
Abstract
We report a facile synthetic method for the preparation of titanium–tantalum oxide by means of a modified Adam’s method. This new method allowed obtaining Ti0.8Ta0.2O2 with a high surface area (234 m2 g−1), to be used as catalyst support for Pd nanoparticles. Cyclic voltammetry and linear sweep voltammetry measurements confirm the noticeable oxygen reduction reaction (ORR) activities of the Pd/Ti0.8Ta0.2O2 electrocatalyst in alkaline electrolytes, along with a high-selectivity towards a 4e− pathway. The good ORR performance for the Pd/Ti0.8Ta0.2O2 could arise from both the strong metal-support interaction and the contribution of the Ti0.8Ta0.2O2 in facilitating the ORR process, acting as co-catalyst. However, the stability of this catalyst seems insufficient for practical applications.
Similar content being viewed by others

Introduction
Oxygen reduction is the most challenging electrochemical reaction for many electrochemical devices, such as fuel cells and metal-air batteries [1,2,3,4,5]. The high potential of this reaction (1.23 V vs. reversible hydrogen electrode, RHE) forces the use of highly active and stable catalysts, being Pt-based ones the most employed [6,7,8,9,10]. However, it is widely known the high cost of this metal; thus, several strategies are sought to replace it [11,12,13]. There has been a huge increment of papers dealing with alternatives to Pt, like the use of other noble metals like Pd, Ag, Au and/or their alloys [14,15,16,17,18,19,20] or the use of non-noble metal catalysts, usually based on transition metals disperse onto a carbon matrix (phthalocyanines, ferrocenes, Co-based catalysts, doped-graphene, etc.) [12, 21,22,23,24]. Additionally, the dispersion of noble metal nanoparticles on high surface area carbon supports is acknowledge as a good strategy to reduce the amount of expensive active phases [25,26,27,28]. However, under the conditions of a fuel cell or a metal-air battery, carbon-based catalysts suffer from the corrosion of the support [29, 30].
There has been a great interest in developing alternative supports, such as the ones based on titanium dioxide. TiO2 has been recognized as a versatile material, easy to produce and with a wide variety of applications (solar cells, degradation of organic pollutants, electrocatalysts supports, etc.) [31,32,33,34,35]. It features relatively low cost, non-toxicity, photo-stability, and inertness [31, 34]. However, depending on the application, increasing the electrical conductivity of TiO2 is necessary. Heating TiO2 in a reducing atmosphere or doping with cations of higher valence (transition metals) are strategies usually pursued to enhance the electrical conductivity; however, at the expense of the specific surface area [34,35,36,37,38]. Several authors have introduced different dopants such as Nb, W, V, Ta, etc., by different methods to enhance the electronic conductivity maintaining an adequate surface area [34, 35, 37, 39, 40]. For example, Beauger et al. synthesized TiO2 aerogels and xerogels doped with Nb, V and Ta as alternative to conventional carbon black supports for PEMFCs [36]. Wang et al. obtained TaNbTiO2 and C–TaNbTiO2 as hybrid supports for Pt–Pd nanoparticles for the ORR [34]. Stassi et al. [41] supported a Pt–Co alloy on Ta-doped TiO2 and Siracusano et al. studied bared and doped TiO2 as catalysts supports for fuel cells [42]. In all cases, doped-TiO2 showed an increased resistance to corrosion.
The different properties of TiO2, such as surface area, crystallographic structure, etc., depend on the synthesis method. There have been many synthetic methods studied in literature, like sol–gel processes, hydrothermal and solvothermal routes, reverse microemulsion methods, etc. [33, 40].
Herein, we propose a new synthetic method to prepare TiO2 doped with Ta, having a porous structure, to be used as the support for Pd nanoparticles. The prepared Pd supported on TiTa-oxide electrocatalyst was investigated, in the present work, for the oxygen reduction reaction in alkaline media. Pd nanoparticles have been already demonstrated as a suitable catalyst for the ORR in basic electrolytes [14, 43,44,45]. The advantages of this method are the easy scalability and simplicity, leading to the preparation of a highly porous TiO2-based support doped with Ta, what increases its electrical conductivity. The stability and activity towards the ORR in alkaline electrolyte of the Pd/TiTa-oxide electrocatalyst was compared to a commercial Pd/C based material.
Experimental
Materials and methods
Titanium–tantalum oxides were synthesized by the Adams fusion method using the procedure modified by Marshall [46, 47]. TiCl4 (98%, Fluka) and TaCl5 (99.8%, Sigma-Aldrich) metal precursors were added to isopropanol (99.5%, Sigma-Aldrich) to obtain a total metal concentration of 0.08 M. This solution was magnetically stirred at room temperature for 1 h to ensure the complete dissolution of the precursors. Then NaNO3 (99.0%, Sigma-Aldrich), previously grounded in ball-milling, was added to the isopropanol solution under vigorous stirring. The slurry was then heated at 90 °C under constant stirring, until obtaining a humid paste, which was dried in a ventilated oven at 90 °C for 24 h. The dry salt was then placed in a furnace at 500 °C for 30 min. The fused salt oxide was washed with distilled water to remove the remaining salts, filtered and dried in an oven at 80 °C for 12 h.
The so-obtained TiTa-oxide was employed as the support for Pd nanoparticles synthesized by a sulphite complex methodology. Firstly, a Pd-sulphite complex was prepared by reaction of PdCl2 (99.9%, Strem Chemicals) with sodium bisulfite (99.995%, Aldrich). Once obtained, the Pd-sulphite salt was first dissolved in acidic solution and subsequently decomposed with H2O2 (40% p/v, Titolchimica) to form a colloidal dispersion of PdOx that was impregnated on the oxide support. The as-formed catalyst was reduced in H2 atmosphere (10 wt% in Ar) at 25 °C to obtain Pd metal nanoparticles supported on the TiTa-oxide with a loading of 60 wt% of Pd [45].
Physico-chemical characterization
Several characterization techniques were employed to investigate the different physico-chemical features of both the support, TiTa-oxide, and the catalyst, Pd/TiTa-oxide. X-ray diffraction (XRD) was performed with Cu Kα radiation operating at 40 kV and 20 mA in a Philips X-pert 3710 X-ray diffractometer. The diffraction patterns were fitted to Joint Committee on Powder Diffraction Standards (JCPDS). The peaks broadening was used to calculate the crystallite size by the Debye–Scherrer equation after correction for the instrumental broadening. The morphology of the samples and their composition were studied by scanning electron microscopy (SEM) and energy dispersive X-ray (EDX) analysis, carried out by a FEI XL30 SFEG microscope. The instrument was operated at 25 kV and the EDX probe was used to determine the bulk elemental composition of the samples. Transmission electron microscopy (TEM) analysis was performed with a FEI CM12 microscope by depositing some drops of the samples dispersed in isopropyl alcohol on carbon film-coated Cu grids. The specific surface area of the oxide support was calculated by the Brunauer–Emmett–Teller (BET) equation and nitrogen adsorption–desorption isotherms, measured at − 196 °C, using an ASAP 2020 M Micrometrics.
Electro-chemical characterization
The electrochemical studies were carried out using a standard three-electrode cell and an Autolab potentiostat/galvanostat with GPES software and connected to a rotating disk electrode. A glassy carbon electrode (GC, 5 mm in diameter) was used as the working electrode, a platinum mesh was used as the counter and an Hg|HgO electrode was used as the reference electrode. Typically, 5 mg of the catalyst was dispersed in 5 mL of a mixture of isopropanol (99.5%, Sigma-Aldrich) and water (Milli Q water, 18.2 MΩ cm) in a 3:1 v/v ratio and sonicated for 30 min. 30 wt% of Nafion (5%, Ion Power) was added as polymer binder. Then, 15 µL of the catalyst ink was dropped onto the glassy carbon electrode to obtain a metal loading of 50 µg cm−2 of Pd and allowed to dry at room temperature for 15 min to obtain a uniform carbon film. All electrochemical experiments were carried out at room temperature and ambient pressure using 1 M KOH (90%, Sigma-Aldrich) as the electrolyte solution. Linear sweep voltammetries from 0.65 V vs. RHE to 0.25 V vs. RHE were performed at a scan rate of 5 mV s−1 at different rotation rate: 100, 200, 400, 1000, 1600 and 2500 rpm, bubbling pure O2 in the electrolyte. Before the measurements, the electrode was repeatedly potentiodynamically swept, with a scan rate of 100 mV s−1, from 0 to 1.2 V vs RHE in the deaerated (degassed with He) 1 M KOH solution until a steady voltammogram was obtained. The durability of the TiTaOx-based catalyst was assessed by potential cycling between 0.6 and 1.2 V vs. RHE, bubbling He, until 5000 cycles were completed. The activity of the catalyst was evaluated before and after the cycles by linear sweep voltammetries at different rotation speeds (as previously detailed).
The Pd/TiTa-oxide catalyst was compared to a commercial catalyst employing a carbonaceous support, 30% Pd/C (E-TEK).
Results and discussion
The oxide support was initially characterized in terms of structure, morphology and surface area. The textural parameters for the TiTa-oxide support, obtained by N2 physisorption, are shown in Table 1. The specific surface area, determined applying the BET equation to the adsorption–desorption isotherms (shown in Fig. 1), is 234 m2 g−1. TiTa-oxide shows the characteristic features of a type IV isotherm, mesoporous solid [48]. The hysteresis loop at high P/Po is associated with capillary condensation taking place in mesopores. The mesoporosity of the oxide support favors the diffusion of reagents towards the catalytic centers. The calcination temperature is mild enough to not compromise the textural properties of the support.

N2-adsorption/desorption isotherm for the TiTa-oxide support
Figure 2 shows SEM images of the TiTa-oxide support at different magnification. The support presents a granulated morphology, with a mixture of small and large particles agglomerates. However, a porous structure is observed. The EDX analysis was employed to investigate the elemental bulk composition of the doped-oxide support, being Ti:Ta = 80.5:19.5, in agreement with the nominal one. From now on, TiTa-oxide will be expressed as Ti0.8Ta0.2O2, considering the ratio obtained by EDX.

SEM images for the TiTa-oxide support obtained at different magnification: (a) ×1500, (b) ×3500 and (c) ×8000
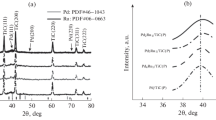
Figure 3 presents the X-ray diffraction patterns for the Ti0.8Ta0.2O2 support and the corresponding Pd catalyst. As evidenced from the graph, Ti0.8Ta0.2O2 is not entirely crystalline (peaks are not clearly defined); however, the main phase observed is anatase. The calcination temperature employed during the synthesis, 500 °C, is not high enough to fully crystallize the amorphous oxide obtained after the synthesis. Pd/Ti0.8Ta0.2O2 catalyst shows the typical peaks of the Pd crystallographic structure, faced-centered cubic, with main reflections at 40°, 47°, 68°, 82° and 86° (JCPDS Card No. 46-1043). Crystallite size was calculated by applying the Scherrer’s equation to the peak at 68º (corresponding to the (220) plane). The catalyst showed a crystallite size of 4.5 nm, which is higher than that of the commercial 30% Pd/C (E-TEK) [49], used for the electrochemical comparison, with crystallite size equal to 3.4 nm. The difference in the crystal size might be ascribed to the different metallic loading, being 60 wt% for the Pd/Ti0.8Ta0.2O2 catalyst, in comparison to Pd/C (E-TEK), with a 30 wt%.

XRD diffraction patterns for Ti0.8Ta0.2O2 and Pd/Ti0.8Ta0.2O2 catalyst
TEM micrographs for the Pd-based catalyst supported on Ti0.8Ta0.2O2 are shown in Fig. 4 at low (Fig. 4a) and high (Fig. 4b) magnification. Figure 4a shows the Ti0.8Ta0.2O2 (light spots) fully covered with Pd particles (dark spots). The high metallic loading (60 wt%) employed in the synthesis of the Pd/Ti0.8Ta0.2O2 catalyst is responsible for this agglomeration. Titanium–tantalum based oxides are not so electronically conductive as carbon materials, even though doping with Ta increases the conductivity of the material [36]. It has already been proved that doping TiO2 with cations of a higher valence, such as Ta, favors the presence of oxygen vacancies that will increase the electronic conductivity, due to the concentration of positively charged defects in the lattice [4, 33]. In any case, to favour the electronic percolation also through the metallic nanoparticles, the Pd/Ti0.8Ta0.2O2 was synthesized with a high metallic loading (60 wt%), determined by SEM–EDX. Figure 4b shows an image at higher magnification in which metallic Pd particles and the Ti0.8Ta0.2O2 support are clearly observed. The average Pd particle size, determined from TEM images, is about 5 nm. On the other hand, the Pd/C commercial catalyst shows an average particle size of 4.3 nm [49].

TEM micrographs for Pd/Ti0.8Ta0.2O2 at (a) low and (b) high magnification
The behavior of the Pd/Ti0.8Ta0.2O2 catalyst in comparison to Pd/C (E-TEK) was investigated for the oxygen reduction reaction (ORR) in an alkaline solution with a rotating disk electrode (RDE). Titanium-based materials have been widely studied in literature as supports for noble metal catalysts such as Pt or Au and mainly in acid media [50,51,52]. Recently, there has been a growing interest of this type of catalysts for alkaline media [53, 54], also in combination with carbon materials [55, 56]. Up to our knowledge, Pd supported on Ta-doped titanium oxide has not been studied in alkaline electrolytes until now. For example, Elezovic et al. studied a Nb–TiO2 supported Pt catalyst in comparison with Vulcan supported one, showing similar catalytic activity towards oxygen reduction. Maltanava et al. studied the electrocatalytic activity of both bare high-ordered TiO2 nanotubes (TNTs) and gold nanoparticles (Au NPs) loaded TNTs toward ORR. They determined that the overpotential of O2 reduction on the surface of Au NPs with a definite size increases with increasing the annealing temperature of TiO2 support. In general, it has been thoroughly proved that titanium-based supports seem to enhance the catalytic activity of noble metals towards the ORR in alkaline media.
The electrochemically active surface area (ECSA) was first evaluated in a deaerated 1 M KOH solution by cyclic voltammetry from 0.05 to 1.2 V vs. RHE, at a scan rate of 100 mV s−1 (not shown) as previously described in [45]. Briefly, ECSA was determined from the integration of the peak related to Pd-oxide reduction (between 0.4 and 0.8 V vs. RHE), assuming 405 µC cm−2 for the reduction of a monolayer of Pd-oxide [45].
The ECSA for the Pd/Ti0.8Ta0.2O2 catalyst was of 25.8 m2 g−1, whereas for the Pd/C was 69.7 m2 g−1, significantly higher due to the lower concentration of metal (30 wt %) on the carbonaceous support and, thus, a better nanoparticle dispersion [49].
Figure 5 shows the polarization curves for both catalysts in an O2-saturated 1 M KOH solution at 1600 rpm. Pd/C (E-TEK) and Pd/Ti0.8Ta0.2O2 present a very similar performance towards the ORR. The carbon-based catalyst shows a slightly better onset potential and limiting current density, what might be due to its higher ECSA. However, the presence of Ti0.8Ta0.2O2 may favour the activity of this catalyst towards the ORR in alkaline electrolyte, allowing to obtain a similar performance. It has been proved that the catalytic activity toward the ORR of TiO2-based catalysts depends on both the structural properties and the fabrication procedure. For example, spray-deposited anatase and single crystal rutile type TiO2 exhibit a 4e − pathway in alkaline electrolytes; whereas amorphous titanium oxides exhibit 2e − pathway [32, 33]. Sacco et al. determined that anatase TiO2 nanotubes were as active for the ORR in alkaline media as commercial Pt-catalysts [33]. Mentus et al. demonstrated the activity of anodically grown TiO2 towards the ORR in alkaline media, reaching values of current density around 6–7 mA cm−2. Pei et al. showed that TiO2 single crystals [particularly, (001) high-energy facets] are highly active towards the ORR in alkaline media [57]. Anatase-type TiO2 is also active towards ORR, but not as much as the former. Pei et al. ascribed the enhanced activity to the oxygen vacancies in non stoichiometric TiO2 [57]. In our case, doping TiO2 with Ta creates oxygen vacancies that could contribute to the enhancement of the activity towards the ORR. The crystallographic structure of our Ti0.8Ta0.2O2, in the prevalent form of anatase, also contributes to the enhancement of the activity. With an engineered design of the crystallographic orientation of TaTi-oxide crystals, this activity could be further enhanced, as demonstrated by Pei et al. [57] or Liu et al. [58]. On the other hand, a review conducted by Trasatti et al. [59] showed that the activity towards the ORR of noble metals supported on titanium-based supports is increased due to the strong metal support interaction (SMSI) effect, a fact that has been proved by other authors in more recent papers [34, 41, 60]. In summary, both the intrinsic activity of titanium-based materials towards the ORR in alkaline media and the SMSI effect might be responsible for the significant activity of our Pd/Ti0.8Ta0.2O2 catalyst.

Polarization curves in an O2-saturated 1 M KOH solution at ω = 1600 rpm; scan rate 5 mV s−1. Pd loading on the glassy carbon electrode 50 μg cm−2 of Pd
Figure 6 presents Koutecky–Levich (K–L) plots obtained for both catalysts. The K–L plots show a linear behavior of the inverse of the current density with the reciprocal of the square root of the rotation speed. By applying the Levich equation [61] to the linear fitting of the experimental data, from the slope (B) we calculate the transferred electron number (n). The diffusion coefficient of oxygen in the electrolyte (D), the cinematic viscosity of the electrolyte (ν), and the bulk concentration of oxygen in the electrolyte (C*) are taken from Ref. [62]. Both catalysts follow a 4e − pathway, being 3.8 e − for Pd/Ti0.8Ta0.2O2 and 3.9 e − for commercial Pd/C.

(a) Levich plots for the Pd/Ti0.8Ta0.2O2 and (b) Pd/C (E-TEK) catalysts. Potentials are referred to the RHE and currents are per unit of geometric area. Electrolyte solution is a 1.0 M KOH solution at 25 °C
The assessment of the stability was carried out by potential cycling between 0.6 and 1.2 V vs. RHE, shown in Fig. 7. Both samples present a significant decay in performance, determined by the reduction of the area of the peak at ca. 0.7 V vs. RHE.

Cyclic voltagrams from 0.6 to 1.2 V vs RHE used as accelerated degradation test for (a) Pd/C (E-TEK) and (b) Pd/TiTaOx; scan rate 100 mV s−1. 1.0 M KOH solution at 25 °C as electrolyte
The activity (Fig. 8) and the ECSA (Fig. 9) were evaluated before and after the 5000 cycles. The decay in performance for the Pd/Ti0.8Ta0.2O2 catalyst is remarkable, losing around 54 mV in terms of onset potential and a 25% in terms of limiting current density, as can be ascertained from Fig. 8. However, Pd/C E-TEK, even though possessing a carbon support, widely known for its tendency to corrosion, suffers a lower decay in performance of around 16 mV in terms of onset potential and around 16% in terms of limiting current density. This is also corroborated by the ECSA losses, shown in Fig. 9. Pei et al. ascribed the poorer stability of TiO2 polycrystals to their anatase–rutile mixed crystal phase [57]. In our case, both the anatase crystallographic structure and the fact of being a semi-crystalline material could be responsible for the poor stability of the titanium-based support.

Polarization curves at the beginning of the test (BoT) and at the end of the test (EoT) in an O2-saturated 1 M KOH solution at ω = 1600 rpm; scan rate 5 mV s−1. Pd loading on the glassy carbon electrode: 50 μg cm−2 of Pd. 1.0 M KOH solution at 25 °C as electrolyte

Cyclic voltagrams from 0 to 1.2 V vs RHE before (blue line) and after (red line) the accelerated degradation test for (a) Pd/C and (b) Pd/Ti0.8Ta0.2O2; scan rate 100 mV s−1. 1.0 M KOH solution at 25 °C as electrolyte
Cyclic voltamograms in Fig. 9 show the H-adsorption/desorption peaks around 0.2 V vs. RHE both in the anodic and the cathodic sense. The Pd oxidation peak is visible in the range 1.0–1.2 V vs. RHE (anodic sense) and the reduction of the Pd-oxide formed is visible at around 0.7 V vs. RHE (cathodic sense). The size of this peak is significantly higher for the commercial catalyst in comparison to the TiTaOx-based one, indicating a higher ECSA for the Pd/C (E-TEK), as previously described. Besides, in the case of the commercial catalyst (Fig. 9a), there is a shoulder at 0.6 V vs RHE that could be ascribed to the oxidation of the carbon support. After 5000 cycles, the width of the voltammogram (red line) is severely reduced for both catalysts. ECSA calculated from the charge associated to the reduction of the Pd-oxide peak (0.7 V vs RHE) was 7.6 m2 g−1 for the Pd/C (E-TEK) catalyst (Fig. 9a) and 0.44 m2 g−1 for Pd/Ti0.8Ta0.2O2 (Fig. 9b). This explains the considerable decay in performance towards the ORR measured by linear sweep voltammetry in previously shown Fig. 8. From these results, it is clear that, although Ti0.8Ta0.2O2 synthesized by this new method presents a good activity for the ORR, this support is not stable enough for a practical application.
Conclusions
A new method for the synthesis of Ta-doped titanium oxides was proposed. The Adam’s method permits obtaining considerable amounts of oxide in a reproducible way with a good compromise between surface area and crystallinity.
Pd nanoparticles were supported on the as-prepared Ti0.8Ta0.2O2 and studied as a catalyst for the oxygen reduction reaction in alkaline media. The activity of Pd/Ti0.8Ta0.2O2 was very similar to that of a commercial Pd/C catalyst. The activity of the titanium-based catalyst was ascribed to both the strong metal support interaction effect and to the intrinsic activity of the Ti0.8Ta0.2O2 towards the ORR in alkaline solution. Stability tests were carried out by potential cycling. Results determined that the Ti0.8Ta0.2O2 synthesized by this new method is not stable, leading to a loss of performance of the catalyst of a 25% in terms of current density. The lack of stability of the support was attributed to the crystallographical structure, being semi-crystalline anatase less stable than other crystallographic phases of titanium-based materials. Future studies will center on the study of the calcination temperature, to increase both conductivity and crystallinity notwithstanding the excellent textural properties of the oxide support.
References
Neburchilov, V., Wang, H., Martin, J.J., Qu, W.: A review on air cathodes for zinc–air fuel cells. J. Power Sources 195, 1271–1291 (2010). https://doi.org/10.1016/j.jpowsour.2009.08.100
Dresp, S., Luo, F., Schmack, R., Kühl, S., Gliech, M., Strasser, P.: An efficient bifunctional two-component catalyst for oxygen reduction and oxygen evolution in reversible fuel cells, electrolyzers and rechargeable air electrodes. Energy Environ. Sci. 9, 2020–2024 (2016). https://doi.org/10.1039/C6EE01046F
Narayan, S.R., Manohar, A.K., Mukerjee, S.: Bi-Functional Oxygen Electrodes—Challenges and Prospects. Electrochem. Soc. Interface. Summer 24, 65–69 (2015)
Jörissen, L.: Bifunctional oxygen/air electrodes. J. Power Sources 155, 23–32 (2006). https://doi.org/10.1016/j.jpowsour.2005.07.038
Caramia, V., Bozzini, B.: Materials science aspects of zinc-air batteries: a review. Mater. Renew. Sustain. Energy 3, 28 (2014). https://doi.org/10.1007/s40243-014-0028-3
Gasteiger, H.A., Kocha, S.S., Sompalli, B., Wagner, F.T.: Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B Environ. 56, 9–35 (2005). https://doi.org/10.1016/j.apcatb.2004.06.021
Van Der Vliet, D., Wang, C., Debe, M., Atanasoski, R., Markovic, N.M., Stamenkovic, V.R.: Platinum-alloy nanostructured thin film catalysts for the oxygen reduction reaction. Electrochim. Acta 56, 8695–8699 (2011). https://doi.org/10.1016/j.electacta.2011.07.063
Wang, Y., Leung, D.Y.C., Xuan, J., Wang, H.: A review on unitized regenerative fuel cell technologies, part-A: unitized regenerative proton exchange membrane fuel cells. Renew. Sustain. Energy Rev. 65, 961–977 (2016). https://doi.org/10.1016/j.rser.2016.07.046
Mani, P., Srivastava, R., Strasser, P.: Dealloyed binary PtM3 (M = Cu Co, Ni) and ternary PtNi3 M (M = Cu Co, Fe, Cr) electrocatalysts for the oxygen reduction reaction: performance in polymer electrolyte membrane fuel cells. J. Power Sources 196, 666–673 (2011). https://doi.org/10.1016/j.jpowsour.2010.07.047
Sebastián, D., Serov, A., Artyushkova, K., Gordon, J., Atanassov, P., Aricò, A.S., Baglio, V.: High performance and cost-effective direct methanol fuel cells: Fe-N-C methanol-tolerant oxygen reduction reaction catalysts. Chemsuschem 9, 1986–1995 (2016). https://doi.org/10.1002/cssc.201600583
Bashyam, R., Zelenay, P.: A class of non-precious metal composite catalysts for fuel cells. Nature 443, 63–66 (2006). https://doi.org/10.1038/nature05118
Chen, Z., Higgins, D., Yu, A., Zhang, L., Zhang, J., Heller, A., Hui, S.Q., Zhang, J.J., Ota, K., Campbell, S.A., Dahn, J.R., Olson, T., Pylypenko, S., Atanassov, P., Ustinov, E.A.: A review on non-precious metal electrocatalysts for PEM fuel cells. Energy Environ. Sci. 4, 3167 (2011). https://doi.org/10.1039/c0ee00558d
Karim, N.A., Kamarudin, S.K.: An overview on non-platinum cathode catalysts for direct methanol fuel cell. Appl. Energy 103, 212–220 (2013). https://doi.org/10.1016/j.apenergy.2012.09.031
McKerracher, R., Alegre, C., Baglio, V., Aricò, A.S., Ponce de León, C., Mornaghini, F., Rodlert, M., Walsh, F.C.: A nanostructured bifunctional Pd/C gas-diffusion electrode for metal-air batteries. Electrochim. Acta 174, 508–515 (2015). https://doi.org/10.1016/j.electacta.2015.06.001
Miller, H.A., Lavacchi, A., Vizza, F., Marelli, M., Di Benedetto, F., D’Acapito, F., Paska, Y., Page, M., Dekel, D.R.: A Pd/C-CeO 2 Anode catalyst for high-performance platinum-free anion exchange membrane fuel cells. Angew. Chemie Int. Ed. 55, 6004–6007 (2016). https://doi.org/10.1002/anie.201600647
Félix-Navarro, R.M., Beltrán-Gastélum, M., Reynoso-Soto, E.A., Paraguay-Delgado, F., Alonso-Nuñez, G., Flores-Hernández, J.R.: Bimetallic Pt–Au nanoparticles supported on multi-wall carbon nanotubes as electrocatalysts for oxygen reduction. Renew. Energy. 87, 31–41 (2016). https://doi.org/10.1016/j.renene.2015.09.060
Macak, J.M., Schmidt-Stein, F., Schmuki, P.: Efficient oxygen reduction on layers of ordered TiO2 nanotubes loaded with Au nanoparticles. Electrochem. Commun. 9, 1783–1787 (2007). https://doi.org/10.1016/j.elecom.2007.04.002
Shao, M.: Palladium-based electrocatalysts for hydrogen oxidation and oxygen reduction reactions. J. Power Sources 196, 2433–2444 (2011). https://doi.org/10.1016/j.jpowsour.2010.10.093
Lo Vecchio, C., Alegre, C., Sebastián, D., Stassi, A., Aricò, A.S., Baglio, V.: Investigation of supported Pd-based electrocatalysts for the oxygen reduction reaction: performance. Mater. (Basel). 8, 7997–8008 (2015). https://doi.org/10.3390/ma8125438
Abo Zeid, E.F., Ibrahem, I.A.: Preparation, characterization and electrocatalytic activity for oxygen reduction reaction in PEMFCs of bimetallic PdNi nanoalloy. Mater. Renew. Sustain. Energy 6, 19 (2017). https://doi.org/10.1007/s40243-017-0103-7
Hong, W.T., Risch, M., Stoerzinger, K.A., Grimaud, A., Suntivich, J., Shao-Horn, Y.: Toward the rational design of non-precious transition metal oxides for oxygen electrocatalysis. Energy Environ. Sci. 8, 1404–1427 (2015). https://doi.org/10.1039/C4EE03869J
Jaouen, F., Proietti, E., Lefèvre, M., Chenitz, R., Dodelet, J.-P., Wu, G., Chung, H.T., Johnston, C.M., Zelenay, P.: Recent advances in non-precious metal catalysis for oxygen-reduction reaction in polymer electrolyte fuelcells. Energy Environ. Sci. 4, 114–130 (2011). https://doi.org/10.1039/C0EE00011F
Cao, R., Lee, J.-S.S., Liu, M., Cho, J.: Recent progress in non-precious catalysts for metal-air batteries. Adv. Energy Mater. 2, 816–829 (2012). https://doi.org/10.1002/aenm.201200013
Alegre, C., Busacca, C., Di Blasi, O., Antonucci, V., Aricò, A.S., Di Blasi, A., Baglio, V.: A combination of CoO and Co nanoparticles supported on electrospun carbon nanofibers as highly stable air electrodes. J. Power Sources 364, 101–109 (2017). https://doi.org/10.1016/j.jpowsour.2017.08.007
Antolini, E.: Carbon supports for low-temperature fuel cell catalysts. Appl. Catal. B Environ. 88, 1–24 (2009). https://doi.org/10.1016/j.apcatb.2008.09.030
García, G., Roca-Ayats, M., Lillo, A., Galante, J.L., Peña, M.A., Martínez-Huerta, M.V.: Catalyst support effects at the oxygen electrode of unitized regenerative fuel cells. Catal. Today 210, 67–74 (2013). https://doi.org/10.1016/j.cattod.2013.02.003
S. L. Suib, F. Maillard, N. Job, M. Chatenet, Chapter 14—Approaches to Synthesize Carbon-Supported Platinum-Based Electrocatalysts for Proton-Exchange Membrane Fuel Cells, New Futur. Dev. Catal., 2013: pp. 407–428. https://doi.org/10.1016/b978-0-444-53880-2.00019-3
S.L. Suib, F. Maillard, N. Job, M. Chatenet, Chapter 17 – Basics of PEMFC Including the Use of Carbon-Supported Nanoparticles, in: New Futur. Dev. Catal., 2013: pp. 401–423. https://doi.org/10.1016/b978-0-444-53874-1.00018-4
Maass, S., Finsterwalder, F., Frank, G., Hartmann, R., Merten, C.: Carbon support oxidation in PEM fuel cell cathodes. J. Power Sources 176, 444–451 (2008). https://doi.org/10.1016/j.jpowsour.2007.08.053
Castanheira, L., Dubau, L., Mermoux, M., Berthomé, G., Caqué, N., Rossinot, E., Chatenet, M., Maillard, F.: Carbon corrosion in proton-exchange membrane fuel cells: from model experiments to real-life operation in membrane electrode assemblies. ACS Catal. 4, 2258–2267 (2014). https://doi.org/10.1021/cs500449q
Cabello, G., Davoglio, R.A., Pereira, E.C.: Microwave-assisted synthesis of anatase-TiO2 nanoparticles with catalytic activity in oxygen reduction. J. Electroanal. Chem. 794, 36–42 (2017). https://doi.org/10.1016/j.jelechem.2017.04.004
Mentus, S.V.: Oxygen reduction on anodically formed titanium dioxide. Electrochim. Acta 50, 27–32 (2004). https://doi.org/10.1016/j.electacta.2004.07.009
Sacco, A., Garino, N., Lamberti, A., Pirri, C.F., Quaglio, M.: Anodically-grown TiO2 nanotubes: effect of the crystallization on the catalytic activity toward the oxygen reduction reaction. Appl. Surf. Sci. 412, 447–454 (2017). https://doi.org/10.1016/j.apsusc.2017.03.224
Wang, Y.-J., Wilkinson, D.P., Neburchilov, V., Song, C., Guest, A., Zhang, J.: Ta and Nb co-doped TiO2 and its carbon-hybrid materials for supporting Pt–Pd alloy electrocatalysts for PEM fuel cell oxygen reduction reaction. J. Mater. Chem. A. 2, 12681 (2014). https://doi.org/10.1039/C4TA02062F
Lv, H., Zhang, G., Hao, C., Mi, C., Zhou, W., Yang, D., Li, B., Zhang, C.: activity of IrO 2 supported on tantalum-doped TiO2 electrocatalyst for solid polymer electrolyte water electrolyzer. RSC Adv. 7, 40427–40436 (2017). https://doi.org/10.1039/C7RA06534E
Beauger, C., Testut, L., Berthon-Fabry, S., Georgi, F., Guetaz, L.: Doped TiO2 aerogels as alternative catalyst supports for proton exchange membrane fuel cells: a comparative study of Nb, v and Ta dopants. Microporous Mesoporous Mater. 232, 109–118 (2016). https://doi.org/10.1016/j.micromeso.2016.06.003
Stodolny, M., Laniecki, M.: Synthesis and characterization of mesoporous Ta2O5-TiO2 photocatalysts for water splitting. Catal. Today 142, 314–319 (2009). https://doi.org/10.1016/j.cattod.2008.07.034
Siracusano, S., Baglio, V., D’Urso, C., Antonucci, V., Aricò, A.S.: Preparation and characterization of titanium suboxides as conductive supports of IrO2 electrocatalysts for application in SPE electrolysers. Electrochim. Acta 54, 6292–6299 (2009). https://doi.org/10.1016/j.electacta.2009.05.094
C. Hao, H. Lv, B. Li, H. Xin, J. Ma, Investigation of mesoporous vanadium doped TiO2 support for anode catalyst of SPE electrolyzer, Taiyangneng Xuebao/Acta Energiae Solaris Sin. 34 (2013) 1464–1470. http://www.scopus.com/inward/record.url?eid=2-s2.0-84886859703&partnerID=tZOtx3y1
Cavaliere, S., Subianto, S., Savych, I., Jones, D.J., Rozière, J.: Electrospinning: designed architectures for energy conversion and storage devices. Energy Environ. Sci. 4, 4761 (2011). https://doi.org/10.1039/c1ee02201f
Stassi, A., Gatto, I., Baglio, V., Passalacqua, E., Aricò, A.S.: Oxide-supported PtCo alloy catalyst for intermediate temperature polymer electrolyte fuel cells. Appl. Catal. B Environ. 142–143, 15–24 (2013). https://doi.org/10.1016/j.apcatb.2013.05.008
Siracusano, S., Stassi, A., Modica, E., Baglio, V., Aricò, A.S.S.: Preparation and characterisation of Ti oxide based catalyst supports for low temperature fuel cells. Int. J. Hydrogen Energy 38, 11600–11608 (2013). https://doi.org/10.1016/j.ijhydene.2013.04.161
McKerracher, R.D., Figueredo-Rodríguez, H.A., Ponce de León, C., Alegre, C., Baglio, V., Aricò, A.S., Walsh, F.C.: A high-performance, bifunctional oxygen electrode catalysed with palladium and nickel–iron hexacyanoferrate. Electrochim. Acta 206, 127–133 (2016). https://doi.org/10.1016/j.electacta.2016.04.090
Alegre, C., Modica, E., Lo Vecchio, C., Sebastián, D., Lázaro, M.J., Aricò, A.S., Baglio, V.: Carbon nanofibers as advanced Pd catalyst supports for the air electrode of alkaline metal-air batteries. Chempluschem. 80, 1384–1388 (2015). https://doi.org/10.1002/cplu.201500120
Alegre, C., Modica, E., Lo Vecchio, C., Siracusano, S., Aricò, A.S., Baglio, V.: Pd supported on Ti-suboxides as bifunctional catalyst for air electrodes of metal-air batteries. Int. J. Hydrogen Energy 41, 19579–19586 (2016). https://doi.org/10.1016/j.ijhydene.2016.03.095
Marshall, A., Børresen, B., Hagen, G., Tsypkin, M., Tunold, R.: Preparation and characterisation of nanocrystalline IrxSn1 − xO2 electrocatalytic powders. Mater. Chem. Phys. 94, 226–232 (2005). https://doi.org/10.1016/j.matchemphys.2005.04.039
Siracusano, S., Van Dijk, N., Payne-Johnson, E., Baglio, V., Aricò, A.S.: Nanosized IrOx and IrRuOx electrocatalysts for the O2 evolution reaction in PEM water electrolysers. Appl. Catal. B Environ. 164, 488–495 (2015). https://doi.org/10.1016/j.apcatb.2014.09.005
Sing, K.S.W.: Reporting physisorption data for gas, solid systems with special reference to the determination of surface area and porosity (Recommendations,1984). Pure Appl. Chem. 57, 603–619 (1985). https://doi.org/10.1351/pac198557040603
Rivera Gavidia, L., Sebastián, D., Pastor, E., Aricò, A., Baglio, V.: Carbon-supported Pd and PdFe alloy catalysts for direct methanol fuel cell cathodes. Materials (Basel). 10, 580 (2017). https://doi.org/10.3390/ma10060580
Bauer, A., Chevallier, L., Hui, R., Cavaliere, S., Zhang, J., Jones, D., Rozière, J.: Synthesis and characterization of Nb-TiO2 mesoporous microsphere and nanofiber supported Pt catalysts for high temperature PEM fuel cells. Electrochim. Acta 77, 1–7 (2012). https://doi.org/10.1016/J.ELECTACTA.2012.04.028
Huang, D., Zhang, B., Bai, J., Zhang, Y., Wittstock, G., Wang, M., Shen, Y.: Pt catalyst supported within TiO2 mesoporous films for oxygen reduction reaction. Electrochim. Acta 130, 97–103 (2014). https://doi.org/10.1016/J.ELECTACTA.2014.02.115
Kim, J.-H., Ishihara, A., Mitsushima, S., Kamiya, N., Ota, K.-I.: Catalytic activity of titanium oxide for oxygen reduction reaction as a non-platinum catalyst for PEFC. Electrochim. Acta 52, 2492–2497 (2007). https://doi.org/10.1016/j.electacta.2006.08.059
Chanmanee, W., de Tacconi, N.R., Rajeshwar, K., Lin, W.-Y., Nikiel, L., Wampler, W.A.: Photocatalytically generated trimetallic (Pt-Pd-Au/C-TiO2) nanocomposite electrocatalyst. J. Electrochem. Soc. 159, F226–F233 (2012). https://doi.org/10.1149/2.038207jes
Tammeveski, K., Tenno, T., Rosental, A., Talonen, P., Johansson, L.-S., Niinistö, L.: The reduction of oxygen on Pt-TiO[sub 2] coated Ti electrodes in alkaline solution. J. Electrochem. Soc. 146, 669 (1999). https://doi.org/10.1149/1.1391660
Tiido, K., Alexeyeva, N., Couillard, M., Bock, C., MacDougall, B.R., Tammeveski, K.: Graphene–TiO2 composite supported Pt electrocatalyst for oxygen reduction reaction. Electrochim. Acta 107, 509–517 (2013). https://doi.org/10.1016/J.ELECTACTA.2013.05.155
Jukk, K., Kozlova, J., Ritslaid, P., Sammelselg, V., Alexeyeva, N., Tammeveski, K.: Sputter-deposited Pt nanoparticle/multi-walled carbon nanotube composite catalyst for oxygen reduction reaction. J. Electroanal. Chem. 708, 31–38 (2013). https://doi.org/10.1016/J.JELECHEM.2013.09.009
Pei, D.-N., Gong, L., Zhang, A.-Y., Zhang, X., Chen, J.-J., Mu, Y., Yu, H.-Q.: Defective titanium dioxide single crystals exposed by high-energy 001 facets for efficient oxygen reduction. Nat. Commun. 6, 8696 (2015). https://doi.org/10.1038/ncomms9696
Liu, S., Yu, J., Jaroniec, M.: Anatase TiO2 with dominant high-energy 001 facets: synthesis, properties, and applications. Chem. Mater. 23, 4085–4093 (2011). https://doi.org/10.1021/cm200597m
A. Więckowski, Interfacial electrochemistry : theory, experiment, and applications, Marcel Dekker, 1999
Park, K.-W., Seol, K.-S.: Nb-TiO2 supported Pt cathode catalyst for polymer electrolyte membrane fuel cells. Electrochem. Commun. 9, 2256–2260 (2007). https://doi.org/10.1016/j.elecom.2007.06.027
Castegnaro, M.V., Paschoalino, W.J., Fernandes, M.R., Balke, B., Alves, M.C.M., Ticianelli, E.A., Morais, J.: Pd–M/C (M = Pd, Cu, Pt) Electrocatalysts for Oxygen reduction reaction in alkaline medium: correlating the electronic structure with activity. Langmuir 33, 2734–2743 (2017). https://doi.org/10.1021/acs.langmuir.7b00098
Dumitru, A., Mamlouk, M., Scott, K.: Effect of different chemical modification of carbon nanotubes for the oxygen reduction reaction in alkaline media. Electrochim. Acta 135, 428–438 (2014). https://doi.org/10.1016/j.electacta.2014.04.123
Acknowledgements
The research leading to these results has received funding from the ``Accordo di Programma CNR-MiSE, Gruppo tematico Sistema Elettrico Nazionale e Progetto: Sistemi elettrochimici per l’accumulo di energia’’.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Publisher's Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Alegre, C., Siracusano, S., Modica, E. et al. Titanium–tantalum oxide as a support for Pd nanoparticles for the oxygen reduction reaction in alkaline electrolytes. Mater Renew Sustain Energy 7, 8 (2018). https://doi.org/10.1007/s40243-018-0114-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40243-018-0114-z