 2 of the study and were included in both the PK and safety analysis sets. Overall, the mean age of subjects was 41.9 years, and the majority were male (87.5%) and predominantly Caucasian (87.5%). Subject demographics were comparable between genotypes (Table S1, ESM Online Resource 4).
3.2.2 PK of Siponimod and Selected Metabolites: Part 1
The mean plasma concentration–time profile and mean PK parameters of a single oral dose of siponimod 0.25 mg in CYP2C9*1/*1, CYP2C9*2/*3 and CYP2C9*3/*3 subjects in part 1 are presented in Fig. 2a and Table 2, respectively. The PK of siponimod in healthy subjects with the CYP2C9*1/*1 genotype was consistent with historical study data of a single oral dose of siponimod 0.25 mg (unpublished data).

Arithmetic mean (SD) plasma concentration–time profiles of siponimod and its metabolites in healthy subjects with the CYP2C9*1/*1, CYP2C9*2/*3 and CYP2C9*3/*3 genotypes after a single oral dose of siponimod 0.25 mg in Part 1 (linear scale; inset: semi-logarithmic scale) [Study B]. a Siponimod plasma concentration. b Metabolite M3 (LNL925) plasma concentration. c Metabolite M5 (LNL931) plasma concentration. CYP cytochrome P450, SD standard deviation
The mean AUCs were approximately two and fourfold greater in subjects with the CYP2C9*2/*3 and CYP2C9*3/*3 genotypes, respectively, and the mean Cmax increased by 21 and 16%, respectively, versus subjects with the CYP2C9*1/*1 genotype. The median Tmax of 5 h and 4 h in subjects with the CYP2C9*2/*3 and CYP2C9*3/*3 genotypes, respectively, was comparable with that for the CYP2C9*1/*1 genotype (4 h), whereas the T½ was prolonged to 50.9 h and 126 h, respectively, versus the CYP2C9*1/*1 genotype (28.1 h). The mean CL/F of siponimod was reduced in the CYP2C9*2/*3 (1.7 L/h) and CYP2C9*3/*3 (0.9 L/h) genotypes compared with CYP2C9*1/*1 (3.6 L/h).
The mean plasma concentration–time profile and geometric mean PK parameters of the metabolites M3 and M5 following a single oral dose of siponimod 0.25 mg in part 1 are presented in Fig. 2b, c, respectively, and in Table 2. The Cmax and AUC of M3 and M5 were lower in subjects with the CYP2C9*2/*3 and CYP2C9*3/*3 genotypes versus the CYP2C9*1/*1 genotype. The PK of M3 and M5 in subjects with the CYP2C9*1/*1 genotype was consistent with historical study data of a single oral dose of siponimod 0.25 mg [6].
For M3, compared with the CYP2C9*1/*1 genotype, the mean AUC∞, AUClast and Cmax decreased by 12, 66 and 49%, respectively, in subjects with the CYP2C9*2/*3 genotype, and by 23, 76 and 88%, respectively, in subjects with the CYP2C9*3/*3 genotype. The median Tmax was two- and fourfold greater in CYP2C9*2/*3 and CYP2C9*3/*3 subjects, respectively, compared with the CYP2C9*1/*1 genotype, and the T½ was prolonged (54.8 and 96 h, respectively, vs. 32.9 h for CYP2C9*1/*1).
For M5, compared with the CYP2C9*1/*1 genotype, the mean AUClast decreased by 38 and 95% in CYP2C9*2/*3 and CYP2C9*3/*3 subjects, respectively, and Cmax by 47 and 74%, respectively. The half-life was prolonged in CYP2C9*2/*3 (105 h), and was noncalculable in CYP2C9*3/*3 versus CYP2C9*1/*1 (37.9 h).
3.2.3 PK of Siponimod and Selected Metabolites: Part 2
Following administration of siponimod 0.5 mg on day 3 (preceded by 0.25 mg doses on days 1 and 2), the Cmax of siponimod in subjects with the CYP2C9*2/*3 and CYP2C9*3/*3 genotypes was similar to the Cmax observed in the dose titration study [15] for the same dosing regimen. Subjects with the CYP2C9*2/*3 and CYP2C9*3/*3 genotypes were comparably exposed to siponimod, with a mean Cmax of 6.86 and 7.54 ng/mL and a mean AUC24 of 125 and 148 h*ng/mL, respectively. The median Tmax was delayed (both 6 h) versus the Tmax observed in historical data (1.35 h) (unpublished data), and the AUC24 on day 3 was 1.4- and 1.7-fold higher for CYP2C9*2/*3 and CYP2C9*3/*3 subjects, respectively, versus the dose titration study data.
For M3, the mean Cmax was 4.6-fold higher and AUC24 was 4.4-fold higher in CYP2C9*2/*3 subjects versus CYP2C9*3/*3 subjects. The median M3 Tmax was 8 h for both genotypes, i.e. slightly earlier compared with part 1 (12 h for CYP2C9*2/*3 subjects and 24 h for CYP2C9*3/*3 subjects).
For M5, the mean Cmax and AUC24 were four to fivefold higher in subjects with the CYP2C9*2/*3 genotypes compared with the CYP2C9*3/*3 genotype. The median Tmax was 10 h in CYP2C9*2/*3 genotypes and 7 h in CYP2C9*3/*3 genotypes.
3.2.4 Safety
Of the 24 subjects, four (16.7%) reported at least one AE during part 1 of the study, while three (25%) of 12 subjects experienced at least one AE during part 2 of the study. Headache was the most prevalent AE (8.3% in both parts). Only one AE was considered to be possibly related to siponimod (somnolence). The low incidence of AEs did not allow for any meaningful comparison across genotype or phenotype groups.
Most AEs were of mild intensity. Two AEs (conjunctivitis and viral rash) were of moderate intensity and resolved on treatment. With the exception of two events (skin discolouration and temporomandibular joint syndrome), all other AEs were resolved without any need for treatment by the end-of-study visit. No deaths or SAEs were reported during the study, and no systematic, clinically relevant alterations of laboratory, vital signs or ECG data were identified. No clinically significant changes were observed with respect to body weight and vital signs throughout the study.
4 Discussion
The study in healthy subjects with the CYP2C9*1/*1 genotype found that coadministration with fluconazole led to a twofold increase in siponimod AUC, together with a 50% increase in T½, and a minor increase in Cmax compared with siponimod alone. In another study conducted in healthy subjects with polymorphic variants of CYP2C9, siponimod AUC was approximately twofold and fourfold greater in the CYP2C9*2/*3 and CYP2C9*3/*3 genotypes, respectively, and mean Cmax was increased by < 25% compared with the CYP2C9*1/*1 genotype.
For a drug that is a substrate of a polymorphic enzyme or transporter, the difference in drug exposures between EM and PM genotypes would generally represent the most extreme change that could be caused by a strong inhibitor of that pathway. An alternative to a genotype-specific PK study is to administer the investigational drug to EMs with and without concomitant administration of a known strong inhibitor of the metabolic pathway [16]. This interaction approach is considered even more attractive when the prevalence of the PM genotype is very low, as for the CYP2C9*3/*3 genotype. However, in the absence of any strong CYP2C9 inhibitor in clinical use, this alternative approach could not be used to estimate the siponimod PK in CYP2C9 PMs. Two separate studies were therefore conducted to fully characterise the role of the CYP2C9 pathway on siponimod PK [3].
As polymorphic variations in the CYP2C9 enzyme may lead to increased exposure of siponimod due to reduced metabolic clearance, Study A prescreened CYP2C9 genotypes and excluded subjects who were not homozygous for the CYP2C9*1 allele (wild-type) to reduce the intersubject variability in PK parameters and reduce sample size. Study B included subjects with the CYP2C9*2/*3 and CYP2C9*3/*3 genotypes because the largest impact on PK and drug metabolism through CYP2C9 polymorphism is expected in these populations [17], as indicated by in vitro metabolism results and predicted with SimCYP PBPK modelling [3]. In Caucasians, the prevalence of CYP2C9*1/*1 ranges from 62 to 65%; CYP2C9*1/*2 from 20 to 24%; CYP2C9*2/*2 from 1 to 2%; CYP2C9*1/*3 from 9 to 12%; CYP2C9*2/*3 from 1.4 to 1.7%; and CYP2C9*3/*3 from 0.4 to 0.5% [18, 19].
The selection of dose in both studies was based on the PK data generated from the single (unpublished data) and multiple-ascending dose [1] range studies in healthy subjects and was supported by the SimCYP simulations study (SimCYP Ltd) [3]. Single doses of siponimod up to 25 mg were well tolerated and showed favourable safety profiles in healthy subjects in a previous study (unpublished data). In Study A, a dose of siponimod 4 mg was administered as this dose represented a relevant therapeutic dose level for siponimod in multiple sclerosis (MS) at the time this study was conducted. The selected dose provided a sufficient safety margin for an up to fivefold increased systemic exposure due to an interaction with fluconazole (similar exposure as with a single-dose maximum tolerated dose [MTD] of 25 mg). Based on the SimCYP simulation model, steady-state concentration of fluconazole was reached within 3 days following oral doses of 200 mg twice daily on day 1 and 200 mg administered once daily after day 2 [3]. This was consistent with the fluconazole prescribing information indicating that administration of a loading dose (on day 1) of twice the usual daily dose results in plasma concentrations close to steady-state by the second day [20]. To achieve a rapid steady-state plasma concentration of fluconazole, a loading dose of twice daily on the first day of therapy was recommended [21]. A two to threefold increase in siponimod exposure (AUC∞) with fluconazole treatment was expected based on the results of SimCYP simulations [3]. In part 1 of Study B, the selected dose of 0.25 mg was 100-fold lower than the MTD defined in the phase I, single-ascending dose range study in healthy volunteers (unpublished data). In part 2, the target dose of 0.5 mg on day 3 was preceded by 0.25 mg once daily over 2 days in order to attenuate the bradyarrhythmic effects of siponimod, thereby mimicking the first three dosing days of the dose titration regimen used in clinical studies in patients with MS. The SimCYP simulations predicted that siponimod exposure was 4.5-fold higher (based on AUC∞) in subjects with a CYP2C9*3/*3 versus CYP2C9*1/*1 genotype [3]. The therapeutically relevant dose of siponimod in MS, as investigated in pivotal studies, is 2 mg [22, 23]. In view of the dose-linear PK, the 0.5 mg dose in PM subjects was therefore expected to have a similar exposure compared with the therapeutic dose of 2 mg.
Coadministration of a single dose of siponimod and fluconazole at steady-state in healthy subjects (EMs: CYP2C9*1/*1 genotype) led to a twofold increase in plasma AUC of siponimod and an approximately 50% increase in terminal T½. As expected, these results show that fluconazole did not affect the absorption phase of siponimod, and CYP2C9 inhibition only influenced the elimination phase. The results observed in this study were in line with, and confirmed, the predictions obtained through SimCYP simulations [3]. Other enzymes and transporters might also play minor roles in siponimod disposition and hence inhibition of these pathways by fluconazole could have also partly contributed to the increase in siponimod exposure.
Although the expression and activity of the CYP2C9 enzyme is lower overall in the gut than in the liver, the surface area of the proximal small intestine is large, and intestinal CYP2C9 may well contribute to the first-pass metabolism of their substrate drugs [24]. The first-pass effect of siponimod was estimated by SimCYP to be minor (in the range of 5% of the administered dose only (unpublished data). In the DDI study (Study A), the Cmax of siponimod was slightly increased by 10% in the presence of fluconazole. It cannot be excluded that this slight increase could be due to the inhibition of the first-pass effect through fluconazole inhibition of the CYP2C9 enzyme expressed in the duodenum and small intestine.
As siponimod displays dose-proportional and time-independent PK exposure, the observed magnitude increase on exposure in single-dose studies can therefore be extrapolated to steady-state conditions (repeat dose) [25]. SimCYP PBPK modelling [3] was carried out under single-dose and steady-state conditions. Both simulation conditions predicted similar exposure ratios for siponimod under fluconazole inhibition (unpublished data). Therefore, the impact of coadministration of fluconazole on the repeat-dose PK of siponimod (steady state) is predicted to result in an approximately twofold increase in mean AUCtau,ss, similar to the single-dose situation (based on AUC∞).
In a multiple-ascending dose study, siponimod doses up to 20 mg administered daily for 28 days were well tolerated, demonstrating a favourable safety profile at all doses [1]. Siponimod exposure was increased by twofold in the presence of fluconazole, which provided an adequate safety margin of approximately fivefold considering a siponimod therapeutic dose of 2 mg once daily. The increased exposure of siponimod in the presence of moderate CYP2C9 inhibitors is therefore considered not to be associated with safety and tolerability risks for short-term combination treatment. However, for longer coadministration periods, the reduced CYP2C9 metabolic activity may be compensated by reducing the dose of siponimod when administered in the presence of a moderate CYP2C9/CYP3A inhibitor (e.g. fluconazole).
Some key drug-metabolising enzymes, such as CYPs, are known to be modulated by systemic proinflammatory cytokines released during infection or inflammation, resulting in alteration in biotransformation and elimination of small-molecule substrates of the affected CYPs [26]. Systemic levels of interleukin (IL)-6, a potent proinflammatory cytokine, have been found to be elevated in patients with various systemic inflammatory diseases, including MS [27]. A cocktail clinical DDI study conducted in patients with rheumatoid arthritis reported that the administration of an anti-IL-6 receptor monoclonal antibody reversed IL-6-induced suppression of CYP enzyme activity, including CYP2C9 [28]. The influence of disease state on siponimod exposure was investigated in two clinical studies in MS patients and in two population PK evaluations. No significant differences in the PK of MS patients and healthy subjects were observed (unpublished data). Based on these results, systemic IL-6 release is unlikely to significantly affect siponimod PK in patients with MS.
The genetic variation in the CYP2C9 enzyme affects the metabolism of siponimod. After a single oral dose of siponimod 0.25 mg, the AUC∞ and AUClast of siponimod were approximately two and fourfold higher in PMs with the CYP2C9*2/*3 and CYP2C9*3/*3 genotypes, respectively, compared with CYP2C9 EMs (CYP2C9*1/*1). The results observed in the present study were in line with the predictions obtained through the SimCYP simulation method [3]. Due to the reduced CYP2C9 enzymatic activity in PMs, the siponimod plasma T½ was prolonged in subjects with the CYP2C9*2/*3 and CYP2C9*3/*3 genotypes (50.9 and 126 h, respectively) compared with EMs (CYP2C9*1/*1: 28.1 h). The metabolite M3 accounts for approximately 28% of the exposure to siponimod, whereas M5 represents only approximately 2.5% of the parent plasma exposure [6]. Both M3 and M5 have weak pharmacological activity on the S1P1 receptor only (M5 is approximately 470-fold and M3 is approximately 10,000-fold less potent than siponimod on the S1P1 receptor). The metabolite M5 results from metabolism primarily via CYP2C9, with a minor contribution from CYP3A4. A decrease in M3 and M5 exposure is therefore expected when the metabolite activity of CYP2C9 is decreased in CYP2C9 PMs (unpublished data). As expected in M3 and M5 metabolites, Cmax and AUC were markedly lower in the CYP2C9*2/*3 and CYP2C9*3/*3 genotypes compared with the CYP2C9*1/*1 genotype, with a maximum 95% reduction in AUC for the M5 CYP2C9*3/*3 genotype compared with the CYP2C9*1/*1 genotype, confirming the major role of CYP2C9 in siponimod metabolism.
The magnitude of the effect observed in both studies is consistent with the major contribution of the CYP2C9 pathway to total metabolic clearance (fm; 79%) determined in vitro [3].
For drugs where PG is important for PK variability, pharmaceutical companies are encouraged by regulators to assess if enzyme polymorphism may lead to a different benefit–risk in certain genetic subpopulations [29]. These investigations are aimed at evaluating whether exposure in genetic subpopulations is different to such an extent that this would require a change in the posology of the drug for the specific subpopulation to achieve an exposure that is shown to be effective and safe.
Based on the results of Study B on the CYP2C9 genetic polymorphism, a siponimod genotype-based dosing may be considered for individuals with certain genetic polymorphisms, to adjust for the reduced CYP2C9 metabolic activity and avoid potential long-term safety risks of chronic higher exposure.
Final recommendations on the concomitant use of siponimod and CYP2C9/3A4 inhibitors, and on the use of siponimod in SPMS patients with different CYP2C9 genetic polymorphisms, will be made considering all pertinent clinical and in silico data.
5 Conclusions
These two studies quantitatively describe how siponimod metabolism by CYP2C9 can be modulated by exogenous factors such as the CYP2C9 inhibitor fluconazole and by innate CYP2C9 genotypes. Coadministration of siponimod with fluconazole, a moderate CYP2C9 inhibitor, produced no changes in the absorption phase but increased the elimination phase of siponimod in healthy subjects (EMs; CYP2C9*1/*1), resulting in a twofold increase in siponimod exposure. The study on the CYP2C9 genetic polymorphism indicates that reduced CYP2C9 enzymatic activity in healthy PMs (CYP2C9*2/*3 and CYP2C9*3/*3 genotypes) does not affect its absorption but prolongs the elimination of siponimod, resulting in a two and fourfold increase in siponimod exposure in CYP2C9*2/*3 and CYP2C9*3/*3 genotypes, respectively. These findings corroborate the results predicted previously through the SimCYP simulation model for a CYP2C9 inhibitor and the CYP2C9 genetic polymorphism [3].
References
Gergely P, Nuesslein-Hildesheim B, Guerini D, Brinkmann V, Traebert M, Bruns C, et al. The selective sphingosine 1-phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate. Br J Pharmacol. 2012;167:1035–47.
Kappos L, Bar-Or A, Cree BA, Fox RJ, Giovannoni G, Gold R, et al. Siponimod versus placebo in secondary progressive multiple sclerosis: a double-blind randomised, phase 3 study. Lancet. 2018;391(10127):1263–73.
Jin Y, Borell H, Gardin A, Ufer M, Huth F, Camenisch G. In vitro studies and in silico predictions of fluconazole and CYP2C9 genetic polymorphism impact on siponimod metabolism and pharmacokinetics. Eur J Clin Pharmacol. 2018;74:455–64.
Shakeri-Nejad K, Aslanis V, Veldandi UK, Mooney L, Pezous N, Brendani B, et al. Effects of therapeutic and supratherapeutic doses of siponimod (BAF312) on cardiac repolarization in healthy subjects. Clin Ther. 2015;37:2489–505.
Shakeri-Nejad K, Aslanis V, Veldandi UK, Gardin A, Zaehringer A, Dodman A, et al. Pharmacokinetics, safety, and tolerability of siponimod (BAF312) in subjects with different levels of hepatic impairment: a single-dose, open-label, parallel-group study. Int J Clin Pharmacol Ther. 2017;55:41–53.
Gardin A, Dodman A, Kalluri S, Neelakantham S, Tan X, Legangneux E, et al. Pharmacokinetics, safety, and tolerability of siponimod (BAF312) in subjects with severe renal impairment: a single-dose, open-label, parallel-group study. Int J Clin Pharmacol Ther. 2017;55:54–65.
Glaenzel U, Jin Y, Nufer R, Li W, Schroer K, Adam-Stitah S, et al. Metabolism and disposition of siponimod, a novel selective S1P1/S1P5 agonist, in healthy volunteers and in vitro identification of human cytochrome P450 enzymes involved in its oxidative metabolism. Drug Metab Dispos. 2018;46:1001–13.
US FDA. Guidance for Industry Drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendation, 2012. Draft guidance. 2013. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf. Accessed 30 Aug 2017.
European Medicines Agency. Guideline on the investigation of drug interactions. 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf. Accessed 30 Aug 2017.
Human Cytochrome P450 (CYP) Allele Nomenclature Committee. CYP2C9 allele nomenclature. 2016. www.cypalleles.ki.se/cyp2c9.htm. Accessed 30 Aug 2017.
Hirota T, Eguchi S, Ieiri I. Impact of genetic polymorphisms in CYP2C9 and CYP2C19 on the pharmacokinetics of clinically used drugs. Drug Metab Pharmacokinet. 2013;28:28–37.
Van Booven D, Marsh S, McLeod H, Carrillo MW, Sangkuhl K, Klein TE, et al. Cytochrome P450 2C9-CYP2C9. Pharmacogenet Genom. 2010;20:277–81.
WMA Declaration of Helsinki. Ethical Principles for Medical Research Involving Human Subjects. 2013. https://www.wma.net/wp-content/uploads/2016/11/DoH-Oct2013-JAMA.pdf. Accessed 30 Aug 2017.
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Guidance for Industry Investigator Responsibilities—Protecting the Rights, Safety, and Welfare of Study Subjects. 2009. http://www.fda.gov/downloads/Drugs/…/Guidances/UCM187772.pdf. Accessed 30 Aug 2017.
Legangneux E, Gardin A, Johns D. Dose titration of BAF312 attenuates the initial heart rate reducing effect in healthy subjects. Br J Clin Pharmacol. 2013;75:831–41.
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Center for Devices and Radiological Health (CDRH). Guidance for industry—clinical pharmacogenomics: premarket evaluation in early-phase clinical studies and recommendations for labelling. 2013. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM337169.pdf. Accessed 30 Aug 2017.
Kumar V, Brundage RC, Oetting WS, Leppik IE, Tracy TS. Differential genotype dependent inhibition of CYP2C9 in humans. Drug Metab Dispos. 2008;36:1242–8.
Kirchheiner J, Brockmöller J. Clinical consequences of cytochrome P450 2C9 polymorphisms. Clin Pharmacol Ther. 2005;77:1–16.
Scott SA, Khasawneh R, Peter I, Kornreich R, Desnick RJ. Combined CYP2C9, VKORC1 and CYP4F2 frequencies among racial and ethnic groups. Pharmacogenomics. 2010;11:781–91.
Hardman JG, Limbird LE, Gilman AG. Goodman & Gilman’s the pharmacological basis of therapeutics. New York: McGraw-Hill; 2001.
Olkkola KT, Ahonen J, Neuvonen PJ. The effects of the systemic antimycotics, itraconazole and fluconazole, on the pharmacokinetics and pharmacodynamics of intravenous and oral midazolam. Anesth Analg. 1996;82:511–6.
Selmaj K, Li DK, Hartung HP, Hemmer B, Kappos L, Freedman MS, et al. Siponimod for patients with relapsing-remitting multiple sclerosis (BOLD): an adaptive, dose-ranging, randomised, phase 2 study. Lancet Neurol. 2013;12:756–67.
Kappos L, Li DK, Stüve O, Hartung HP, Freedman MS, Hemmer B, et al. Safety and efficacy of siponimod (BAF312) in patients with relapsing-remitting multiple sclerosis: dose-blinded, randomized extension of the Phase 2 BOLD study. JAMA Neurol. 2016;73:1089–98.
Läpple F, von Richter O, Fromm MF, Richter T, Thon KP, Wisser H, et al. Differential expression and function of CYP2C isoforms in human intestine and liver. Pharmacogenetics. 2003;13:565–75.
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Guidance for Industry Investigator Responsibilities—protecting the rights, safety, and welfare of study subjects. 2017. https://www.fda.gov/downloads/drugs/guidances/ucm292362.pdf. Accessed 22 Jun 2018.
Morgan ET. Impact of infectious and inflammatory disease on cytochrome P450-mediated drug metabolism and pharmacokinetics. Clin Pharmacol Ther. 2009;85:434–8.
Janssens K, Slaets H, Hellings N. Immunomodulatory properties of the IL-6 cytokine family in multiple sclerosis. Ann N Y Acad Sci. 2015;1351:52–60.
Zhuang Y, de Vries DE, Xu Z, Marciniak SJ Jr, Chen D, Leon F, et al. Evaluation of disease-mediated therapeutic protein-drug interactions between an anti-interleukin-6 monoclonal antibody (sirukumab) and cytochrome P450 activities in a phase 1 study in patients with rheumatoid arthritis using a cocktail approach. J Clin Pharmacol. 2015;55:1386–94.
European Medicines Agency. Guideline on the use of pharmacogenetic methodologies in the pharmacokinetic evaluation of medicinal products. 2011. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/02/WC500121954.pdf. Accessed 6 Nov 2017.
Acknowledgements
The authors thank the subjects and participating centres involved in the study. They would also like to acknowledge Anuja Shah of Novartis Healthcare Pvt. Ltd. Hyderabad, India, for providing medical writing support, which encompassed preparing the manuscript, formatting, referencing, preparing tables and figures, incorporating authors’ revisions, finalising and submission, all under the direction of the authors and Sivaram Vedantam (Novartis Healthcare Pvt. Ltd.) for review support. In keeping with the guidelines of the International Committee of Medical Journal Editors, all authors have contributed significantly to the study and were thoroughly involved in the critical review of the manuscript for important intellectual content. All authors have reviewed and approved the final draft for submission.
Author information
Authors and Affiliations
Contributions
AG, MU and KSN conceptualised and designed the studies, and analysed and interpreted the study data. EL was involved in the conceptualisation and design of the studies, and supervising the research. GR was involved in study design and study execution. YJ participated in the conception of the studies and in silico predictions of PK outcomes with different dose regimens. ZS was involved in the study execution, data analysis and the preparation of the study report. PP conceptualised and designed the studies, analysed and interpreted the study data, and conducted statistical analysis of the data. WL was involved in the study execution and the acquisition and analysis of data. All authors made significant contributions to data interpretation and review of the manuscript.
Corresponding author
Ethics declarations
Availability of Data and Material
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Funding
This study was funded by Novartis Pharma AG, Basel, Switzerland.
Conflict of interest
Anne Gardin, Eric Legangneux, Yi Jin, Parasar Pal, Wenkui Li, and Kasra Shakeri-Nejad are employees of Novartis. Mike Ufer, Gianluca Rossato, Zhenzhong Su were employees of Novartis at the time of the conduct of this reasearch. They have no other potential conflicts of interest that might be relevant to the content of this manuscript.
Research Involving Human Participants
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gardin, A., Ufer, M., Legangneux, E. et al. Effect of Fluconazole Coadministration and CYP2C9 Genetic Polymorphism on Siponimod Pharmacokinetics in Healthy Subjects. Clin Pharmacokinet 58, 349–361 (2019). https://doi.org/10.1007/s40262-018-0700-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-018-0700-3