
Abstract
Ulcerative colitis (UC) is an inflammatory bowel disease (IBD) of unknown etiology, probably caused by a combination of genetic and environmental factors. The treatment of patients with active UC depends on the severity, localization and history of IBD medication. According to the classic step-up approach, treatment with 5-aminosalicylic acid compounds is the first step in the treatment of mild to moderately active UC. Corticosteroids, such as prednisolone are used in UC patients with moderate to severe disease activity, but only for remission induction therapy because of side effects associated with long-term use. Thiopurines are the next step in the treatment of active UC but monotherapy during induction therapy in UC patients is not preferred because of their slow onset. Therapeutic drug monitoring (TDM) of the pharmacologically active metabolites of thiopurines, 6-thioguanine nucleotide (6-TGN), has proven to be beneficial. Thiopurine S-methyltransferase (TMPT) plays a role in the metabolic conversion pathway of thiopurines and exhibits genetic polymorphism; however, the clinical benefit and relevance of TPMT genotyping is not well established. In patients with severely active UC refractory to corticosteroids, calcineurin inhibitors such as ciclosporin A (CsA) and tacrolimus are potential therapeutic options. These agents usually have a rather rapid onset of action. Monoclonal antibodies (anti-tumor necrosis factor [TNF] agents, vedolizumab) are the last pharmacotherapeutic option for UC patients before surgery becomes inevitable. Body weight, albumin status and antidrug antibodies contribute to the variability in the pharmacokinetics of anti-TNF agents. Additionally, the use of concomitant immunomodulators (thiopurines/methotrexate) lowers the rate of immunogenicity, and therefore the concomitant use of anti-TNF therapy with an immunomodulator may confer some advantage compared with monotherapy in certain patients. TDM of anti-TNF agents could be beneficial in patients with primary nonresponse and secondary loss of response. The potential benefit of applying TDM during vedolizumab treatment has yet to be determined.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
The ulcerative colitis treatment armamentarium includes 5-aminosalicylic acid compounds, corticosteroids, thiopurines, calcineurin inhibitors and monoclonal antibodies (mAbs). |
Therapeutic drug monitoring (TDM) of thiopurines, calcineurin inhibitors and mAbs is being applied in clinical practice. |
Factors associated with pharmacokinetics can be used when applying TDM, e.g. genetic polymorphism for thiopurines or tacrolimus, and body weight, albumin serum concentrations and antidrug antibodies for infliximab (IFX) therapy. |
1 Introduction
Ulcerative colitis (UC) is an inflammatory bowel disease (IBD) probably caused by a combination of genetic and environmental factors [1]. The severity of UC can be classified as mild, moderate or severe depending on clinical (e.g. daily number of stools with or without blood, urgency), biochemical and endoscopic findings [2, 3]. According to the Montreal classification, the disease extent determined at endoscopy can be classified into three subgroups: involvement limited to the rectum (E1), involvement limited to the distal colon not exceeding the splenic flexure (E2), or involvement exceeding the splenic flexure (E3) [4]. The precise etiology of UC is not known and ‘cure’ of this disease is not yet possible. Therefore, the primary therapeutic goal is to induce and maintain remission of the inflammation [5]. The main treatment goals for UC include clinical remission, i.e. relief of symptoms, in combination with mucosal healing. According to the classic step-up approach, treatment of UC starts with administration of 5-aminosalicylic acid (5-ASA) compounds, followed by short-term use of corticosteroids, immunomodulators and biologicals (Fig. 1). As a counterpart to the step-up approach, treatment with immunomodulators and/or biologicals can be applied as therapy of first choice according to the top-down approach [6, 7]. Patients with acute extensive disease activity (Montreal classification E3) could potentially benefit from a top-down approach [7]. Therapeutic drug monitoring (TDM) is increasingly used in the treatment of UC to maximize the clinical benefit of therapeutic agents [8,9,10,11]. TDM is defined as measuring a drug concentration, interpreting this value using pharmacokinetic principles, followed by making decisions about possible dose adjustments or dosing intervals in order to optimize treatment [12]. This review provides an overview of the pharmacokinetic and pharmacodynamic considerations in the treatment of UC.

Classical step-up approach for ulcerative colitis
2 Methods
PubMed was searched for studies including only humans, using the following terms: (“inflammatory bowel disease” [Mesh] OR “ulcerative colitis” [Mesh]) AND (“pharmacokinetic*” OR “pharmacodynamic*”). Articles were selected based on relevance, and additional articles were obtained from their reference lists.
3 5-Aminosalicylic Acid (5-ASA) Compounds
The prodrug sulfasalazine was the first oral 5-ASA compound developed for the treatment of UC. After oral administration, sulfasalazine is split by colonic bacteria with the liberation of the pharmacologically active 5-ASA (mesalazine) and the pharmacologically inactive sulfapyridine [13]. Since sulfapyridine is responsible for several side effects, mesalazine is preferably administered as a direct-acting agent or as a prodrug coupled to other vehicles, in the case of olsalazine. In the treatment of UC, 5-ASA compounds are used to both induce and maintain remission [14]. In patients with mild to moderately active distal UC, topical treatment with mesalazine suppositories or enemas is the preferred initial treatment [5, 15, 16]. Combined oral and topical 5-ASA treatment is preferred in patients with mild to moderately active left-sided colitis [5].
3.1 Pharmacokinetic and Pharmacodynamic Considerations
After oral and rectal administration of mesalazine, a considerable amount of the drug is excreted unabsorbed with the faeces [17]. Mesalazine is unstable in gastric acid and rapidly absorbed in the small intestine, with approximately 20% reaching the terminal ileum and colon [18, 19]. With pH-dependent coatings and controlled-release formulations, premature absorption of mesalazine is prevented until the distal part of the bowel is reached. Absorbed mesalazine undergoes presystemic N-acetylation in intestinal mucosa and the liver. This pharmacologically inactive N-acetylated mesalazine is mainly excreted into the urine [17, 20]. After rectal administration, 10–35% of the drug is absorbed [17], whereas after oral administration, depending on the dose and the type of formulation used, 15–67% of the drug is absorbed and excreted into the urine [21]. Pharmacokinetic parameters are listed in Table 1.
Mesalazine is metabolized locally by the intestinal N-acetyltransferase 1 (NAT1), whereas sulfapyridine, as part of the prodrug sulfasalazine, is metabolized by the hepatic N-acetyltransferase 2 (NAT2) [16]. Both NAT1 and NAT2 genotypes do not predict response for mesalazine and sulfasalazine, respectively, in UC patients [23].
5-ASA acts locally on the colonic epithelium and has multiple anti-inflammatory effects [24]. Although the exact mechanism of action remains to be elucidated, the main mechanism includes inhibition of cyclooxygenase and lipoxygenase, subsequently leading to reduced production of prostaglandins and leukotrienes, respectively [25]. Additionally, the nuclear receptor peroxisome proliferators-activated receptor ligand-γ (PPAR-γ) is activated by 5-ASA. PPAR-γ is a transcription factor that modulates inflammatory responses by inhibiting the production of tumor necrosis factor (TNF), amongst others [18].
After oral and rectal administration, systemic concentrations of 5-ASA are low (< 0.5 µg/ml) compared with intraluminal colonic concentrations, and have been shown to increase with higher oral 5-ASA doses [17, 21, 24, 26,27,28]. Systemic exposure to 5-ASA was shown to be lower after rectal administration than after oral administration of the same dose [28]. This might be due to the physiological absence of specific uptake transporters in intestinal mucosa. Lower systemic exposure after rectal administration could potentially reduce drug toxicity in comparison with oral drug delivery systems. Rectal mesalazine has been shown to be more effective, with clinical improvement rates superior to oral mesalazine in patients with distal UC [29]. To evaluate the concentration–effect relationship of 5-ASA, local tissue measurement of 5-ASA is preferred because of the topical mechanism of action [21]. Increasing the oral dose of delayed-release mesalazine from 1.2 to 2.4 g daily has been associated with an increase in rectal 5-ASA tissue concentrations [21]. Furthermore, an inverse relationship between 5-ASA tissue concentrations and disease activity has been shown [30,31,32]. Likewise, lower mucosal levels of soluble interleukin (IL) receptor 2 were found, a marker of mucosal inflammation [32]. This suggests that the dose-related effect of 5-ASA may depend on the concentration in the mucosa.
With sulfasalazine, multiple dosing per day was required to minimalize the risk of adverse events because of the toxic metabolite sulfapyridine. With the development of sulfapyridine-free 5-ASA compounds, clinical trials followed the same pharmacokinetic principle and the compounds were also administered by multiple dosing; however, a multiple dosing regimen is associated with nonadherence [33]. Kruis et al. showed that once-daily dosing of mesalazine granules was as equally effective and safe as dosing three times per day [34]. Additionally, Multi-Matrix System mesalazine (MMX®, LIALDA®, Asacol HD®) allows high-dose once-daily dosing and is proven to be well tolerated and effective for the treatment of mild to moderately active UC administered as 2.4 or 4.8 g/day [35,36,37]. Recently, high concentration tablets of mesalazine once daily were shown to be as equally effective as multiple doses of mesalazine daily to induce and maintain mucosal healing [38].
4 Corticosteroids
Corticosteroids are potent anti-inflammatory drugs that are used for the treatment of various autoimmune and inflammatory diseases. Remission induction therapy with corticosteroids is appropriate in IBD patients with moderate to severe activity, and in patients with mild activity who are intolerant or refractory to 5-ASA compounds [5, 39]. According to the European Crohn’s and Colitis Organisation (ECCO) guidelines, an appropriate regimen for moderately active disease is administration of oral prednisolone 40 mg/day for 1 week, lowering the daily dose by 5 mg each week and resulting in an 8-week course [5]. For patients admitted with severe UC, initial treatment with intravenous corticosteroids is recommended, with methylprednisone 40–60 mg/day as a bolus infusion. Corticosteroids should not be used as maintenance treatment because of the high probability of developing adverse events and corticosteroid-dependent disease [40, 41].
Remission rates are significantly higher in UC patients receiving oral corticosteroids compared with placebo [42]. Corticosteroids give a remission rate of up to 73% and a number needed to treat for remission of 2 in mild disease, and are therefore considered as effective for remission induction therapy [43]. Corticosteroids bind to the glucocorticoid receptor, and the mechanism of action is based on inhibition of protein transcription and synthesis by affecting the stability of messenger RNA. Corticosteroids downregulate the production of the transcription factor NF-κB and many proinflammatory cytokines, such as IL-1, IL-6 and TNF [44, 45].
4.1 Pharmacokinetic and Pharmacodynamic Considerations
Corticosteroids can be administered via different routes: intravenous, oral, and rectal formulations are available. Prednisone and budesonide are the most commonly used corticosteroids for the treatment of UC.
Prednisolone is the pharmacologically active metabolite of prednisone. Both compounds are rapidly absorbed after oral administration and maximum plasma concentrations are reached after 1–3 h [46]. Prednisolone and prednisone are metabolically interconvertible and are present in plasma in a 10:1 ratio, respectively. Because of the rapid metabolism of prednisone to prednisolone, prednisone bioavailability is generally measured in terms of plasma levels of prednisolone and is approximately 70% [47]. Direct administration of prednisolone bypasses the conversion of prednisone to prednisolone in the liver and is favorable in patients with liver diseases. Only the unbound fraction of prednisolone is biologically active by diffusing into target tissues and cells, and is clinically most relevant [48]. Pharmacokinetic parameters of corticosteroids are listed in Table 2.
Dose-dependent pharmacokinetics have been demonstrated for prednisolone [48]. Both clearance and volume of distribution have been shown to increase with increasing prednisolone concentrations. This dose-dependent clearance of prednisolone might be explained by its concentration-dependent binding to the plasma proteins albumin (low affinity) and transcortin (high affinity) [49, 50]. Higher doses of prednisone saturate the binding to transcortin, and proportionally more unbound prednisolone will be present, resulting in an increased total clearance. For IBD patients, protein binding is decreased in patients with active disease compared with remission, probably due to decreased plasma albumin concentrations in active disease [51].
Budesonide is a locally acting corticosteroid with considerable presystemic elimination. It has a low systemic bioavailability (10%) and thereby exhibits lower systemic toxicity compared with prednisone [19, 52,53,54,55,56]. Budesonide has shown to be efficacious and safe in the treatment of UC [49,50,51]. It is metabolized in the liver via the cytochrome P450 (CYP) 3A4 system into two pharmacologically inactive metabolites, which are primarily excreted via the kidneys [52, 53]. Since the standard formulation of budesonide is completely absorbed in the gastrointestinal tract, controlled-release formulations for oral intake have been developed. Rectal administration of budesonide has also been demonstrated to be an effective therapeutic option in patients with distal UC [54, 55]. Strong CYP3A4 inhibitors, such as ketoconazole, will inhibit the metabolism of budesonide, thereby increasing the area under the curve (AUC) of budesonide [56].
High remission rates and a low number needed to treat have been reported for IBD patients receiving corticosteroids. Nevertheless, approximately 30% of IBD patients have a poor or absent clinical response to corticosteroids [57]. Abnormalities of the glucocorticoid receptor might be the cause of an absent response to corticosteroids. Low affinity of the glucocorticoid receptor for corticosteroids has been shown in a small prospective trial with UC patients who did not respond to intravenous corticosteroid therapy [58]. The cause of this low affinity could not be defined, but accelerated intracellular catabolism of the corticosteroid could result in a decrease in binding between the corticosteroid and the glucocorticoid receptor [58]. Second, this low affinity may be caused by the existence of polymorphisms of the glucocorticoid receptor. However, the association between polymorphisms involving the glucocorticoid receptor and response to corticosteroid treatment in UC patients has not yet been demonstrated [57, 59]. Additionally, in corticosteroid-naive IBD patients, Schottelius et al. showed an increased expression of the glucocorticoid receptor and a decreased affinity to this receptor compared with a healthy control group [60]. Deregulation of the glucocorticoid receptor might therefore be of influence on response to corticosteroid treatment in UC patients, but the exact mechanism remains to be elucidated.
5 Thiopurines
Thiopurines were traditionally used for the treatment of acute leukaemia [61], however their use has now become quite common for the treatment of UC, despite relatively limited clinical trial evidence. Thiopurines are slow-acting drugs, taking up to 6 months to reach a therapeutic effect in these patients. Hence, thiopurines cannot be used as induction therapy. In active UC, treatment with thiopurines has been effective in combination with corticosteroid induction because of the delayed therapeutic effect [5]. Thiopurines can also be used in combination with anti-TNF agents to induce and maintain remission [62]. For corticosteroid-dependent UC, azathioprine (AZA) was more effective than 5-ASA in achieving clinical and endoscopic remission [63, 64].
5.1 Pharmacokinetic and Pharmacodynamic Considerations
Thiopurines include the orally administered prodrugs AZA, mercaptopurine (MP), and thioguanine (TG). TG is considered to be an unconventional thiopurine that is only being used in a few countries when patients do not respond to AZA or MP due to inefficacy or side effects.
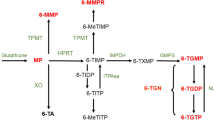
After absorption, glutathione S-transferase converts 90% of AZA to 6-mercaptopurine (6-MP) in erythrocytes [65, 66]. The metabolism of AZA is shown in Fig. 2. 6-MP is metabolized by one of three different enzymes. Xanthine oxidase (XO) and thiopurine S-methyltransferase (TPMT) are catabolic enzymes leading to the inactive metabolites 6-thiouric acid (6-TUA) and 6-methylmercaptopurine (6-MMP), respectively [67]. The metabolite 6-MMP has been related to thiopurine-induced liver toxicity [68]. Metabolism of 6-MP by the anabolic enzyme hypoxanthine–guanine phosphoribosyltransferase (HPRT) eventually leads to the pharmacologically active 6-thioguanine nucleotide (6-TGN) [69]. The low bioavailability (5–37%) of 6-MP is a result of the high first-pass effect caused by the rapid and extensive metabolism of XO into inactive metabolites [70]. The metabolism of 6-TG differs from AZA and 6-MP because 6-TG is metabolized by HPRT directly into 6-TGN, whereas 6-MP nucleotide is oxidized in a rate-limiting step to thioxanthine nucleotide prior to 6-TGN formation [71]. In summary, 6-TGN is responsible for the therapeutic effect, whereas 6-MMP causes hepatotoxicity. Pharmacokinetic parameters of 6-MP are listed in Table 3.

Thiopurine metabolism. 6-TG thioguanine, 6-TGN 6-thioguanine nucleotide, 6-MMP 6-methylmercaptopurine, 6-MP 6-mercaptopurine, 6-TIMP 6-thioinosine monophosphate, 6-TUA 6-thiouric acid, GST glutathione S-transferase, HPRT hypoxanthine–guanine phosphoribosyltransferase, IMPDH inosine 5-monophosphate dehydrogenase, TPMT thiopurine S-methyltransferase, XO xanthine oxidase
After administration of AZA and MP, TPMT competes with XO and HPRT, thereby determining the amount of 6-TGN metabolites that are formed. The gene coding for TPMT exhibits polymorphism, which causes high interpatient variability in exposure to 6-TGN. There are several known genetic polymorphisms related to decreased TMPT activity, and thereby increased 6-TGN concentrations, resulting in side effects [72]. A meta-analysis by Liu et al. showed an association between TPMT polymorphisms, thiopurine-induced overall side effects and bone marrow toxicity [73].
The mechanism of action of thiopurines is still not clear and it is therefore not entirely known why thiopurines are slow-acting. Incorporation of 6-TGN as false purine analogues into DNA and RNA is responsible for the cytotoxic and immunosuppressive effects of thiopurines. After incorporation, DNA damage, cell-cycle arrest and apoptosis occur, resulting in the inhibition of nucleotide and protein synthesis, leading to inhibition of inflammatory gene expression [69]. Apoptosis induction requires co-stimulation with CD28, which is mediated by specific blockade of Rac1 activation through binding of AZA-generated 6-thioguanine triphosphate (6-Thio-GTP) to Rac1 instead of GTP [3]. This mechanism of action might explain a delay in clinical efficacy.
6-TGN concentrations are measured in erythrocytes by the Lennard or Dervieux–Boulieu chromatographic method [74]. Since measurement of 6-TGN concentrations results in 2.6-fold higher levels when measured by the Dervieux–Boulieu method compared with the Lennard method, the type of assay used should be taken into account when interpreting 6-TGN concentrations. Higher 6-TGN levels have been associated with clinical response and remission in IBD patients [68, 75]. 6-TGN concentrations, measured by the Lennard method, of > 230 pmol/8 × 108 have been related to better clinical outcomes in terms of clinical response rates, and 6-TGN concentrations > 450 pmol/8 × 108 are associated with myelotoxicity [68, 76, 77]. Allopurinol influences the pharmacokinetics of thiopurines by increasing production of 6-TGN. Allopurinol inhibits XO and shunts the metabolic pathway of thiopurines towards the production of 6-TGN [78]. The original dose of thiopurine should then be reduced by at least 50% to prevent toxic 6-TGN concentrations. Likewise, combination treatment with 5-ASA can be considered in order to increase 6-TGN concentrations. The metabolic pathway of thiopurine shifts with the addition of 5-ASA, and 6-TGN concentrations consequently increase [79,80,81,82].
A population pharmacokinetic model has been published for pediatric patients with acute lymphoblastic leukaemia treated with 6-MP [83] that showed large interpatient variability in the clearance of pharmacologically active 6-TGN and toxic 6-MMP metabolites. In the population pharmacokinetic model, TMPT genotype was shown to influence the metabolic transformation rate of 6-MP into 6-TGN. This suggests that pharmacogenetically guided dosing of thiopurines is beneficial in optimizing individual treatment. To our knowledge, no population pharmacokinetic models are currently available describing pharmacokinetics of thiopurines in UC patients.
TDM has shown to be useful during treatment with thiopurines because of the small therapeutic range of 6-TGN concentrations balancing between therapeutic concentrations and toxicity [84]. Patients reach steady-state after approximately 4 weeks, therefore TDM within the first 4 weeks is not useful, except when a patient’s nonadherence is suspected. Next to measurement of 6-TGN concentrations, toxic 6-MMP concentrations can be useful when using TDM.
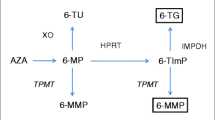
When applying TDM in nonresponders, patients with normal 6-TGN levels are likely refractory to treatment with thiopurines. Patients with low concentrations of both 6-TGN and 6-MMP are most likely underdosed or not compliant. Patients with low 6-TGN levels and high 6-MMP levels are suggested to be metabolically shunting away from 6-TGN production, where more TMPT converts 6-MP to the toxic 6-MMP compared with less 6-MP that is converted to the active 6-TGN by HPRT [85]. The addition of allopurinol can have added value in these patients with a skewed metabolic pathway. An overview of applying TDM in nonresponders is given in Fig. 3. After 4 weeks, steady state is again reached and 6-TGN and 6-MMP concentrations should be re-analysed.

Therapeutic drug monitoring strategy for thiopurines. 6-TGN 6-thioguanine nucleotide, 6-MMP 6-methylmercaptopurine
TPMT genotyping could be considered before starting therapy to identify patients at risk for developing bone marrow toxicity related to 6-MP use [68, 86, 87]. However, there is no consensus regarding the merit of genotyping for TPMT deficiency and related myelotoxicity since TPMT genotype is not the only cause of myelotoxicity [72, 88].
6 Calcineurin Inhibitors
Ciclosporin A (CsA) binds to calcineurin, an enzyme involved in signal transduction of T-lymphocytes and expression of the proinflammatory IL-2 [89]. Tacrolimus is another calcineurin inhibitor that is currently used for the prevention of transplant rejection. Tacrolimus in complex with immunophilin binds to calcineurin, thereby inhibiting phosphatase activity. Inhibition of phosphatase activity results in reduced cytokine production, in particular IL-2, which results in suppressed T-cell proliferation [90]. The introduction of CsA as an immunosuppressive drug after organ (e.g. liver, kidney, lung) transplantation dramatically changed survival rates for this patient population. Intravenously administered CsA has also shown to be beneficial in patients with severe UC [91,92,93]. Monotherapy with CsA can be a short-term alternative in patients with acute severe UC, refractory to corticosteroids, when a rapid onset is preferred as a bridge to thiopurines or vedolizumab [5]. Oral tacrolimus has proven to be safe and effective as remission-induction therapy for patients with steroid refractory UC [94, 95]. In clinical practice, oral tacrolimus is used as both remission-induction and maintenance therapy in UC patients. Tacrolimus is a more potent immunosuppressant than CsA, and, in contrast to CsA, which is only administered intravenously, tacrolimus can be administered orally, usually twice daily.
6.1 Pharmacokinetic and Pharmacodynamic Considerations
CsA and tacrolimus are highly lipophilic compounds with limited solubility. Absorption mainly occurs in the small intestine [96,97,98,99]. For tacrolimus, a bioavailability of 20–25% has been reported [98, 99]. For CsA, bioavailability ranged widely, with values from 11.4 to 87.4% in renal transplant patients [100]. Although most pharmacokinetic studies with CsA have been performed in transplant patients, the pharmacokinetics of CsA have also been studied in 12 patients with Crohn’s disease (CD) after single intravenous and oral administration [101]. This study reported a lower median bioavailability (23.7%) than the average value estimated from a large transplant patient population (30%) [102]. After oral administration of a single dose of CsA in five healthy subjects, a low mean bioavailability was also reported (18%) [103]. Pharmacokinetic parameters of CsA and tacrolimus are listed in Table 4. The influence of food on the bioavailability of CsA has been under discussion, but intake of a high-fat breakfast increased the bioavailability of CsA in healthy volunteers compared with fasting [104]. With the introduction of a microemulsion formula (Neoral®), variability in absorption of CsA could be reduced, and this formula provided a more predictable and consistent concentration–time profile [105, 106]. In an open randomized study with 48 healthy volunteers, the relative bioavailability of the microemulsion formula of CsA compared with the commercial formula ranged from 174 to 239% [107]. For the IBD patient population, the pharmacokinetic parameters of CsA microemulsion were similar to healthy volunteers and other patient populations treated with CsA microemulsion [108].
After absorption, CsA and tacrolimus are metabolized in the liver and small intestine by CYP3A enzymes. Both compounds are substrates for P-glycoprotein (P-gp) [109,110,111,112,113,114]. In the gut, P-gp, also known as multidrug-resistant protein 1, functions as a biological barrier transporting toxins and xenobiotics out of the cell [115]. The predominant metabolite of tacrolimus is 13-O-demethyl-tacrolimus, which has immunosuppressive activity [112]. Up to 95% of tacrolimus is excreted predominantly into faeces as metabolites, and a negligible amount of unchanged drug can be found in both urine and faeces [116]. CsA is primarily excreted into the bile [117].
Drugs metabolized via the CYP3A metabolic pathway interfere with CsA and tacrolimus metabolism, which can result in altered blood concentrations. Genetic polymorphism of CYP3A genes could potentially explain the widely ranging inter- and intravariability in absorption and clearance of CsA. However, a relationship between CYP3A4 polymorphism and required dose adjustments has not been found [118,119,120,121]. Polymorphism in ABCB1, the gene encoding for P-gp, and CYP3A5 polymorphism have been suggested to affect the pharmacokinetics of tacrolimus in UC patients [122, 123]. In Asian UC patients, CYP3A5*1 polymorphism (CYP3A5 ‘expression’) has been associated with lower tacrolimus trough concentrations and lower clinical remission rates at week 4 compared with patients without CYP3A5*1 [122]. In 89 IBD (mostly UC) patients treated with tacrolimus, the influence of polymorphism in both CYP3A5 and ABCB1 were studied [123]. Variants in ABCB1 polymorphism, particular at position 3435 (ABCB1 3435C>T), showed an association with short-term clinical remission. Genotyping for tacrolimus can be of interest to individualize tacrolimus therapy, but implementation in clinical practice would be costly and would likely not affect the decision to start tacrolimus.
Because of the large interindividual variability and narrow therapeutic range, TDM for CsA has great potential for improving clinical outcomes and has been extensively described for patients after renal transplantation. Trough samples were originally taken as a surrogate for exposure (AUC) to reduce toxicity in these patients. However, CsA samples at trough did not predict rejection-free survival and was therefore considered as a suboptimal clinical predictor. Subsequent studies identified AUC for the first 12 h (AUC12) as a more sensitive predictor. Because monitoring of AUC12 is rather impractical, CsA concentrations 2 h after administration are considered best to use when applying TDM in transplant patients [124,125,126,127,128,129]. Although CsA concentrations 2 h after administration are measured in renal transplant patients, in patients with an UC exacerbation it is recommended to measure trough concentrations at days 2 and 4 after the start of CsA. For intravenous and orally administered CsA, trough concentrations between 250 and 450 ng/ml, and 150 and 250 ng/ml, are aimed for, respectively. For IBD patients with a dose of 4 mg/kg/day, steady-state trough concentrations were between 300 and 400 ng/ml [130]. UC patients receiving a dose of 2 mg/kg and achieving trough concentrations between 150 and 250 ng/ml have shown similar efficacy and less adverse events [131, 132].
The extensive binding of tacrolimus to erythrocytes is saturable, and hence concentration-dependent. The blood–plasma ratio for tacrolimus ranges from 15:1 to 35:1, and can vary even more depending on drug concentration and haematocrit values [98, 111, 133, 134]. The blood–plasma ratio will decrease with higher drug concentrations and lower haematocrit values. Because tacrolimus binds extensively to erythrocytes, drug levels are routinely being measured in whole blood [99]. In plasma, tacrolimus binds to α1-acid glycoprotein and albumin with high affinity; its unbound fraction is < 1%. Unbound tacrolimus is thought to mediate the pharmacological effects [133,134,135]. In a randomized, placebo-controlled, dose-finding study, corticosteroid-refractory active UC patients received placebo or oral tacrolimus administered twice daily over a 2-week period, followed by an open-label 10-week extension [94]. Based on tacrolimus whole blood trough levels, patients were assigned to either a high (10–15 ng/ml) or low (5–10 ng/ml) trough concentration group. The clinical response rate (improvement in disease activity index score ≥ 4 points, all categories improved) at 2 weeks was higher in patients with the high troughs compared with low troughs (68.4 vs. 38.1%, respectively). Consequently, in daily practice, the target range of tacrolimus trough levels ranges from 10 to 15 ng/ml, since this range is associated with improved clinical outcomes in UC. Blood trough levels of tacrolimus can be affected by fasting, and absorption of tacrolimus decreases by the presence of food [136, 137]. The maximum concentration (Cmax) after oral administration is up to fourfold higher during fasting compared with a fed condition.
7 Biologicals
7.1 Anti-Tumor Necrosis Factor Agents
According to the classic therapeutic step-up approach, use of monoclonal antibodies (mAbs) is considered to be the last medical treatment option before surgery. However, in severe UC, IFX can also offer an early treatment option. MAbs targeting TNF cover the majority of the registered therapeutic mAbs used in the treatment of UC. Approved therapeutic mAbs for UC are of the immunoglobulin (Ig) G1 antibody type consisting of a Fab fragment and a Fc-portion. IFX, an intravenously administered chimeric IgG1 antibody, was the first anti-TNF agent approved for the treatment of UC. Adalimumab (ADL) and golimumab (GLM) are both fully human, subcutaneously administered IgG1 antibodies registered for the treatment of UC. These agents are effective as induction as well as maintenance treatment in UC [138,139,140,141,142]. A meta-analysis conducted by Thorlund et al. showed superiority compared with placebo for all three anti-TNF agents in terms of achieving clinical response and remission after 6–8 weeks [143]. Additionally, a more recent meta-analysis showed that anti-TNF agents were more effective than placebo for both inducing (45 vs. 30%) and maintaining mucosal healing (33 vs. 18%) [144].
7.1.1 Pharmacokinetic and Pharmacodynamic Considerations
Therapeutic mAbs are administered parenterally since oral administration of mAbs is challenged by their high molecular weight (approximately 150 kDa), hydrophilicity, and gastric degradation. Despite these drawbacks, a novel oral anti-TNF mAb is under development and has proven efficacy in both IBD mouse models and a first-in-human trial in UC patients [145, 146]. Intravenous administration of a mAb provides instantaneously high systemic concentrations, which is beneficial in severe UC. After subcutaneous administration of the mAb, absorption takes place via lymphatic drainage, and the Cmax is reached after several days [147]. Systemic absorption and presystemic catabolism of subcutaneously administered mAbs determine their bioavailability [147]. Reported bioavailabilities for ADL after subcutaneous administration range from 50 to 100%, and a bioavailability of 51% is reported for GLM [147, 148]. Additionally, no differences were found in bioavailability after administration of GLM in the upper arm, thigh or abdomen [148]. Pharmacokinetic parameters of IFX, ADL, and GLM are listed in Table 5. Because of their high molecular weight and hydrophilicity, there is limited distribution of mAbs into peripheral tissue. A volume of distribution of 4.5–6 L has been reported for IFX at steady state, approximately equal to the extracellular fluid volume [149]. The exact mechanisms by which mAbs are eliminated from the body are not fully understood, but the primary route is via proteolytic catabolism after receptor-mediated endocytosis in the reticuloendothelial system (RES). Additionally, loss of IFX into stool has been demonstrated in patients with moderate to severely active UC, with highest faecal IFX concentrations found in patients with primary nonresponse [150]. Elimination via the faeces has not yet been demonstrated for other biologicals.
IgG1 mAbs interact with the neonatal Fc receptor (FcRn), thereby increasing their half-life (Fig. 4). FcRn is functionally expressed in monocytes, macrophages, dendritic cells, and endothelial cells [151, 152]. After endocytosis into vascular endothelial cells, FcRn binds the IgG antibody within the acidic environment of the endosome. Binding to FcRn results in protection from degradation, thereby prolonging half-life. The IgG antibody is returned to the cell surface and dissociates from FcRn in a physiologic environment (pH 7.4) and is released into the systemic circulation.

FcRn recycling mechanism. IgG immunoglobulin G, FcRn neonatal Fc receptor
In patients with active IBD, increased levels of TNF have been found in blood, epithelial tissue and stool [153, 154]. TNF is a pro-inflammatory cytokine that is produced by macrophages and monocytes [155]. TNF is a trimer and appears as both soluble (sTNF)- and membrane (mTNF)-bound TNF. IFX binds to both trimeric and monomeric forms of TNF; the latter may prevent association of these subunits to form the bioactive trimeric TNF [156]. TNF is produced as mTNF, from which sTNF is released by proteolytic cleavage [157]. Both sTNF and mTNF production is increased in patients with IBD, and both forms play a role in driving intestinal inflammation [157]. Binding of sTNF to type 1 (TNF-R1) and type 2 TNF receptors (TNF-R2) mediates multiple biologic effects. Binding to TNF-R1 leads to apoptosis and NF-κB activation and translocation to the nucleus. TNF-R2 has a role in circulating blood leukocytes and can lead to NF-κB signalling [158, 159]. mTNF also binds to TNF-R1 and TNF-R2, but its biological activity is probably assigned to binding to TNF-R2 [160]. Both TNF-R1 and TNF-R2 are cell surface receptors and are expressed on almost all nucleated cells [161]. Polymorphisms in genes encoding TNF receptors have been studied, and Pierik et al. found that the TNFR2 587G allele was more frequently present in UC. However, the TNF receptor polymorphisms did not predict clinical response to IFX treatment [159].
Higher serum trough concentrations of anti-TNF agents during maintenance treatment are associated with improved clinical outcomes (i.e. clinical response, clinical remission and mucosal healing) in UC patients. In contrast, low(er) concentrations have been demonstrated to be associated with the formation of antidrug antibodies (ADAs). Serum trough IFX concentrations at steady-state between 3 and 7 µg/ml have been related to improved clinical outcomes [9, 138, 162,163,164,165,166,167,168,169,170].
For ADL, serum trough concentrations > 7.1 µg/ml are associated with mucosal healing, with the increased rate of mucosal healing reaching a plateau at 12 µg/ml in IBD patients [171]. For GLM, less data are available with regard to optimal serum trough concentrations. To date, there is no consensus about what the therapeutic range of GLM should be. Detrez et al. performed a prospective trial to define the therapeutic range of GLM in UC patients. A cut-off value of 2.6 µg/ml at week 6 was associated with partial clinical response at 14 weeks (defined as clinical improvement despite persistent rectal blood loss) [172].
Although efficacy for anti-TNF agents has been proven, up to 30% of patients have no clinical improvement after initiation of anti-TNF therapy (primary nonresponse), and up to 50% of patients lose response over time (secondary nonresponse) [173]. TDM may therefore be applied in order to prevent primary and secondary nonresponse [11, 174,175,176,177]. To understand the mechanisms behind the phenomenon of primary and secondary nonresponse, understanding of factors that influence the pharmacokinetics and pharmacodynamics of mAbs is most important. Population pharmacokinetic modeling has provided insight into factors associated with the clearance of mAbs by identifying variability between and within patients.
For IFX, several population pharmacokinetic analyses have been performed to identify and quantify important patient characteristics influencing the pharmacokinetics in UC patients. First, the relationship between the presence of antibodies against IFX and an increased clearance of IFX in patients with UC has repeatedly been shown [178,179,180]. Second, baseline serum albumin concentrations have been found to be inversely and strongly related to clearance [178]. Like IFX, albumin also interacts with FcRn, thereby prolonging half-life (Fig. 4). Lower albumin concentrations, representing a more severe disease status, could reflect a lower number of FcRns, thereby explaining an increased clearance of IFX. This inverse relationship has also been reported by Buurman et al. [180]. However, due to the low variability in albumin levels of patients included in that study, this effect was not large enough to be incorporated into their final population pharmacokinetic model.
Third, body weight has been identified as a covariate influencing volume of distribution [178, 179]. Since volume of distribution of IFX is similar to the extracellular fluid volume, and this increases with body weight, the effect of body weight on volume of distribution is expected. This shows the need for weight-based dosing of IFX in order to allow equivalent exposure to IFX among patients with different body weights.
Elevated C-reactive protein (CRP), as a marker of disease activity, has been associated with lower IFX trough concentrations and loss of response to IFX in IBD patients [181, 182]. For patients with rheumatoid arthritis, higher baseline CRP has been associated with an increased clearance; however, CRP is not always elevated in patients with mild to moderately active UC [183]. This might explain why CRP is often not recognized as a significant covariate associated with increased clearance of IFX in UC patients. In addition, albumin might already explain most of the variability seen in the clearance of IFX due to disease activity.
For ADL, no population pharmacokinetic analyses have been performed to identify factors influencing pharmacokinetics in UC patients. Nevertheless, Ternant et al. were the first to report a quantitative description of the pharmacokinetics of ADL in patients with CD [184], and showed a 5.5-fold increase in clearance in the presence of antibodies to ADL. Sharma et al. also identified the influence of antibodies to ADL, and additionally reported body weight as a significant covariate influencing both clearance and volume of distribution in a paediatric CD patient population treated with ADL [185].
To identify factors influencing the pharmacokinetics of GLM, a post hoc analysis of the PURSUIT trial was performed [186]. Body weight, serum albumin and antibodies against GLM were found as influencing factors on the pharmacokinetics of GLM.
The presence of ADAs can be persistent or transient (i.e. transient ADAs disappear over time) and neutralizing or non-neutralizing [163, 187, 188]. Neutralizing ADAs directly prevent an anti-TNF agent from binding TNF by binding to the TNF-binding fragment (Fab) of the anti-TNF agent or by steric hindrance. Non-neutralizing ADAs also form a complex with anti-TNF agents, but do not prevent binding of the anti-TNF agent to TNF; however, the pharmacokinetics of the anti-TNF agents might still be altered [188]. Different assays can be used to assess the ADA status of a patient. Most assays cannot distinguish between neutralizing and non-neutralizing ADAs, and quantitative results cannot be compared between different assays or different drugs. Most of these assays are not drug tolerant and are therefore not able to detect ADAs in the presence of the drug [189]; however, drug-tolerant assays are able to detect antibodies in the presence of the drug [190]. A recent paper by Van Stappen et al. evaluated the clinical relevance of a drug-tolerant assay [191], and concluded that a drug-tolerant assay allows closer follow-up of the effect of dose intensification on ADA concentrations and the differentiation between transient and persistent ADAs. However, no clinical benefit was shown for the use of a drug-tolerant assay during maintenance treatment with IFX.
Next to ADA formation, concomitant treatment with immunomodulators, such as thiopurines or methotrexate (MTX), can also influence the pharmacokinetics of anti-TNF mAbs [192, 193]. Higher IFX trough concentrations have been found in IBD patients receiving both IFX and concomitant immunomodulator treatment compared with the IFX monotherapy group (7.5 vs. 4.6 µg/ml, p = 0.04) [194]. The addition of an immunomodulator to IBD patients receiving anti-TNF monotherapy, with loss of response due to immunogenicity, has been shown to result in undetectable ADAs, increased serum drug concentrations and regained clinical response [193]. In addition, the UC SUCCESS trial showed significantly higher rates of corticosteroid-free remission (39.7 vs. 22.1%) and mucosal healing (62.8 vs. 54.6%) at week 16 in UC patients receiving IFX/AZA combination therapy compared with patients receiving IFX alone [62]. In clinical practice, the initiation of anti-TNF therapy should be combined with an immunomodulator to prevent the formation of detectable ADAs.
Only a part of the interpatient variability of clearance of anti-TNF agents is explained by body weight, ADAs and disease activity (albumin). Unexplained interpatient variability may be reduced by TDM. Dashboard systems (software packages that integrate information from multiple components into a single display for clinical use) are currently being developed to implement adaptive dosing strategies [195]. Dashboard systems for clinical decision support use Bayesian approaches in which a priori information is combined with a posteriori information to predict future concentrations [196]. A recent paper by Wojciechowski et al. showed the potential benefits of model-based dosing for IFX [197]. They compared several dosing strategies by simulating a virtual population with time-varying covariates. This in silico assessment showed that model-based approaches were superior to other strategies (label recommendations, TDM-guided stepwise dosing, TDM-guided proportional dosing). The application of model-based dosing during the maintenance phase resulted in more patients reaching target trough concentrations, and additionally decreased interpatient variability in IFX serum concentrations; however, this should be confirmed by a large prospective trial.
The randomized controlled TAXIT trial included 263 adult IBD patients with stable response to maintenance IFX therapy [198]. After dose optimization, patients received either clinically based dosing or concentration-based dosing based on an algorithm. Concentration-based dosing was not superior to clinically-based dosing regarding clinical and biochemical remission after 1 year, but was associated with fewer flares. Recently, the TAILORIX trial has been published comparing concentration-based dosing with adaptive dosing based on symptoms alone [199]. Anti-TNF-naive CD patients starting on IFX were included, and the percentage of patients with sustained corticosteroid-free clinical remission from weeks 22 through 54 was assessed. Although the study was stated to be underpowered to show the benefit of TDM, it was concluded that concentration-based IFX dosing was not superior to dosing based on symptoms alone. In conclusion, the prospective TAXIT and TAILORIX trials were not able to show the merit of TDM in IFX maintenance treatment; however, when a patient is lacking or losing clinical response to IFX, TDM provides useful information for the clinician. With low serum trough concentrations, the proven concentration–effect relationship for IFX justifies increasing the dose or decreasing the dosing interval in order to (re)gain clinical response. In addition, detectable ADAs can be overcome by the addition of an immunomodulator. When a patient is lacking or losing response and TDM shows sufficiently high serum trough levels of IFX, switching to another therapy should be considered. Potential benefit of TDM during the induction phase remains an interesting concept, but little is known about the optimal therapeutic window for IFX during induction phase. Papamichael et al. showed that IFX trough concentrations of 28.3, 15, and 2.1 µg/ml are associated with short-term mucosal healing at weeks 2, 6, and 14, respectively [200]. On the contrary, TDM can also be applied for dose de-escalation. Most mAbs do not have a maximum tolerated dose resulting in toxic concentrations, but high-dose IFX may be associated with more serious adverse events requiring hospitalization [201]. Regarding costs, in CD patients with secondary loss of response to IFX, algorithm-based dosing using serum IFX concentrations was shown to reduce average treatment costs in the long-term and without any negative effect on clinical efficacy [202, 203].
7.2 Vedolizumab
The latest addition to the biological treatment armamentarium for UC is vedolizumab, which was registered by the EMA and US FDA in 2014. Vedolizumab is a humanized IgG1 mAb that targets α4ß7-integrine, which is present on T cells in the lamina propria [204]. Binding of vedolizumab to α4ß7 prevents binding of α4ß7 to mucosal vascular addressin cell adhesion molecule 1 (MAdCAM-1), which is expressed on endothelial cells [205]. Prevention of the α4ß7/MAdCAM-1 interaction blocks the infiltration of α4ß7+ cells into the gastrointestinal mucosa and gut-associated lymphoid tissue, suppressing gut inflammation [205, 206]. The pivotal GEMINI-1 trial showed efficacy of vedolizumab in UC, with a greater proportion of patients achieving clinical response, clinical remission, and mucosal healing compared with placebo [207]. Vedolizumab is administered intravenously as a fixed dose of 300 mg at weeks 0, 2 and 6, followed by infusion every 8 weeks during the maintenance phase.
7.2.1 Pharmacokinetics and Pharmacodynamics
As an IgG mAb, vedolizumab exhibits the same pharmacokinetic principles as anti-TNF mAbs. The half-life of vedolizumab (25.5 days) is relatively long compared with other therapeutic mAbs [149, 208]. In theory, it takes four to five times a half-life before a drug reaches steady state and has maximal potential effect. It also takes four to five times the half-life before the drug is fully eliminated from the body after discontinuation of treatment. Pharmacokinetic parameters of vedolizumab are listed in Table 6. Elimination of vedolizumab has been described by parallel linear and nonlinear elimination [208]. The linear elimination represents a nonsaturable pathway, such as Fc-mediated elimination that occurs at higher therapeutic concentrations. The nonlinear elimination might be the result of clearance by saturable, target-mediated mechanisms such as receptor-mediated endocytosis occurring at lower concentrations. This target-mediated drug disposition has previously been reported for therapeutic mAbs showing affected pharmacokinetics by high-target affinity [209, 210].
A positive relationship between vedolizumab exposure and efficacy (clinical response and clinical remission) has been revealed for UC patients [207, 211]. Clinical remission rates at week 6 were comparable for patients with vedolizumab trough concentration < 16 µg/ml and the placebo group [211]. The GEMINI trial showed numerically higher clinical response and clinical remission rates at week 6 in UC patients with higher vedolizumab trough concentrations compared with patients without clinical response or remission [207]. Almost 30% of patients with vedolizumab trough concentrations at week 6 of between 0 and 16.7 µg/ml achieved clinical response. With vedolizumab trough concentrations between 16.7 and 24.8 µg/ml, 47% of patients achieved a clinical response. Rosario et al. confirmed this finding in a post hoc analysis of all treated patients from the GEMINI trials. They demonstrated that higher baseline serum albumin, lower faecal calprotectin and no previous anti-TNF agent exposure were associated with an increased remission probability in UC patients [211]. A population pharmacokinetic analysis of patients with CD and UC treated with vedolizumab showed similar pharmacokinetic parameters between CD and UC patients [208]. Both body weight and albumin serum concentrations were influencing factors on clearance of vedolizumab. In this post hoc study, concomitant use of immunomodulators was not found to be of influence on the clearance of vedolizumab.
The pharmacodynamics of vedolizumab were assessed in a randomized controlled trial in patients with active UC [212]. Patients were randomly assigned to a low-dose group (0.5 mg/kg), a high-dose group (2.0 mg/kg) or placebo. In both the low- and high-dose groups, saturation of α4ß7 on more than 90% of T-cells in the peripheral circulation was observed at weeks 4 and 6.
With regard to immunogenicity, ADAs for vedolizumab were detected in 4% of patients participating in the GEMINI-1 (UC patients) and GEMINI-2 (CD patients) trials during the first year of treatment. The use of concomitant immunomodulators reduced the rate of ADAs from 4 to 3%, a nonrelevant difference from a clinical point of view [213]. Based on data from the ongoing GEMINI long-term safety study, the immunogenicity rate does not appear to increase over time [213].
The potential benefit of applying TDM during vedolizumab treatment has to be determined as an optimal therapeutic range has not yet been defined. The results of a prospective observational trial with CD and UC patients showed an association between vedolizumab trough concentrations < 18.5 μg/ml at week 6 and the need for extended therapy within the first 6 months [214]. Nevertheless, adjustments in vedolizumab dosing are limited to adjustments in dosing interval since a fixed dose of 300 mg is used for all patients. After evaluation of a weight-based dose (6 mg/kg) in healthy volunteers, higher vedolizumab exposure was seen in patients with higher body weight [215]. Weight-adjusted dosing in these patients overcompensated for exposure of vedolizumab, and subsequent phase III studies were therefore conducted with fixed-dose regimens. Although only extreme values of body weight were of clinically relevant influence on the clearance of vedolizumab, Rosario et al. have shown a clear association between higher bodyweight and increased clearance [208]. This finding might support the use of vedolizumab dosing based on body weight, rather than a fixed dose, to optimize vedolizumab treatment.
8 Conclusion
An overview is provided of the pharmacokinetic and pharmacodynamic clinical considerations in the treatment of UC patients. Treatment of UC is discussed according to the classic step-up approach. For patients with mild to moderately active distal colitis, 5-ASA compounds are the preferred initial treatment. Combination treatment with both oral and topical 5-ASA is recommended in patients with mild to moderately active left-sided colitis. The efficacy of 5-ASA can be assessed by measuring local tissue concentrations via biopsies taken at endoscopy. Since this is rather impractical, this has no implications for clinical practice. Short-term use of corticosteroids, such as prednisone and budesonide, is appropriate in UC patients with moderate to severely active disease and in patients with mild activity who are intolerant or refractory to 5-ASA compounds. Treatment with CsA or tacrolimus can be a short-term alternative in patients with acute severe UC, refractory to corticosteroids, when a rapid onset is preferred as a bridge to the next treatment step. However, proven, effective, oral treatment with tacrolimus is a more potent calcineurin inhibitor compared with CsA and tacrolimus exhibits a less variable pharmacokinetic profile. For optimal clinical efficacy, trough concentrations for tacrolimus should be 10–15 ng/ml. Treatment with thiopurines is the next step in the step-up strategy of the treatment of active UC. 6-TGN concentrations > 230 pmol/8 × 108 (Lennard method) are associated with increased clinical response and remission rates, whereas 6-TGN concentrations > 450 pmol/8 × 108 (Lennard method) are associated with myelotoxicity. Lastly, treatment of UC has been greatly improved since the introduction of mAbs. In particular, the introduction of anti-TNF agents such as IFX and ADL led to higher clinical and endoscopic remission rates. However, a proportion of patients do not respond to induction treatment (primary nonresponders) and secondary loss of response is also not uncommon. Primary nonresponse to anti-TNF therapy can be associated with underdosing in the acute phase of disease, often combined with faecal loss through the ulcerated, ‘leaky’ gut. Secondary loss of response can be seen in patients with low serum drug levels, often in combination with the formation of ADAs. These factors, among factors associated with the clearance of mAbs, can be used in applying TDM. Since patients with lower albumin concentrations show higher IFX clearance, adjustments in dosing or dosing interval should be taken into account in patients with low albumin concentrations. The same applies for patients with detectable ADAs, where adjustments in dosing and dosing interval should be taken into account to potentially overcome these detectable ADAs. Additionally, the addition of concomitant immunomodulators (thiopurines/MTX) can overcome this detectable presence of ADAs. Vedolizumab, the latest addition to the biological treatment armamentarium for UC, is a humanized IgG1 mAb that targets α4ß7-integrine, which is present on T-cells in the lamina propria. Because of this gut-specific type of action, vedolizumab can be an option for UC patients not responding to anti-TNF therapy, or patients with recurrent infections due to the overall immunosuppressive side effect of anti-TNF therapy.
The focus of this review was the step-up strategy for the treatment of UC patients; however, in some patients, a top-down strategy should be the preferred option. This more aggressive approach, starting immediately with anti-TNF treatment, preferably in combination with an immunomodulator, might be needed in patients with acute severe UC. The benefit of early treatment with vedolizumab is still being investigated.
References
Magro F, Gionchetti P, Eliakim R, Ardizzone S, Armuzzi A, Barreiro-de Acosta M, et al. Third European evidence-based consensus on diagnosis and management of ulcerative colitis. Part 1: definitions, diagnosis, extra-intestinal manifestations, pregnancy, cancer surveillance, surgery, and ileo-anal pouch disorders. J Crohn’s Colitis. 2017;11(6):649–70.
Truelove S, Witts L. Cortisone in ulcerative colitis. BMJ. 1955;2:1041–8.
Inflammatory Hanauer S, Disease Bowel. N Eng J Med. 1996;334:841–8.
Satsangi J, Silverberg MS, Vermeire S, Colombel J-F. The Montreal classification of inflammatory bowel disease: controversies, consensus, and implications. Gut. 2006;55(6):749–53.
Harbord M, Eliakim R, Bettenworth D, Karmiris K, Katsanos K, Kopylov U, et al. Third European evidence-based consensus on diagnosis and management of ulcerative colitis. Part 2: Current Management. J Crohns Colitis. 2017;11:769–84.
Rogler G. Top-down or step-up treatment in Crohn’s disease? Dig Dis. 2013;31(1):83–90.
Sandborn WJ. Step-up versus top-down therapy in the treatment of ulcerative colitis. Gastroenterol Hepatol (N Y). 2007;3(1):16–7.
Oude Munnink T, Henstra M, Segerink L, Movig K, Brummelhuis-Visser P. Therapeutic drug monitoring of monoclonal antibodies in inflammatory and malignant disease: translating TNF-α experience to oncology. Clin Pharmacol Ther. 2016;99(4):419–31.
Baert F. Is there a role for therapeutic drug monitoring of anti-TNF monoclonal antibodies in inflammatory bowel disease. Dig Dis. 2015;33(Suppl 1):70–7.
Mould D. Why therapeutic drug monitoring is needed for monoclonal antibodies and how do we implement this? Clin Pharmacol Ther. 2016;99(4):351–4.
Stroh M, Lum B. Should therapeutic drug monitoring for monoclonal antibodies remain the exception or become the norm? Clin Pharmacol Ther. 2016;100(3):215–7.
Touw DJ, Neef C, Thomson AH, Vinks AA. Cost-effectiveness of therapeutic drug monitoring: a systematic review. Ther Drug Monit. 2005;27(1):10–7.
Peppercorn MA, Goldman P. The role of intestinal bacteria in the metabolism of salicylazosulfapyridine. J Pharmacol Exp Ther. 1972;181(3):555–62.
Ford AC, Achkar J-P, Khan KJ, Kane SV, Talley NJ, Marshall JK, et al. Efficacy of 5-aminosalicylates in ulcerative colitis: systematic review and meta-analysis. Am J Gastroenterol. 2011;106(4):601–16.
Gionchetti P, Rizzello F, Venturi A, Ferretti M, Brignola C, Miglioli M, et al. Comparison of oral with rectal mesalazine in the treatment of ulcerative proctitis. Dis Colon Rectum. 1998;41(1):93–7.
Marshall JK, Thabane M, Steinhart AH, Newman JR, Anand A, Irvine EJ. Rectal 5-aminosalicylic acid for induction of remission in ulcerative colitis. Cochrane Database Syst Rev. 2010;(1):CD004115.
Klotz U, Maier KE. Pharmacology and pharmacokinetics of 5-aminosalicylic acid. Dig Dis Sci. 1987;32(12):46–50.
Criscuoli V, Modesto I, Orland A, Cottone M. Mesalazine for the treatment of inflammatory bowel disease. Expert Opin Pharmacother. 2013;14(12):1669–78.
Haagen Nielsen O, Bondesen S. Kinetics of 5-aminosalicylic acid after jejunal instillation in man. Br J Clin Pharmacol. 1983;16:738–40.
Bondesen S. Intestinal fate of 5-aminosalicylic acid: regional and systemic kinetic studies in relation to inflammatory bowel disease. Pharmacol Toxicol. 1997;81:3–28.
Hussain FN, Ajjan RA, Riley SA. Dose loading with delayed-release mesalazine: a study of tissue drug concentrations and standard pharmacokinetic parameters. Br J Clin Pharmacol. 2000;49(4):323–30.
Quetglas EG, Armuzzi A, Fiorine G, Barnscheid L, Danese S. Review article: the pharmacokinetics and pharmacodynamics of drugs used in inflammatory bowel disease treatment. Eur J Clin Pharmacol. 2015;71:773–99.
Ricart E, Taylor WR, Loftus EV, O’Kane D, Weinshilboum RM, Tremaine WJ, et al. N-acetyltransferase 1 and 2 genotypes do not predict response or toxicity to treatment with mesalamine and sulfasalazine in patients with ulcerative colitis. Am J Gastroenterol. 2002;97(7):1763–8.
Campieri M, Lanfranchi GA, Boschi S, Brignola C, Bazzocchi G, Gionchetti P, et al. Topical administration of 5-aminosalicylic acid enemas in patients with ulcerative colitis. Stud Rectal Absorpt Excretion Gut. 1985;26:400–5.
Kaiser GC, Yan F, Polk DB. Mesalamine Blocks tumor necrosis factor growth inhibition and nuclear factor kB activation in mouse colonocytes. Gastroenterology. 1999;116(3):602–9.
Christensen LA, Fallingborg J, Jacobsen BA, Abildgaard K, Rasmussen HH, Hansen SH, et al. Comparative bioavailability of 5-aminosalicylic acid from a controlled release preparation and an azo-bond preparation. Aliment Pharmacol Ther. 1994;8(3):289–94.
Staerk Laursen L, Stokholm M, Bukhave K, Rask-Madsen J, Lauritsen K, Sterk Laursen M, Stokholm K, Lauritsen DL. Disposition of 5-aminosalicylic acid by olsalazine and three mesalazine preparations in patients with ulcerative colitis: comparison of intraluminal colonic concentrations, serum values, and urinary excretion. Gut. 1990;31:1271–6.
Dilger K, Trenk D, Rössle M, Cap M, Zähringer A, Wacheck V, et al. A clinical trial on absorption and N-acetylation of oral and rectal mesalazine. Eur J Clin Invest. 2007;37(7):558–65.
Cohen RD, Woseth DM, Thisted RA, Hanauer SB. A meta-analysis and overview of the literature on treatment options for left-sided ulcerative colitis and ulcerative proctitis. Am J Gastroenterol. 2000;95:1263–76.
Naganuma M, Iwao Y, Ogata H, Inoue N, Funakoshi S, Yamamoto S, et al. Measurement of colonic mucosal concentrations of 5-aminosalicylic acid is useful for estimating its therapeutic efficacy in distal ulcerative colitis: comparison of orally administered mesalamine and sulfasalazine. Inflamm Bowel Dis. 2001;7(3):221–5.
Frieri G, Pimpo MT, Palumbo GC, Onori L, Viscido A, Latella G, et al. Rectal and colonic mesalazine concentration in ulcerative colitis: oral vs. oral plus topical treatment. Aliment Pharmacol Ther. 1999;13(11):1413–7.
Frieri G, Giacomelli R, Pimpo M, Palumbo G, Passacantando A, Pantaleoni G, et al. Mucosal 5-aminosalicylic acid concentration inversely correlates with severity of colonic inflammation in patients with ulcerative colitis. Gut. 2000;47:410–4.
Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther. 2001;23(8):1296–310.
Kruis W, Kiudelis G, Rácz I, Gorelov IA, Pokrotnieks J, Horynski M, et al. Once daily versus three times daily mesalazine granules in active ulcerative colitis: a double-blind, double-dummy, randomised, non-inferiority trial. Gut. 2009;58(2):233–40.
D’Haens G, Hommes D, Engels L, Baert F, Van Der Waaij L, Connor P, et al. Once daily MMX mesalazine for the treatment of mild-to-moderate ulcerative colitis: a phase II, dose-ranging study. Aliment Pharmacol Ther. 2006;24(7):1087–97.
Kamm MA, Sandborn WJ, Gassull M, Schreiber S, Jackowski L, Butler T, et al. Once-daily, high-concentration MMX mesalamine in active ulcerative colitis. Gastroenterology. 2007;132(1):66–75 (quic 432–3).
Sandborn WJ, Kamm MA, Lichtenstein GR, Lyne A, Butler T, Joseph RE. MMX Multi Matrix System mesalazine for the induction of remission in patients with mild-to-moderate ulcerative colitis: a combined analysis of two randomized, double-blind, placebo-controlled trials. Aliment Pharmacol Ther. 2007;26(2):205–15.
D’Haens GR, Sandborn WJ, Zou G, Stitt LW, Rutgeerts PJ, Gilgen D, et al. Randomised non-inferiority trial: 1600 mg versus 400 mg tablets of mesalazine for the treatment of mild-to-moderate ulcerative colitis. Aliment Pharmacol Ther. 2017;46(3):292–302.
Kornbluth A, Sachar DB. Practice Parameters Committee of the American College of Gastroenterology. Ulcerative Colitis Practice Guidelines in Adults: American College of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol. 2010;105(3):501–23.
Stein RB, Hanauer SB. Comparative tolerability of treatments for inflammatory bowel disease. Drug Saf. 2000;23(5):429–48.
Aceituno M, García-Planella E, Heredia C, Zabana Y, Feu F, Domènech E, et al. Steroid-refractory ulcerative colitis: predictive factors of response to cyclosporine and validation in an independent cohort. Inflamm Bowel Dis. 2008;14(3):347–52.
Ford AC, Bernstein CN, Khan KJ, Abreu MT, Marshall JK, Talley NJ, et al. Glucocorticosteroid Therapy in inflammatory bowel disease: systematic review and meta-analysis. Am J Gastroenterol. 2011;106(4):590–9.
Bebb JR, Scott BB. Systematic review: how effective are the usual treatments for ulcerative colitis? Aliment Pharmacol Ther. 2004;20(2):143–9.
Barnes PJ. Molecular mechanisms and cellular effects of glucocorticosteroids. Immunol Allergy Clin North Am. 2005;25(3):451–68.
Silverman J, Otley A. Budesonide in the treatment of inflammatory bowel disease. Expert Rev Clin Immunol. 2011;7(4):419–28.
Schwab M, Klotz U. Pharmacokinetic considerations in the treatment of inflammatory bowel disease. Clin Pharmacokinet. 2001;40(10):723–51.
Ferry JJ, Horvath AM, Bekersky I, Heath EC, Ryan CF, Colburn WA. Relative and absolute bioavailability of prednisone and prednisolone after separate oral and intravenous doses. J Clin Pharmacol. 1988;28(1):81–7.
Frey BM, Frey FJ. Clinical pharmacokinetics of prednisone and prednisolone. Clin Pharmacokinet. 1990;19(2):126–46.
Rose JQ, Yurchak AM, Jusko WJ. Dose dependent pharmacokinetics of prednisone and prednisolone in man. J Pharmacokinet Biopharm. 1981;9(4):389–417.
Bergrem H, Grottum P, Rugstad HE. Pharmacokinetics and protein binding of prednisolone after oral and intravenous administration. Eur J Clin Pharmacol. 1983;24:415–9.
Milsap RL, George DE, Szefler SJ, Murray KA, Lebenthal E, Jusko WJ. Effect of inflammatory bowel disease on absorption and disposition of prednisolone. Dig Dis Sci. 1983;28(2):161–8.
Jönsson G, Aström A, Andersson P. Budesonide is metabolized by cytochrome P450 3A (CYP3A) enzymes in human liver. Drug Metab Dispos. 1995;23(1):137–42.
Dahlberg E, Thalén A, Brattsand R, Gustafsson JA, Johansson U, Roempke K, et al. Correlation between chemical structure, receptor binding, and biological activity of some novel, highly active, 16 alpha, 17 alpha-acetal-substituted glucocorticoids. Mol Pharmacol. 1984;25(1):70–8.
Danielsson Å, Löfberg R, Persson T, Salde L, Schiöler R, Suhr O, et al. A steroid enema, budesonide, lacking systemic effects for the treatment of distal ulcerative colitis or proctitis. Scand J Gastroenterol. 1992;27(1):9–12.
Bar-Meir S, Fidder HH, Faszczyk M, Bianchi Porro G, Sturniolo GC, Mickisch O, et al. Budesonide foam vs. hydrocortisone acetate foam in the treatment of active ulcerative proctosigmoiditis. Dis Colon Rectum. 2003;46(7):929–36.
Edsbäcker S, Andersson T. Pharmacokinetics of budesonide (Entocort EC) capsules for Crohn’s disease. Clin Pharmacokinet. 2004;43(12):803–21.
Maltese P, Palma L, Sfara C, De Rocco P, Latiano A, Palmieri O, et al. Glucocorticoid resistance in Crohn’s disease and ulcerative colitis: an association study investigating GR and FKBP5 gene polymorphisms. Pharmacogenom J. 2011;12(10):432–8.
Shimada T, Hiwatashi N, Yamazaki H, Kinouchi Y, Toyota T. Relationship between glucocorticoid receptor and response to glucocorticoid therapy in ulcerative colitis. Dis Colon Rectum. 1997;40(10 Suppl):S54–8.
Mwinyi J, Wenger C, Eloranta JJ, Kullak-Ublick GA. Glucocorticoid receptor gene haplotype structure and steroid therapy outcome in IBD patients. World J Gastroenterol. 2010;16(31):3888–96.
Schottelius A, Wedel S, Weltrich R, Rohde W, Buttgereit F, Schreiber S, et al. Higher expression of glucocorticoid receptor in peripheral mononuclear cells in inflammatory bowel disease. Am J Gastroenterol. 2000;95:1994–9.
Hitchings GH, Elion GB. The chemistry and biochemistry of purine analogs. Ann N Y Acad Sci. 1954;60(2):195–9.
Panaccione R, Ghosh S, Middleton S, Márquez JR, Scott BB, Flint L, et al. Combination therapy with infliximab and azathioprine is superior to monotherapy with either agent in ulcerative colitis. Gastroenterology. 2014;146(392–400):e3.
Chebli LA, de Chaves LDM, Pimentel FF, Guerra DM, de Barros RMF, Gaburri PD, et al. Azathioprine maintains long-term steroid-free remission trough 3 years in patients with steroid-dependent ulcerative colitis. Inflamm Bowel Dis. 2010;16(4):613–9.
Ardizzone S, Maconi G, Russo A, Imbesi V, Colombo E, Bianchi Porro G, et al. Randomised controlled trial of azathioprine and 5-aminosalicylic acid for treatment of steroid dependent ulcerative colitis. Gut. 2006;55:47–53.
Marinaki AM, Ansari A, Duley JA, Arenas M, Sumi S, Lewis CM, et al. Adverse drug reactions to azathioprine therapy are associated with polymorphism in the gene encoding inosine triphosphate pyrophosphatase (itpase). Pharmacogenetics. 2004;14(3):181–7.
Eklund BI, Moberg M, Bergquist J, Mannervik B. Divergent activities of human glutathione transferases in the bioactivation of azathioprine. Mol Pharmacol. 2006;70(2):747–54.
Krynetski EY, Krynetskaia NF, Yanishevski Y, Evans WE. Methylation of mercaptopurine, thioguanine, and their nucleotide metabolites by heterologously expressed human thiopurine S-methyltransferase. Mol Pharmacol. 1995;47(6):1141–7.
Dubinsky MC, Lamothe SP, Yang HY, Targan SR, Sinnett D, Or YT, et al. Pharmacogenomics and metabolite measurement for 6-mercaptopurine therapy in inflammatory bowel disease. Gastroenterology. 2000;118(4):705–13.
Coskun M, Steenholdt C, De Boer NK, Haagen Nielsen O. Pharmacology and optimization of thiopurines and methothrexate in inflammatory bowel disease. Clin Pharmacokinet. 2016;55:257–74.
Ding TL, Benet LZ. Comparative bioavailability and pharmacokinetic studies of azathioprine and 6-mercaptopurine in the rhesus monkey. Drug Metab Dispos. 1979;7(6):373–7.
Lancaster DL, Patel N, Lennard L, Lilleyman JS. 6-Thioguanine in children with acute lymphoblastic leukaemia: infuence of food on parent drug pharmacokinetics and 6-thioguanine nucleotide concentrations. Br J Clin Pharmacol. 2001;51(6):531–9.
Lennard L. TPMT in the treatment of Crohn’s disease with azathioprine. Gut. 2002;51:143–6.
Liu Y-P, Wu H-Y, Yang X, Xu H-Q, Li Y-C, Shi D-C, et al. Association between thiopurine s-methyltransferase polymorphisms and thiopurine-induced adverse drug reactions in patients with inflammatory bowel disease: a meta-analysis. PLoS One. 2015;10(3):e0121745.
Shipkova M, Armstrong VW, Wieland E, Oellerich M. Differences in nucleotide hydrolysis contribute to the differences between erythrocyte 6-thioguanine nucleotide concentrations determined by two widely used methods. Clin Chem. 2003;49(2):260–8.
Osterman MT, Kundu R, Lichtenstein GR, Lewis JD. Association of 6-thioguanine nucleotide levels and inflammatory bowel disease activity: a meta-analysis. Gastroenterology. 2006;130(4):1047–53.
Cuffari C, Hunt S, Bayless T. Utilisation of erythrocyte 6-thioguanine metabolite levels to optimise azathioprine therapy in patients with inflammatory bowel disease. Gut. 2001;48(5):642–6.
Cuffari C, Theoret Y, Latour S, Seidman G, Seidman EG, Theoret PY, et al. 6-Mercaptopurine metabolism in Crohn’s disease: correlation with efficacy and toxicity. Gut. 1996;39(39):401–6.
Sparrow MP, Hande SA, Friedman S, Lim WC, Reddy SI, Cao D, et al. Allopurinol safely and effectively optimizes tioguanine metabolites in inflammatory bowel disease patients not responding to azathioprine and mercaptopurine. Aliment Pharmacol Ther. 2005;22(5):441–6.
Van Bodegraven AA, Mulder CJ. Indications for 5-aminosalicylate in inflammatory bowel disease: Is the body of evidence complete? World J Gastroenterol World J Gastroenterol. 2006;12(38):6115–23.
Hande S, Wilson-Rich N, Bousvaros A, Zholudev A, Maurer R, Banks P, et al. 5-Aminosalicylate therapy is associated with higher 6-thioguanine levels in adults and children with inflammatory bowel disease in remission on 6-mercaptopurine or azathioprine. Inflamm Bowel Dis. 2006;12(4):251–7.
Szumlanski C, Weinshilboum R. Sulphasalazine inhibition of thiopurine methyltransferase: possible mechanism for interaction with 6-mercaptopurine and azathioprine. Br J Clin Pharmacol. 1995;39(4):456–9.
De Graaf P, De Boer N, Wong D, Karner S, Jharap B, Hooymans P, et al. Influence of 5-aminosalicylic acid on 6-thioguanosine phosphate metabolite levels: a prospective study in patients under steady thiopurine therapy. Br J Pharmacol. 2010;160:1083–91.
Hawwa AF, Collier PS, Millership JS, McCarthy A, Dempsey S, Cairns C, et al. Population pharmacokinetic and pharmacogenetic analysis of 6-mercaptopurine in paediatric patients with acute lymphoblastic leukaemia. Br J Clin Pharmacol. 2008;66(6):826–37.
Gilissen LP, Derijks LJ, Bos LP, Bus PJ, Hooymans PM, Engels LG. Therapeutic drug monitoring in patients with inflammatory bowel disease and established azathioprine therapy. Clin Drug Investig. 2004;24(8):479–86.
Aberra FN, Lichtenstein GR. Review article: monitoring of immunomodulators in inflammatory bowel disease. Aliment Pharmacol Ther. 2005;21(4):307–19.
Black AJ, McLeod HL, Capell HA, Powrie RH, Matowe LK, Pritchard SC, et al. Thiopurine methyltransferase genotype predicts therapy-limiting severe toxicity from azathioprine. Ann Intern Med. 1998;129(9):716.
Cabaleiro T, Roman M, Gisbert JP, Abad-Santos F. Utility of assessing thiopurine S-methyltransferase polymorphisms before azathioprine therapy. Curr Drug Metab. 2012;13(9):1277–93.
Nguyen CM, Mendes MAS, Ma JD. Thiopurine methyltransferase (TPMT) genotyping to predict myelosuppression risk. PLoS Curr. 2011;3:RRN1236.
Wiederrecht G, Lam E, Hung S, Martin M, Sigal N. The mechanism of action of FK-506 and cyclosporin A. Ann N Y Acad Sci. 1993;696:9–19.
Chow DK, Leong RW. The use of tacrolimus in the treatment of inflammatory bowel disease. Expert Opin Drug Saf. 2007;6(5):479–85.
Truelove SC, Jewell DP. Intensive intravenous regimen for severe attacks of ulcerative colitis. Lancet. 1974;303(7866):1067–70.
Lichtiger S, Present DH, Kornbluth A, Gelernt I, Bauer J, Galler G, et al. Cyclosporine in severe ulcerative colitis refractory to steroid therapy. New Engl J Med. 1994;330(26):1841–5.
Lichtiger S, Present DH. Preliminary report: cyclosporin in treatment of severe active ulcerative colitis. Lancet. 1990;336(8706):16–9.
Ogata H, Matsui T, Nakamura M, Iida M, Takazoe M, Suzuki Y, et al. A randomised dose finding study of oral tacrolimus (FK506) therapy in refractory ulcerative colitis. Gut. 2006;55(9):1255–62.
Ogata H, Kato J, Hirai F, Hida N, Matsui T, Matsumoto T, et al. Double-blind, placebo-controlled trial of oral tacrolimus (FK506) in the management of hospitalized patients with steroid-refractory ulcerative colitis. Inflamm Bowel Dis. 2012;18(5):803–8.
Jorga A, Holt DW, Johnston A. Therapeutic drug monitoring of cyclosporine. Transplant Proc. 2004;36(2 Suppl):396S–403S.
Drewe J, Beglinger C, Kissel T. The absorption site of cyclosporin in the human gastrointestinal tract. Br J Clin Pharmacol. 1992;33(1):39–43.
Venkataramanan R, Swaminathan A, Prasar T, Jain A, Zuckerman S, Warty V, et al. Clinical pharmacokinetics of tacrolimus. Clin Pharmacokinet. 1995;29(6):404–30.
Jusko WJ, Thomson AW, Fung J, McMaster P, Wong SH, Zylber-Katz E, et al. Consensus document: therapeutic monitoring of tacrolimus (FK-506). Ther Drug Monit. 1995;17(6):606–14.
Lindholm A, Kahan BD. Influence of cyclosporine pharmacokinetics, trough concentrations, and AUC monitoring on outcome after kidney transplantation. Clin Pharmacol Ther. 1993;54(2):205–18.
Brynskov J, Freund L, Campanini MC, Kampmann JP. Cyclosporin pharmacokinetics after intravenous and oral administration in patients with Crohn’s disease. Scand J Gastroenterol. 1992;27(11):961–7.
Atkinson K, Britton K, Paull P, Farrell C, Concannon A, Dodds A, et al. Detrimental effect of intestinal disease on absorption of orally administered cyclosporine. Transplant Proc. 1983;15(Suppl l):2446–9.
Sandborn WJ, Strong RM, Forland SC, Chase RE, Cutler RE. The pharmacokinetics and colonic tissue concentrations of cyclosporin after IV, oral, and enema administration. J Clin Pharmacol. 1991;31(1):76–80.
Gupta SK, Manfro RC, Tomlanovich SJ, Gambertoglio JG, Garovoy MR, Benet LZ. Effect of food on the pharmacokinetics of cyclosporine in healthy subjects following oral and intravenous administration. J Clin Pharmacol. 1990;30(7):643–53.
Kovarik JM, Mueller EA, Kutz K, Van Bree JB, Tetzloff W. reduced inter- and intraindividual variability in cyclosporine pharmacokinetics from a microemulsion formulation. J Pharm Sci. 1994;83(3):444–6.
Noble S, Markham A. Cyclosporin. A review of the pharmacokinetic properties, clinical efficacy and tolerability of a microemulsion-based formulation (Neoral). Drugs. 1995;50(5):924–41.
Mueller EA, Kovarik JM, van Bree JB, Tetzloff W, Grevel J, Kutz K. Improved dose linearity of cyclosporine pharmacokinetics from a microemulsion formulation. Pharm Res. 1994;11(2):301–4.
Latteri M, Angeloni G, Silveri NG, Manna R, Gasbarrini G, Navarra P. Pharmacokinetics of cyclosporin microemulsion in patients with inflammatory bowel disease. Clin Pharmacokinet. 2001;40(6):473–83.
Kronbach T, Fischer V, Meyer UA. Cyclosporine metabolism in human liver: identification of a cytochrome P-450III gene family as the major cyclosporine-metabolizing enzyme explains interactions of cyclosporine with other drugs. Clin Pharmacol Ther. 1988;43(6):630–5.
Combalbert J, Fabre I, Fabre G, Dalet I, Derancourt J, Cano JP, et al. Metabolism of cyclosporin A. IV. Purification and identification of the rifampicin-inducible human liver cytochrome P-450 (cyclosporin A oxidase) as a product of P450IIIA gene subfamily. Drug Metab Dispos. 1989;17(2):197–207.
Plosker GL. Tacrolimus: a further update of its pharmacology and therapeutic use in the management of organ transplantation. Drugs. 2000;59(2):323–89.
Iwasaki K. Metabolism of tacrolimus (FK506) and recent topics in clinical pharmacokinetics. Drug Metab Pharmacokinet. 2007;22(5):328–35.
Zhang Y, Benet LZ. The Gut as a barrier to drug absorption. Clin Pharmacokinet. 2001;40(3):159–68.
Saeki T, Ueda K, Tanigawara Y, Hori R, Komano T. Human P-glycoprotein transports cyclosporin A and FK506. J Biol Chem. 1993;268(9):6077–80.
Lin JH, Yamazaki M. Role of P-glycoprotein in pharmacokinetics. Clin Pharmacokinet. 2003;42(1):59–98.
Möller A, Iwasaki K, Kawamura A, Teramura Y, Shiraga T, Hata T, Sc Afer A, et al. The disposition of 14C-labeled tacrolimus after intravenous and oral administration in healthy human subjects. Drug Metab Dispos. 1999;27(6):633–6.
Burckart GJ, Starzl TE, Venkataramanan R, Hashim H, Wong L, Wang P, et al. Excretion of cyclosporine and its metabolites in human bile. Transplant Proc. 1986;18(6 Suppl 5):46–9.
Hesselink DA, Van Schaik RHN, Van Der Heiden IP, Van Der Werf M, Smak Gregoor PJH, Lindemans J, et al. Genetic polymorphisms of the CΥP3A4, CΥP3A5, and MDR-1 genes and pharmacokinetics of the calcineurin inhibitors cyclosporine and tacrolimus. Clin Pharmacol Ther. 2003;74(3):245–54.
Rivory LP, Qin H, Clarke SJ, Eris J, Duggin G, Ray E, et al. Frequency of cytochrome P 450 3A4 variant genotype in transplant population and lack of association with cyclosporin clearance. Eur J Clin Pharmacol. 2000;56(5):395–8.
von Ahsen N, Richter M, Grupp C, Ringe B, Oellerich M, Armstrong VW. No influence of the MDR-1 C3435T polymorphism or a CYP3A4 promoter polymorphism (CYP3A4-V allele) on dose-adjusted cyclosporin A trough concentrations or rejection incidence in stable renal transplant recipients. Clin Chem. 2001;47(6):1048–52.
Utecht KN, Hiles JJ, Kolesar J. Effects of genetic polymorphisms on the pharmacokinetics of calcineurin inhibitors. Am J Heal Pharm. 2006;63(23):2340–8.
Hirai F, Takatsu N, Yano Y, Satou Y, Takahashi H, Ishikawa S, et al. Impact of CYP3A5 genetic polymorphisms on the pharmacokinetics and short-term remission in patients with ulcerative colitis treated with tacrolimus. J Gastroenterol Hepatol. 2014;29(1):60–6.
Herrlinger KR, Koc H, Winter S, Teml A, Stange EF, Fellermann K, et al. ABCB1 single-nucleotide polymorphisms determine tacrolimus response in patients with ulcerative colitis. Clin Pharmacol Ther. 2011;89(3):422–8.
Midtvedt K. Therapeutic drug monitoring of cyclosporine. Transplant Proc. 2004;36(2 Suppl):430S–3S.
Halloran PF, Helms LM, Kung L, Noujaim J. The temporal profile of calcineurin inhibition by cyclosporine in vivo. Transplantation. 1999;68(9):1356–61.
Sindhi R, LaVia MF, Paulling E, McMichael J, Burckart G, Shaw S, et al. Stimulated response of peripheral lymphocytes may distinguish cyclosporine effect in renal transplant recipients receiving a cyclosporine + rapamycin regimen. Transplantation. 2000;69(3):432–6.
Mardigyan V, Giannetti N, Cecere R, Besner J-G, Cantarovich M. Best single time points to predict the area-under-the-curve in long-term heart transplant patients taking mycophenolate mofetil in combination with cyclosporine or tacrolimus. J Heart Lung Transplant. 2005;24(10):1614–8.
Cantarovich M, Barkun JS, Tchervenkov JI, Besner JG, Aspeslet L, Metrakos P. Comparison of neoral dose monitoring with cyclosporine through levels versus 2-hr postdose levels in stable liver transplant patients. Transplantation. 1998;66(12):1621–7.
Cantarovich M, Besner JG, Barkun JS, Elstein E, Loertscher R. Two-hour cyclosporine level determination is the appropriate tool to monitor Neoral therapy. Clin Transplant. 1998;12(3):243–9.
Kornbluth A, Present DH, Lichtiger S, Hanauer S. Cyclosporin for severe ulcerative colitis: a user’s guide. Am J Gastroenterol. 1997;92(9):1424–8.
Rayner CK, McCormack G, Emmanuel AV, Kamm MA. Long-term results of low-dose intravenous ciclosporin for acute severe ulcerative colitis. Aliment Pharmacol Ther. 2003;18(3):303–8.
Van Assche G, D’Haens G, Noman M, Vermeire SV, Hiele M, Asnong K, et al. Randomized, double-blind comparison of 4 mg/kg versus 2 mg/kg intravenous cyclosporine in severe ulcerative colitis. Gastroenterology. 2003;125(4):1025–31.
Størset E, Holford N, Midtvedt K, Bremer S, Bergan S, Åsberg A. Importance of hematocrit for a tacrolimus target concentration strategy. Eur J Clin Pharmacol. 2014;70(1):65–77.
Zahir H, McCaughan G, Gleeson M, Nand RA, McLachlan AJ. Factors affecting variability in distribution of tacrolimus in liver transplant recipients. Br J Clin Pharmacol. 2004;57(3):298–309.
Zahir H, Nand RA, Brown KF, Tattam BN, Mclachlan AJ. Validation of methods to study the distribution and protein binding of tacrolimus in human blood. J Pharmacol Toxicol Methods. 2001;46(1):27–35.
Lainesse A, Hussain S, Monif T, Reyar S, Tippabhotla S, Madan A, et al. Bioequivalence studies of tacrolimus capsule under fasting and fed conditions in healthy male and female subjects. Arzneimittelforschung. 2011;58(5):242–7.
Bekersky I, Dressler D, Mekki QA. Effect of low- and high-fat meals on tacrolimus absorption following 5 mg single oral doses to healthy human subjects. J Clin Pharmacol. 2001;41(2):176–82.
Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, Johanns J, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353(23):2462–76.
Gisbert JP, González-Lama Y, Maté J. Systematic review: Infliximab therapy in ulcerative colitis. Aliment Pharmacol Ther. 2007;25(1):19–37.
D’Haens G. Infliximab for ulcerative colitis: finally some answers. Gastroenterology. 2005;128(7):2161–6.
Sandborn WJ, Van Assche G, Reinisch W, Colombel J, D’Haens G, Wolf DC, et al. Adalimumab induces and maintains clinical remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2012;142(2):257–265.e3.
Sandborn WJ, Feagan BG, Marano C, Zhang H, Strauss R, Johanns J, et al. Subcutaneous golimumab maintains clinical response in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014;146(1):96–109.e1.
Thorlund K, Druyts E, Toor K, Mills EJ. Comparative efficacy of golimumab, infliximab, and adalimumab for moderately to severely active ulcerative colitis: a network meta-analysis accounting for differences in trial designs. Expert Rev Gastroenterol Hepatol. 2015;9(5):693–700.
Cholapranee A, Hazlewood GS, Kaplan GG, Peyrin-Biroulet L, Ananthakrishnan AN. Systematic review with meta-analysis: comparative efficacy of biologics for induction and maintenance of mucosal healing in Crohn’s disease and ulcerative colitis controlled trials. Aliment Pharmacol Ther. 2017;45(10):1291–302.
Harris MS, Hartman D, Lemos BR, Erlich EC, Spence S, Kennedy S, et al. AVX-470, an orally delivered anti-tumour necrosis factor antibody for treatment of active ulcerative colitis: results of a first-in-human trial. J Crohn Colitis. 2016;10(6):631–40.
Bhol KC, Tracey DE, Lemos BR, Lyng GD, Erlich EC, Keane DM, et al. AVX-470: a novel oral anti-TNF antibody with therapeutic potential in inflammatory bowel disease. Inflamm Bowel Dis. 2013;19(11):2273–81.
Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93(11):2645–68.
Xu Z, Wang Q, Zhuang Y, Frederick B, Yan H, Bouman-Thio E, et al. Subcutaneous Bioavailability of golimumab at 3 different injection sites in healthy subjects. J Clin Pharmacol. 2010;50(3):276–84.
Klotz U, Teml A, Schwab M. Clinical pharmacokinetics and use of infliximab. Clin Pharmacokinet. 2007;46(8):645–60.
Brandse JF, van den Brink GR, Wildenberg ME, van der Kleij D, Rispens T, Jansen JM, et al. Loss of infliximab into feces is associated with lack of response to therapy in patients with severe ulcerative colitis. Gastroenterology. 2015;149(2):350–355.e2.
Zhu X, Meng G, Dickinson BL, Li X, Mizoguchi E, Miao L, et al. MHC class I-related neonatal Fc receptor for IgG is functionally expressed in monocytes, intestinal macrophages, and dendritic cells. J Immunol. 2001;166(5):3266–76.
Borvak J, Richardson J, Medesan C, Antohe F, Radu C, Simionescu M, et al. Functional expression of the MHC class I-related receptor, FcRn, in endothelial cells of mice. Int Immunol. 1998;10(9):1289–98.
Ngo BP, Farrell C, Barr M, Wolov K, Bailey RM, Mullin J, et al. Tumor necrosis factor blockade for treatment of inflammatory bowel disease: efficacy and safety. Curr Mol Pharmacol. 2010;3(3):145–52.
Stucchi A, Reed K, O’Brien M, Cerda S, Andrews C, Gower A, et al. A new transcription factor that regulates TNF-a Gene Expression, LITAF, Is increased in intestinal tissues from patients with CD and UC. Inflamm Bowel Dis. 2006;12(7):581–7.
Sands BE, Kaplan GG. The role of TNFα in ulcerative colitis. J Clin Pharmacol. 2007;47(8):930–41.
Scallon B, Cai A, Solowski N, Rosenberg A, Song X-Y, Shealy D, et al. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther. 2002;301(2):418–26.
Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14(5):329–42.
Sandborn WJ, Hanauer SB. Antitumor necrosis factor therapy for inflammatory bowel disease: a review of agents, pharmacology, clinical results, and safety. Inflamm Bowel Dis. 1999;5(2):119–33.
Pierik M, Vermeire S, Steen KV, Joossens S, Claessens G, Vlietinck R, et al. Tumour necrosis factor-alpha receptor 1 and 2 polymorphisms in inflammatory bowel disease and their association with response to infliximab. Aliment Pharmacol Ther. 2004;20(3):303–10.
Grell M, Douni E, Wajant H, Löhden M, Clauss M, Maxeiner B, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83(5):793–802.
Vandenabeele P, Declercq W, Beyaert R, Fiers W. Two tumour necrosis factor receptors: structure and function. Trends Cell Biol. 1995;5(10):392–9.
Armuzzi A, Pugliese D, Danese S, Rizzo G, Felice C, Marzo M, et al. Infliximab in steroid-dependent ulcerative colitis: effectiveness and predictors of clinical and endoscopic remission. Inflamm Bowel Dis. 2013;19(5):1065–72.
Ungar B, Chowers Y, Yavzori M, Picard O, Fudim E, Har-Noy O, et al. The temporal evolution of antidrug antibodies in patients with inflammatory bowel disease treated with infliximab. Gut. 2014;63(8):1258–64.
Seow CH, Newman A, Irwin SP, Steinhart AH, Silverberg MS, Greenberg GR. Trough serum infliximab: a predictive factor of clinical outcome for infliximab treatment in acute ulcerative colitis. Gut. 2010;59(1):49–54.
Reinisch W, Feagan BG, Rutgeerts PJ, Adedokun OJ, Cornillie FJ, Diamond R, et al. Infliximab concentration and clinical outcome in patients with ulcerative colitis. Gastroenterology. 2012;142(5):S-114.
Murthy S, Kevans D, Seow CH, Newman A, Steinhart AH, Silverberg MS, et al. Association of serum infliximab and antibodies to infliximab to long-term clinical outcome in acute ulcerative colitis. Gastroenterology. 2012;142(5):S-388.
Adedokun OJ, Sandborn WJ, Feagan BG, Rutgeerts P, Xu Z, Marano CW, et al. Association between serum concentration of infliximab and efficacy in adult patients with ulcerative colitis. Gastroenterology. 2014;147(1296–1307):e5.
Cornillie F, Hanauer SB, Diamond RH, Wang J, Tang KL, Xu Z, et al. Postinduction serum infliximab trough level and decrease of C-reactive protein level are associated with durable sustained response to infliximab: a retrospective analysis of the ACCENT I trial. Gut. 2014;63(11):1721–7.
Vande Casteele N, Khanna R, Levesque BG, Stitt L, Zou GY, Singh S, et al. The relationship between infliximab concentrations, antibodies to infliximab and disease activity in Crohn’s disease. Gut. 2015;64(10):1539–45.
Bortlik M, Duricova D, Malickova K, Machkova N, Bouzkova E, Hrdlicka L, et al. Infliximab trough levels may predict sustained response to infliximab in patients with Crohn’s disease. J Crohn Colitis. 2013;7(9):736–43.
Ungar B, Levy I, Yavne Y, Yavzori M, Picard O, Fudim E, et al. Optimizing anti-TNF-α therapy: serum levels of infliximab and adalimumab are associated with mucosal healing in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2016;14(4):550–7.
Detrez I, Dreesen E, van Stappen T, De Vries A, Brouwers E, Van Assche G, et al. Variability in golimumab exposure: a “Real-Life” observational study in active ulcerative colitis. J Crohn Colitis. 2016;10(5):575–81.
Peyrin-Biroulet L, Deltenre P, de Suray N, Branche J, Sandborn WJ, Colombel JF. Efficacy and safety of tumor necrosis factor antagonists in Crohn’s disease: meta-analysis of placebo-controlled trials. Clin Gastroenterol Hepatol. 2008;6(6):644–53.
Vande Casteele N, Feagan BG, Gils A, Vermeire S, Khanna R, Sandborn WJ, et al. Therapeutic drug monitoring in inflammatory bowel disease: current state and future perspectives. Curr Gastroenterol Rep. 2014;16(4):378.
Khanna R, Sattin BD, Afif W, Benchimol EI, Bernard EJ, Bitton A, et al. Review article: a clinician’s guide for therapeutic drug monitoring of infliximab in inflammatory bowel disease. Aliment Pharmacol Ther. 2013;38(5):447–59.
Ordás I, Feagan BG, Sandborn WJ. Therapeutic drug monitoring of tumor necrosis factor antagonists in inflammatory bowel disease. Clin Gastroenterol Hepatol. 2012;10:1079–87.
Stein BN, Pellish RS, Thompson KD, Baptista V, Siegel CA. Using therapeutic drug monitoring to identify variable infliximab metabolism in an individual patient with ulcerative colitis. J Clin Gastroenterol. 2016;50(1):66–8.
Fasanmade AA, Adedokun OJ, Ford J, Hernandez D, Johanns J, Hu C, et al. Population pharmacokinetic analysis of infliximab in patients with ulcerative colitis. Eur J Clin Pharmacol. 2009;65(12):1211–28.
Ternant D, Aubourg A, Magdelaine-Beuzelin C, Degenne D, Watier H, Picon L, et al. Infliximab pharmacokinetics in inflammatory bowel disease patients. Ther Drug Monit. 2008;30(4):523–9.
Buurman DJ, Maurer JM, Keizer RJ, Kosterink JGW, Dijkstra G. Population pharmacokinetics of infliximab in patients with inflammatory bowel disease: potential implications for dosing in clinical practice. Aliment Pharmacol Ther. 2015;42(5):529–39.
Roblin X, Marotte H, Leclerc M, Del Tedesco E, Phelip JM, Peyrin-Biroulet L, et al. Combination of C-reactive protein, infliximab trough levels, and stable but not transient antibodies to infliximab are associated with loss of response to infliximab in inflammatory bowel disease. J Crohn Colitis. 2015;9(7):525–31.
Brandse JF, Mathôt RA, Van Der Kleij D, Rispens T, Ashruf Y, Jansen JM, et al. Pharmacokinetic features and presence of antidrug antibodies associate with response to infliximab induction therapy in patients with moderate to severe ulcerative colitis. Clin Gastroenterol Hepatol. 2016;14(2):251–8.e1–2.
Solem CA, Loftus Jr EV, Tremaine WJ, Harmsen WS, Zinsmeister AR, Sandborn WJ. Correlation of c reactive protein crp with clinical radiographic and edoscopic activity in inflammatory bowel disease ibd. Gastroenerology. 2004;126(4):A-477.
Ternant D, Karmiris K, Vermeire S, Desvignes C, Azzopardi N, Bejan-Angoulvant T, et al. Pharmacokinetics of adalimumab in Crohn’s disease. Eur J Clin Pharmacol. 2015;71(9):1155–7.
Sharma S, Eckert D, Hyams JS, Mensing S, Thakkar RB, Robinson AM, et al. Pharmacokinetics and exposure-efficacy relationship of adalimumab in pediatric patients with moderate to severe Crohn’s disease: results from a randomized, multicenter, phase-3 study. Inflamm Bowel Dis. 2015;21(4):783–92.
Adedokun OJ, Xu Z, Liao S, Marano C, Strauss R, Zhang H, et al. Sa1935 population pharmacokinetic modeling analysis of golimumab in adult patients with moderately to severely active ulcerative colitis. Gastroenterology. 2016;150(4):S408.
Vande Casteele N, Gils A, Singh S, Ohrmund L, Hauenstein S, Rutgeerts P, et al. Antibody response to infliximab and its impact on pharmacokinetics can be transient. Am J Gastroenterol. 2013;108(6):962–71.
Bendtzen K. Immunogenicity of anti-TNF-α biotherapies: II. Clinical relevance of methods used for anti-drug antibody detection. Front Immunol. 2015;6:109.
Wolbink GJ, Vis M, Lems W, Voskuyl AE, De Groot E, Nurmohamed MT, et al. Development of antiinfliximab antibodies and relationship to clinical response in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54(3):711–5.
Van Schouwenburg PA, Bartelds GM, Hart MH, Aarden L, Wolbink GJ, Wouters D. A novel method for the detection of antibodies to adalimumab in the presence of drug reveals hidden immunogenicity in rheumatoid arthritis patients. J Immunol Methods. 2010;362:82–8.
van Stappen T, Vande Casteele N, van Assche G, Ferrante M, Vermeire S, Gils A. Clinical relevance of detecting anti-infliximab antibodies with a drug-tolerant assay: post hoc analysis of the TAXIT trial. Gut. 2018;67(5):818–26.
Seitz K, Zhou H. Pharmacokinetic drug-drug interaction potentials for therapeutic monoclonal antibodies: reality check. J Clin Pharmacol. 2007;47:1104–18.
Strik AS, van den Brink GR, Ponsioen C, Mathot R, Löenberg M, D’Haens GR. Suppression of anti-drug antibodies to infliximab or adalimumab with the addition of an immunomodulator in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2017;45(8):1128–34.
van Schaik T, Maljaars JPW, Roopram RK, Verwey MH, Ipenburg N, Hardwick JCH, et al. Influence of combination therapy with immune modulators on anti-TNF trough levels and antibodies in patients with IBD. Inflamm Bowel Dis. 2014;20(12):2292–8.
Mould DR, Upton RN, Wojciechowski J. Dashboard systems: implementing pharmacometrics from bench to bedside. AAPS J. 2014;16(5):925–37.
Mould DR, D’Haens G, Upton RN. Clinical decision support tools: the evolution of a revolution. Clin Pharmacol Ther. 2016;99(4):405–18.
Wojciechowski J, Upton R, Mould D, Wiese M, Foster D. Infliximab mainentance dosing in inflammatory bowel disease: an example for in silico assessment of adaptive dosing strategies. AAPS. 2017;19(4):1136–47.
Vande Casteele N, Ferrante M, Van Assche G, Ballet V, Compernolle G, Van Steen K, et al. Trough concentrations of infliximab guide dosing for patients with inflammatory bowel disease. Gastroenterology. 2015;148(7):1320–9.e3.
D’Haens G, Vermeire S, Lambrecht G, Baert F, Bossuyt P, Pariente B, et al. increasing infliximab dose based on symptoms, biomarkers, and serum drug concentrations does not increase clinical, endoscopic, or corticosteroid-free remission in patients with active luminal Crohn’s disease. Gastroenterology. 2018;154(5):1343–1351.e1.
Papamichael K, Van Stappen T, Vande Casteele N, Gils A, Billiet T, Tops S, et al. Infliximab concentration thresholds during induction therapy are associated with short-term mucosal healing in patients with ulcerative colitis. Clin Gastroenterol Hetatol. 2016;14(4):543–9.
Hendler SA, Cohen BL, Colombel J-F, Sands BE, Mayer L, Agarwal S. High-dose infliximab therapy in Crohn’s Disease: clinical experience, safety, and efficacy. J Crohn Colitis. 2015;9(3):266–75.
Steenholdt C, Brynskov J, Thomsen OØ, Munck LK, Fallingborg J, Christensen LA, et al. Individualised therapy is more cost-effective than dose intensification in patients with Crohn’s disease who lose response to anti-TNF treatment: a randomised, controlled trial. Gut. 2014;63:919–27.
Steenholdt C, Brynskov J, Thomsen O, Munck LK, Fallingborg J, Christensen LA, et al. Individualized therapy is a long-term cost-effective method compared to dose intensification in Crohn’s disease patients failing infliximab. Dig Dis Sci. 2015;60(9):2762–70.
Andrew DP, Rott LS, Kilshaw PJ, Butcher EC. Distribution of α4β7 and αEβ7 integrins on thymocytes, intestinal epithelial lymphocytes and peripheral lymphocytes. Eur J Immunol. 1996;26(4):897–905.
Raine T. Vedolizumab for inflammatory bowel disease: changing the game, or more of the same? United Eur Gastroenterol J. 2014;2(5):333–44.
Soler D, Chapman T, Yang L-L, Wyant T, Egan R, Fedyk ER. The binding specificity and selective antagonism of vedolizumab, an anti-α4β7 integrin therapeutic antibody in development for inflammatory bowel diseases. J Pharmacol Exp Ther. 2009;330(3):864–75.
Feagan BG, Rutgeerts P, Sands BE, Hanauer S, Colombel J-F, Sandborn WJ, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369(8):699–710.
Rosario M, Dirks NL, Gastonguay MR, Fasanmade AA, Wyant T, Parikh A, et al. Population pharmacokinetics-pharmacodynamics of vedolizumab in patients with ulcerative colitis and Crohn’s disease. Aliment Pharmacol Ther. 2015;42(2):188–202.
Dua P, Hawkins E, van der Graaf P. A tutorial on target-mediated drug disposition (TMDD) models. CPT Pharmacomet Syst Pharmacol. 2015;4(6):324–37.
Gibiansky L, Gibiansky E, Kakkar T, Ma P, Gibiansky L, Gibiansky E, et al. Approximations of the target-mediated drug disposition model and identifiability of model parameters. J Pharmacokinet Pharmacodyn. 2008;35:573–91.
Rosario M, French JL, Dirks NL, Sankoh S, Parikh A, Yang H, et al. Exposure-Efficacy relationships for vedolizumab induction therapy in patients with ulcerative colitis or Crohn’s disease. J Crohn Colitis. 2017;10:S24.
Feagan BG, Greenberg GR, Wild G, Fedorak RN, Paré P, McDonald JW, et al. Treatment of ulcerative colitis with a humanized antibody to the α4β7 integrin. N Engl J Med. 2005;352(24):2499–507.
Colombel J-F, Sands BE, Rutgeerts P, Sandborn W, Danese S, D’Haens G, et al. The safety of vedolizumab for ulcerative colitis and Crohn’s disease. Gut. 2017;66(5):839–51.
Williet N, Boschetti G, Fovet M, Di Bernado T, Claudez P, Del Tedesco E, et al. Association between low trough levels of vedolizumab during induction therapy for inflammatory bowel diseaseswith need for additional doses within 6 months. Clin Gastroenterol Hepatol. 2017;15(11):1750–1757.e3.
FDA. Center for Drug Evaluation and Research. Addendum to office of clinical pharmacology review. BLA 125476. 2014.
Klotz U, Maier KE, Fischer C, Bauer KH. A new slow-release form of 5-aminosalicylic acid for the oral treatment of inflammatory bowel disease. Biopharmaceutic and clinical pharmacokinetic characteristics. Arzneimittelforschung. 1985;35(3):636–9.
Rasmussen SN, Bondesen S, Hvidberg EF, Honore Hansen S, Binder V, Halskov S, et al. 5-Aminosalicylic acid in a slow-release preparation: bioavailability, plasma level, and excretion in humans. Gastroenterology. 1982;83(1):62–70.
Yu DK, Morrill B, Eichmeier LS, Giesing DH, Weir SJ, Lanman RC, et al. Pharmacokinetics of 5-aminosalicylic acid from controlled-release capsules in man. Eur J Clin Pharmacol. 1995;48(3–4):273–7.
Hussain FN, Ajjan RA, Kapur K, Moustafa M, Riley SA. Once versus divided daily dosing with delayed-release mesalazine: a study of tissue drug concentrations and standard pharmacokinetic parameters. Aliment Pharmacol Ther. 2001;15(1):53–62.
Van Os EC, Zins BJ, Sandborn WJ, Mays DC, Tremaine WJ, Mahoney DW, et al. Azathioprine pharmacokinetics after intravenous, oral, delayed release oral and rectal foam administration. Gut. 1996;39:63–8.
Morse GD, Holdsworth MT, Venuto RC, Gerbasi J, Walshe JJ. Pharmacokinetics and clinical tolerance of intravenous and oral cyclosporine in the immediate postoperative period. Clin Pharmacol Ther. 1988;44(6):654–64.
Brynskov J, Freund M, Campanini MC, Kampmann JP. Cyclosporin pharmacokinetics after intravenous and oral administration in patients with Crohn’s disease. Scand J Gastroenterol. 1992;27(11):961–7.
Aubourg A, Picon L, Lecomte T, Bejan-Angoulvant T, Paintaud G, Ternant D. A robust estimation of infliximab pharmacokinetic parameters in Crohn’s disease. Eur J Clin Pharmacol. 2015;71(12):1541–2.
Shin D, Kim Y, Kim YS, Körnicke T, Fuhr R. A randomized, phase I pharmacokinetic study comparing SB2 and infliximab reference product (Remicade(®)) in healthy subjects. BioDrugs. 2015;29(6):381–8.
Boyle A, Tawadros R, Zhu Y. Comparative pharmacokinetics of single and multiple-dose infliximab in Crohn’s disease patients. Gastroenterology. 2002;122:A-614-5.
Cornillie F, Shealy D, D’Haens G, Geboes K, Van Assche G, Ceuppens J, et al. Infliximab induces potent anti-inflammatory and local immunomodulatory activity but no systemic immune suppression in patients with Crohn’s disease. Aliment Pharmacol Ther. 2001;15(4):463–73.
Park KR, Chung H, Yang SM, Lee S, Yoon SHY, Cho J, et al. A randomized, double-blind, single-dose, two-arm, parallel study comparing pharmacokinetics, immunogenicity and tolerability of branded adalimumab and its biosimilar LBAL in healthy male volunteers. Expert Opin Investig Drugs. 2017;26(5):619–24.
Hyland E, Mant T, Vlachos P, Attkins N, Ullmann M, Roy S, et al. Comparison of the pharmacokinetics, safety, and immunogenicity of MSB11022, a biosimilar of adalimumab, with Humira(®) in healthy subjects. Br J Clin Pharmacol. 2016;82(4):983–93.
Humira® (adalimumab). US prescribing information. North Chicago (IL): Abbott Laboratories; 2007.
Harzallah I, Rigaill J, Williet N, Paul S, Roblin X. Golimumab pharmacokinetics in ulcerative colitis: a literature review. Therap Adv Gastroenterol. 2017;10(1):89–100.
Zhuang Y, Xu Z, Frederick B, de Vries DE, Ford JA, Keen M, et al. Golimumab pharmacokinetics after repeated subcutaneous and intravenous administrations in patients with rheumatoid arthritis and the effect of concomitant methotrexate: an open-label. Randomized Study. Clin Ther. 2012;34(1):77–90.
Rosario M, Timothy Wyant B, Timothy Leach B, Serap Sankoh B, Scholz C, Asit Parikh B, et al. Vedolizumab pharmacokinetics, pharmacodynamics, safety, and tolerability following administration of a single, ascending, intravenous dose to healthy volunteers. Clin Drug Investig. 2016;36:913–23.
Parikh A, Leach T, Wyant T, Scholz C, Sankoh S, Mould DR, et al. Vedolizumab for the treatment of active ulcerative colitis: a randomized controlled phase 2 dose-ranging study. Inflamm Bowel Dis. 2012;18(8):1470–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was provided.
Conflicts of interest
Sophie Berends has received lecture fees from Merck Sharp & Dohme, Johnson & Johnson and Tillotts. Anne Strik has received lecture fees from Biogen, Johnson and Johnson, Merck Sharp & Dohme, Mundipharma, Takeda, and Tillotts. Mark Löwenberg has received speaker fees from Abbvie, Covidien, Dr. Falk Pharma, Ferring Pharmaceuticals, Merck Sharp & Dohme, Receptos, Takeda, Tillotts and Tramedico, and has also received research grants from AbbVie, Merck Sharp & Dohme, Achmea Healthcare and ZonMW. Geert D’Haens has served as an advisor for Abbvie, Ablynx, Amakem, AM Pharma, Avax- ia, Biogen, Bristol-Myers Squibb, Boerhinger Ingelheim, Celgene, Celltrion, Cosmo, Covidien, Ferring, Dr. Falk Pharma, Engene, Galapagos, Gilead, GlaxoSmithKline, Hospira, Immunic, Johnson and Johnson, Lycera, Medimetrics, Millenium/Takeda, Mitsubishi Pharma, Merck Sharp & Dohme, Mundipharma, Novonordisk, Pfizer, Prometheus Laboratories/Nestle, Protagonist, Receptos, Robarts Clinical Trials, Salix, Sandoz, Setpoint, Shire, Teva, Tigenix, Tillotts, Topivert, Versant and Vifor, and has also received speaker fees from Abbvie, Ferring, Johnson and Johnson, Merck Sharp & Dohme, Mundipharma, Norgine, Pfizer, Shire, Millenium/Takeda, Tillotts and Vifor. Ron Mathôt has no conflicts of interest to declare.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Berends, S.E., Strik, A.S., Löwenberg, M. et al. Clinical Pharmacokinetic and Pharmacodynamic Considerations in the Treatment of Ulcerative Colitis. Clin Pharmacokinet 58, 15–37 (2019). https://doi.org/10.1007/s40262-018-0676-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-018-0676-z