
Abstract
Introduction
Infantile nephropathic cystinosis (INC) is an autosomal recessive lysosomal disorder in which patients develop deposits of cystine crystals in their kidneys and corneas from a young age.
Methods
We conducted a retrospective analysis of children with INC seen by ophthalmologists at the Manchester Royal Eye Hospital between 2002 and 2018, to evaluate clinical findings, symptoms and treatment.
Results
Twenty-two children diagnosed with INC from age 0 (prenatally) to 11 years were assessed. All evaluable patients had corneal cystine crystal deposits, and 15 had mild to moderate photophobia. Ten patients had other ocular conditions including blepharitis/chalzion (n = 6), swollen optic nerve (n = 3), punctate epitheliopathy (n = 3), corneal scarring (n = 1),and elevated intraocular pressure (n = 2). Confocal imaging identified nerve abnormalities in two patients (enlarged corneal nerve + abnormal-looking tortuous nerves in one patient and beaded nerves in the sub-basal plexus in the other), both of whom had significant crystal deposition in the anterior stroma. Visual acuity was relatively unaffected. All 22 patients were receiving oral cysteamine, and 21 were applying cysteamine eye drops (galenic preparation of 0.55% concentration, compounded by a hospital pharmacy). Recommended application frequency was at least eight times per day in all patients with dosing information available.
Conclusions
This case series of patients with INC highlights the consistent pattern of corneal cystine crystal deposition, which is universally present from a young age in this condition, and the high incidence of photophobia even in young children. Corneal manifestations of INC persisted despite frequent administration of the hospital pharmacy-made eye drop preparation. Reasons for this lack of efficacy may include the lag period between diagnosis and first prescription of cysteamine eye drops and the difficulty in maintaining rigorous compliance with this treatment. In addition, the challenge for patients of maintaining optimal storage conditions may adversely affect the stability and efficacy of cysteamine within this preparation.
Funding
Editorial assistance was funded by Orphan Europe Ltd.
Similar content being viewed by others


Introduction
Cystinosis is a rare autosomal recessive disorder affecting lysosomal storage of the amino acid cystine [1, 2]. In unaffected individuals, cystine is removed from lysosomes via a lysosomal membrane protein called cystinosin in a transport mechanism involving H+ ions [3, 4]. The cystinosin protein contains 367 amino acids and seven transmembrane domains, and is encoded for by the 12-exon CTNS gene on chromosome 17p13.2 [2,3,4,5,6]. Homozygous or compound heterozygous mutations in this gene result in cystinosis, in which lysosomes full of cystine crystals accumulate within cells, causing tissue damage [7]. Kidneys are particularly affected, but cystine crystal accumulation may also occur in the cornea and conjunctiva, causing photophobia, blepharospasm and ocular discomfort [7,8,9]. Corneal crystal deposits are present in most patients from a young age (12–18 months), and can be one of the earliest extrarenal signs of cystinosis [10,11,12]. As a result of the significant and persistent photophobia, patients are at risk of developing refractory blepharospasm [7]. Some patients with cystinosis show involvement of the posterior segment of the eye with hypopigmentary mottling of the retinal pigment epithelium (RPE) [9]. As patients age and their disease progresses, they may develop other ocular complications such as peripheral corneal neovascularization, band keratopathy, glaucoma and visual impairment [9].
Patients with nephropathic cystinosis usually receive oral cysteamine therapy, which enters the lysosome via an unknown transporter and breaks down cystine to form a mixed cysteine–cysteamine disulphide and cysteamine, which exits the lysosome using intact amino acid transporters [7]. Oral cysteamine (mercaptamine) can slow the progression of many cystinosis complications [13, 14], and has led to an increased life expectancy in affected patients. However, oral cysteamine has limited effects on ocular anterior segment complications because of poor systemic delivery to the avascular cornea [7].
Topical cysteamine applied directly to the eye can reduce corneal crystals in some patients with nephropathic cystinosis [15,16,17,18,19,20], although this has not been a consistent finding [21, 22]. Unfortunately, aqueous cysteamine eye drops are not available in many countries, including the UK, such that drops need to be compounded in a pharmacy [23]. This results in variability of standardisation of drop formulation, potential sterility issues, and supply reliability [23]. In addition, cysteamine is rapidly oxidised to cystamine [9], which is ineffective at reducing corneal crystal density [18]. The poor stability of pharmacy-compounded topical cysteamine preparations means that these drops must be carefully stored to prevent oxidation, and used within a week of being opened [9]. There is evidence of lack of stability of cysteamine eye drops under regular storage conditions, even when the drops are prepared by hospital pharmacies at established expert centres [24].
Because cystinosis patients are now living longer, the management of this metabolic inborn and chronic disorder is becoming more complex [25]. Ocular involvement is present in 100% of patients [12], so the ocular manifestations are increasingly being considered as a significant burden of this chronic disease.
This report describes the clinical presentation of ocular changes, symptoms, treatment and clinical course of 22 current patients with infantile nephropathic cystinosis (INC) who were referred to the paediatric ophthalmology service at the Manchester Royal Eye Hospital between 2002 and 2018 from the North–West of England. All subjects remain under ongoing ophthalmology review at the Manchester Royal Eye Hospital, Manchester University Hospital, NHS Foundation Trust, UK.
Methods
A retrospective review was conducted of the clinical records of patients with a genetically and biochemically confirmed diagnosis of INC, who had been referred to the Manchester Royal Eye Hospital. The clinical history, including age at diagnosis, age at which systemic treatment with cysteamine commenced, age at which treatment with topical cysteamine commenced and by whom it was prescribed, was ascertained from the clinical records. The cystine level in white blood cells was elevated in all patients consistent with the diagnosis of INC, and the dose of oral cysteamine treatment was titrated to maintain levels at less than 1.0 nmol half-cysteine/mg protein. The topical cysteamine preparation used in these patients was a galenic preservative-free preparation of cysteamine hydrochloride (0.55% w/v) eye drops, compounded by Guy’s and St. Thomas’ Hospital Pharmacy in London and provided in 12-mL bottles (Fig. 1). As this was a retrospective review of anonymised patient records, no ethics approval was required and the study was approved by the Manchester University Hospital NHS Foundation Trust Review Board.

Cysteamine eye drops prepared by the Guy’s and St. Thomas’ Hospital pharmacy for cystinosis patients at Manchester Royal Eye Hospital
INC patients attending the paediatric ophthalmology clinic routinely underwent a structured clinical assessment consisting of age-appropriate measure of visual acuity, contrast sensitivity, colour vision and, where indicated, ocular motility assessment. For older children, visual fields were formally assessed with Humphrey 24-2 automated perimetry (Carl Zeiss Meditec AG, Jena, Germany). Patients also underwent ocular assessment of pupil reaction, anterior segment slit lamp biomicroscopy, intraocular pressure measurement with Goldmann tonometry or rebound tonometry with the ICare® rebound tonometer (ICare®, Vantaa, Finland).
The integrity of the ocular surface was assessed on slit lamp biomicroscopy after instillation of topical 1% fluorescein eye drops using a cobalt blue filter. Examination of the optic disc, macula and retinal periphery was carried out in all cases. Imaging of the cornea and anterior segment was undertaken in children old enough to co-operate with slit lamp photography, using a slit beam of 1–2 mm width, 10 mm height, at greater than 60° and 16 times magnification focused on the anterior corneal surface, centred on the middle of the cornea with the patients looking straight ahead. In children too young to co-operate fully with corneal imaging, plain images were taken with diffuse anterior illumination of the ocular surface at 16 times magnification. Corneal crystals deposition on slit lamp imaging was graded according to the Gahl score [26].
Anterior segment swept source optical coherence tomography (OCT) was carried out using the Topcon Triton DRI OCT camera (Topcon Europe Medical BV, Capelle aan den IJssel, the Netherlands) adjusted to capture the cornea and anterior segment.
In older children, in vivo confocal microscopy of the cornea was performed with the HRT3-RCM in vivo confocal microscope system (Heidelberg Engineering GmbH, Heidelberg, Germany). Posterior segment imaging was carried out with OCT scans of the macula and disc using the Topcon Triton OCT camera and ultra-wide-angle fundus imaging with the Optomap California (Optos®, Marlborough, MA, USA) retinal imaging device. Dilated fundal examination was carried out after pharmacological dilation with topical 1% cyclopentolate eye drops in each eye.
All patients were asked to answer a questionnaire related to symptoms of photophobia (patient-assessed symptoms), prescribed topical medications, compliance with topical medications and associated side effects. In addition, patients provided qualitative information on the impact of their ocular treatment on day-to-day life from use of their topical medications. If age and development made it possible, the questionnaires were completed by the affected children themselves with the assistance of their parents/guardians. If the child was too young, the questionnaires were completed by the child’s parent/guardian. Patient-assessed photophobia scores were recorded according to the established grading system developed by Liang and colleagues [27]. Clinicians also assessed photophobia.
Results
This retrospective chart review identified 22 patients diagnosed with INC between 1994 and 2017 and who had attended the paediatric ophthalmology clinic at Manchester Royal Eye Hospital. Twenty-one patients had been referred from the paediatric nephrology department of the Royal Manchester Children’s Hospital, and one patient had been referred from a paediatric nephrologist elsewhere. Sixteen of the patients remained under the care of the paediatric nephrology department and were reviewed within a multidisciplinary team (MDT) involving paediatric ophthalmology, paediatric nephrology and paediatric endocrinology. The other six patients were seen by ophthalmologists at the Manchester Royal Eye Hospital, separately from the MDT clinic.
Age at INC diagnosis ranged from 0 to 11 years; two patients were diagnosed before birth by prenatal DNA testing (Table 1). Renal status was known in 19 patients, and only two of these patients, aged 5 years and 12 years, had renal function within the normal range. Both were genetically confirmed to be homozygous for a pathogenic mutation on CTNS, and the younger child had been diagnosed with prenatal testing. Nine patients had undergone renal transplants, including one who was awaiting a second transplant, and two more patients were awaiting their first transplant.
All 22 patients were receiving oral cysteamine therapy; age at start of systemic cysteamine treatment was available for 19 of these patients, with a median age at commencement of 12 months (range 0.25–123 months). Of these, the majority (17 patients, 89.5%) were started oral cysteamine below the age of 2.5 years. Two patients were diagnosed late, age 4 years and aged 11 years respectively, but commenced oral treatment as soon as confirmation of INC was made.
Age at first ophthalmological examination ranged from 9 months to 11 years (median 31 months). For children born in the 1990s, the delay between diagnosis and first ophthalmic examination was between 4.8 and 9 years, whereas every child with INC born after 2000 had an ophthalmic examination within 3 years of diagnosis. Two children had their first ophthalmic examination before they had been diagnosed with INC.
All but one patient had identifiable corneal crystals (Table 2) and 17 patients had mild to moderate photophobia (Fig. 2). In three patients, the photophobia was associated with epiphora. Other symptoms reported by patients included itching (n = 2), irritation (n = 2), occasional pain (n = 1) and glare (n = 1). Complete ophthalmological examination revealed the presence of other ocular conditions in ten patients (see Table 3). Interestingly, there were a number of patients with external eye disease, specifically blepharitis/chalazia in this cohort possibly indicating association with the systemic disease, which may affect Meibomian gland function. Other complications such as disc swelling and raised intracranial pressure are known associations with INC and require ongoing vigilance to monitor in patients with this condition [8]. Other conditions such as glaucoma or ocular hypertension are known late complications [11] but may be coincidental in the two patients identified in this study. The exact cause of external ophthalmoplegia in one patient, despite investigation for mitochondrial or genetic disease, remains undetermined but was not felt to be related to her INC.

Number of patients experiencing photophobia
The patient who had been diagnosed at age 11 years showed significant corneal crystal deposition (Gahl score 3 in both eyes) and moderate photophobia, accompanied by blepharitis, punctate epitheliopathy, retinal depigmentation in both eyes and full discs.
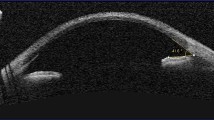
Confocal imaging identified nerve abnormalities in two patients (enlarged corneal nerve + abnormal-looking tortuous nerves in one patient and beaded nerves in the sub-basal plexus in the other); both cases were associated with significant crystal deposition in the anterior stroma (Fig. 3).

a Optomap image of retina of patient with peripheral RPE pigmentary abnormalities; b corneal slit-lamp image of patient with severe grade 3.0 corneal crystal deposition; c Anterior segment swept source OCT showing anterior stromal corneal crystal deposits to a depth of 114 microns (20.8% of the overall depth of cornea); d confocal imaging of cornea in INC. (i) Showing needle-shaped cystine crystals within corneal epithelial layer, (ii) showing beaded nerve abnormalities in cornea of a patient with INC (arrow), (iii) showing denser cystine crystals within the anterior stroma
Visual acuity was mildly reduced in most patients. The logMAR score in either eye ranged from − 0.1 to 0.3 (median 0.1), and five patients had a logMAR score of at most 0.0 in both eyes.
All but one patient was prescribed cysteamine eye drops (one patient refused topical cysteamine drops); these were first prescribed by a nephrologist in 11 patients, all at a frequency of four times a day, and by an ophthalmologist in seven, initially at a frequency of four times a day. The prescriber was not recorded in the other three cases. The eventual dosing frequency was available for seven patients: hourly (n = 2), every 2 h (n = 1), 8 times a day (n = 1) or 10–12 times per day (n = 3). In all cases where the nephrologist first prescribed topical cysteamine, the exact indications for commencement for treatment was unclear, whilst the prescribing of topical cysteamine by ophthalmologists was generally delayed until the patients started to complain of significant photophobia or other ocular symptoms.
Compliance with application of cysteamine drops was good in all except two patients, both of whom reported adverse effects of the eye drops. The remaining patients used cysteamine drops regularly, only occasionally missing doses, although most patients could only manage an eye drop administration frequency of 4–6 times a day. All patients using the cysteamine eye drops found that administration restricted them in some ways because of the need to refrigerate the drops. Most patients described mild stinging, redness and watering after application of the eye drops but were able to tolerate their continued use.
Discussion
This retrospective review of paediatric patients with INC confirms earlier findings that corneal cystine crystal formation is almost universal in these patients and that photophobia is common, despite use of topical cysteamine for many years in a number of patients [28, 29]. One of our patients was too young (9 months old) to accurately assess the amount of corneal crystals, but it is known that crystals start to form within the first year and are visible in all corneas by 16 months of age [26]. Crystals appear to reach maximum density at around 8 years of age [7]. Previous analyses of ocular conditions in patients with INC noted that photophobia evolved and worsened over time, becoming clinically apparent in most patients by the age of 3 or 4 years, causing discomfort from around 10 years of age and becoming severe after the age of 15 years [28, 29]. Our findings support previous reports, with mild to moderate photophobia being present in 72% of patients in our cohort, aged between 9 months and 11 years. Despite extensive and diffuse cystine crystal deposition in our cohort of patients, visual acuity was relatively unaffected. This is consistent with previous reports in patients with INC, which have shown that vision is relatively preserved during childhood, but deteriorates after the age of 20 years [28].
Although this report of a cystinosis case series does not represent a longitudinal analysis of the patients’ condition and care, we noted a promising trend towards more rapid assessment of ocular manifestations of cystinosis in recent years. This may further improve with the development of the MDT which was set up to manage cystinosis patients at our centre in 2017. There have been very few formal analyses of MDT care in cystinosis patients; however, this model of care makes clinical sense for a condition affecting multiple systems, and has been appreciated by cystinosis patients when implemented at other centres [30]. Standardised assessments within the MDT includes systematic imaging of corneas using anterior segment slit-lamp imaging, anterior segment OCT and in vivo confocal microscopy to enable better potential quantitative analysis of crystal deposition within the cornea. This approach enables an objective assessment of response to topical therapy. The additional posterior segment complications can be monitored similarly using OCT imaging of retina and optic nerve as well as wide-angle imaging. These latter techniques are well tolerated and quickly captured. This has enabled us to identify three patients with swollen optic nerves with presumed raised intracranial pressure and one patient with significant retinal pigment epithelial changes, likely to have resulted from a delay in diagnosis and commencement of oral cysteamine treatment.
Most patients in our series were taking oral cysteamine therapy and topical cysteamine eye drops, which may help to relieve symptoms and delay worsening of ocular manifestations of INC [15,16,17,18, 31]. Oral cysteamine does not alleviate corneal crystal deposition because of the avascular nature of the cornea, but may reach the retinal epithelium and help to delay retinal infiltration [28]. It was notable that the diagnosis of INC had been delayed until 11 years of age in one patient in our case series, and this was the only patient that showed pigment dropout in the retinal epithelium of both eyes on fundoscopy in the absence of oral cysteamine treatment.
In our patient cohort, the prescribing of topical cysteamine was variable, with the majority of patients receiving this medication from nephrologists and advised to take the medication four times a day. The reasons for prescribing the topical cysteamine eye drops by nephrologists was unclear. There was, however, a significant lag time between patients receiving oral cysteamine therapy, which coincided with the diagnosis of INC being made, and first commencement of topical cysteamine. Topical administration of cysteamine eye drops, when prescribed by ophthalmologists, was typically delayed until there was clear evidence of symptoms of photophobia and ocular surface discomfort. Nephrologists may have initially deferred prescribing topical cysteamine to ophthalmologists, although latterly, this may have changed with an increased willingness to take it upon themselves to prescribe. The advice to administer topical cysteamine was reinforced and altered within the MDT clinics to more frequent administration (8–10 times a day), such that 64% of patients who were taking cysteamine drops had been advised to use them at least eight times per day. However, most patients found this frequency challenging to sustain. Whilst there is evidence that frequent administration of topical cysteamine has a sustained effect on reducing corneal crystals and that this can be achieved regardless of the age of the patient [26], there is a gap in knowledge as to when it is best to prescribe topical cysteamine, i.e. as soon as crystals are found or when the patient becomes symptomatic. Further study looking at efficacy combined with a validated quality of life survey may provide greater clarity on this issue.
The cysteamine hydrochloride 0.55% prepared by the hospital pharmacy is considered to be less effective when administered at 4–6 times per day [22], and more frequent application is generally advised [15, 21, 32]. The stability of the preservative-free drug formulation is poor and also depends on storage at the correct temperature, which dictates that the drops are used for up to a week before requiring a change. Even storage at a refrigerated temperature has been shown to reduce the efficacy of the eye drop because of oxidation of the cysteamine [24]. In some countries, similar preservative-free local preparations must be used within 24 h of opening [20, 21, 24].
Variable efficacy of aqueous cysteamine eye drops may be due to formulation instability, specifically oxidation of cysteamine, due the absence of antioxidants in the formulations [29]. Other reasons include the genetic heterogeneity of cystinosis [21], insufficient concentration of cysteamine in the drops [16, 21] and poor topical absorption of cysteamine [21, 22]. Aqueous formulations of cysteamine remain for a short period of time on the ocular surface and are rapidly cleared by the patient’s own tears, draining rapidly through the tear ducts. The bioavailability of cysteamine will therefore be limited, mandating frequent administration. Poor compliance associated with frequent administration schedule [9, 15, 32] has been identified as a limiting factor, although some patients do achieve excellent compliance with hourly administration during waking hours. McKenzie et al. investigated the suitability of a variety of gel carriers for ophthalmic delivery of topical cysteamine in laboratory tests. In their analysis, the principle of gel formulations providing good optical properties and good sustained delivery, with significant bioadhesion and long-term stability was demonstrated [33]. As with any retrospective review of patient management, there was no control group of patients that had never received topical cysteamine eye drops in our cohort, so the efficacy of the compounded topical cysteamine could not be proven, but there is good data to suggest that strict compliance to frequent aqueous eye drop administration does reduce corneal crystal load [26].
Recently, a commercial formulation of cysteamine eye drops has been developed and approved by the European Medicines Agency for use in the European Union in 2017 for patients with ocular cystinosis aged 2 years or more [34], allowing physicians to use a licensed preparation instead of pharmacy-prepared aqueous drops. This viscous formulation (Cystadrops; Orphan Europe) maintains contact with the ocular surface for longer than aqueous cysteamine eye drops do, and can be administered four times daily [23, 34]. The viscous formulation may also help with penetration by prolonging the time that the cysteamine is in contact with the ocular surface. In clinical trials, Cystadrops have been shown to be effective in reducing corneal cystine deposits in both adults and children with cystinosis [35, 36]. One of these studies compared Cystadrops with aqueous cysteamine eye drops 0.10% and found that Cystadrops were significantly more effective than aqueous drops for reducing the extent and depth of corneal crystal depositions, and were preferred by patients [36].
The Cystadrops gel formulation has an improved stability profile compared with aqueous drops and it can be stored in the refrigerator without the requirement for freezing. In addition, Cystadrops can be kept at room temperature after the first opening [23, 34]. The expectation of a commercial preparation is more standardised formulation than pharmacy-prepared solutions, and consistent availability [23]. In common with aqueous cysteamine preparations, Cystadrops need to be discarded 7 days after first opening [23, 34].
At present, Cystadrops is not commonly available to be prescribed in the UK, despite the fact that it is licensed and the General Medical Council in the UK, he Royal College of Ophthalmologists and the UK Ophthalmic Pharmacy Group officially recommend prescribing a licensed preparation where it is available to treat a condition [37].
Whilst topical cysteamine remains the main modality of treatment for corneal crystals associated with cystinosis, other therapeutic options may become available in future, including the use of contact lenses impregnated with cysteamine [38] and nanowafers [39] both of which can be placed directly on the cornea and release a sustained and prolonged delivery of the cysteamine to the cornea. Human trials of these modalities are awaited to assess efficacy and acceptability.
Conclusions
This cases series emphasizes the limited efficacy of the hospital compounded topical medication which may be due to a number of factors including challenges with compliance, uncertainty with regard to stability of the hospital compounded formulation, historically variable indications for prescribing resulting in a significant lag period between diagnosis and commencement of topical treatment and the need to gather more data on the quality of life of patients affected by the condition and influence of treatment on this.
This case series of patients with INC highlights the consistent pattern of corneal cystine crystal deposition, which is universally present from a young age in this condition, as well as the high incidence of photophobia even in young children. The data emphasise the importance of thorough ophthalmological examination of patients with INC from a young age. Many patients in this case series had been prescribed cysteamine eye drops by a nephrologist at a frequency of four times a day, whilst there is good evidence in the literature that a more effective dosage regimen for this preparation would be eight times per day or more. Once seen by an ophthalmologist, patients were advised to increase the frequency of eye drop administration to optimize efficacy. As the current aqueous drop treatment is compounded in a hospital pharmacy according to good manufacturing principles, information about stability and characteristics of the topical preparation are unfortunately not available. Despite topical treatment in the majority of patients, ocular manifestations of INC persisted despite regular administration of drops. Causes for this may be due to the lag period between diagnosis and first prescription of cysteamine eye drops, difficulty in maintaining compliance with this treatment, unproven stability and reduced active drug bioavailability. Clinical data on the potential impact of the new viscous formulation of topical cysteamine drops are promising, but real-world data in a case series such as ours would be a useful adjunct to clinical trial findings.
References
Gahl WA, Bashan N, Tietze F, Bernardini I, Schulman JD. Cystine transport is defective in isolated leukocyte lysosomes from patients with cystinosis. Science. 1982;217:1263–5.
Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med. 2002;347:111–21.
Gahl WA, Balog JZ, Kleta R. Nephropathic cystinosis in adults: natural history and effects of oral cysteamine therapy. Ann Intern Med. 2007;147:242–50.
Kalatzis V, Antignac C. New aspects of the pathogenesis of cystinosis. Pediatr Nephrol. 2003;18:207–15.
Town M, Jean G, Cherqui S, et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet. 1998;18:319–24.
Wilmer MJ, Emma F, Levtchenko EN. The pathogenesis of cystinosis: mechanisms beyond cystine accumulation. Am J Physiol Renal Physiol. 2010;299:F905–16.
Gahl W. Cystinosis. In: Avner ED, Harmon WE, Niaudet P, Yoshikawa N, editors. Pediatric nephrology. 6th ed. Berlin Heidelberg: Springer; 2009. p. 1019–2038.
Bishop R. Ocular complications of infantile nephropathic cystinosis. J Pediatr. 2017;183S:S19–21.
Shams F, Livingstone I, Oladiwura D, Ramaesh K. Treatment of corneal cystine crystal accumulation in patients with cystinosis. Clin Ophthalmol. 2014;8:2077–84.
Kaiser-Kupfer MI, Caruso RC, Minkler DS, Gahl WA. Long-term ocular manifestations in nephropathic cystinosis. Arch Ophthalmol. 1986;104:706–11.
Tsilou ET, Rubin BI, Reed GF, Iwata F, Gahl W, Kaiser-Kupfer MI. Age-related prevalence of anterior segment complications in patients with infantile nephropathic cystinosis. Cornea. 2002;21:173–6.
Veys KR, Elmonem MA, Arcolino FO, van den Heuvel L, Levtchenko E. Nephropathic cystinosis: an update. Curr Opin Pediatr. 2017;29:168–78.
Gahl WA, Reed GF, Thoene JG, et al. Cysteamine therapy for children with nephropathic cystinosis. N Engl J Med. 1987;316:971–7.
Markello TC, Bernardini IM, Gahl WA. Improved renal function in children with cystinosis treated with cysteamine. N Engl J Med. 1993;328:1157–62.
Bradbury JA, Danjoux JP, Voller J, Spencer M, Brocklebank T. A randomised placebo-controlled trial of topical cysteamine therapy in patients with nephropathic cystinosis. Eye (Lond). 1991;5(6):755–60.
Iwata F, Kuehl EM, Reed GF, McCain LM, Gahl WA, Kaiser-Kupfer MI. A randomized clinical trial of topical cysteamine disulfide (cystamine) versus free thiol (cysteamine) in the treatment of corneal cystine crystals in cystinosis. Mol Genet Metab. 1998;64:237–42.
Kaiser-Kupfer MI, Fujikawa L, Kuwabara T, Jain S, Gahl WA. Removal of corneal crystals by topical cysteamine in nephropathic cystinosis. N Engl J Med. 1987;316:775–9.
Kaiser-Kupfer MI, Gazzo MA, Datiles MB, Caruso RC, Kuehl EM, Gahl WA. A randomized placebo-controlled trial of cysteamine eye drops in nephropathic cystinosis. Arch Ophthalmol. 1990;108:689–93.
Tsilou ET, Thompson D, Lindblad AS, et al. A multicentre randomised double masked clinical trial of a new formulation of topical cysteamine for the treatment of corneal cystine crystals in cystinosis. Br J Ophthalmol. 2003;87:28–31.
Radojkovic B. Cysteamine eye drops in the treatment of cystinosis—an Australian perspective. J Pharm Pract Res. 2015;45:440–5.
Al-Hemidan A, Shoughy SS, Kozak I, Tabbara KF. Efficacy of topical cysteamine in nephropathic cystinosis. Br J Ophthalmol. 2017;101:1234–7.
MacDonald IM, Noel LP, Mintsioulis G, Clarke WN. The effect of topical cysteamine drops on reducing crystal formation within the cornea of patients affected by nephropathic cystinosis. J Pediatr Ophthalmol Strabismus. 1990;27:272–4.
Lyseng-Williamson KA. Cystadrops (cysteamine hydrochloride 0.55% viscous eye-drops solution) in treating corneal cystine crystal deposits in patients with cystinosis: a profile of its use. Drugs Ther Perspect. 2017;33:195–201.
Reda A, Van Schepdael A, Adams E, et al. Effect of storage conditions on stability of ophthalmological compounded cysteamine eye drops. JIMD Rep. 2018;42:47–51.
Biswas S, Gaviria M, Malheiro L, Marques JP, Giordano V, Liang H. Latest clinical approaches in the ocular management of cystinosis: a review of current practice and opinion from the ophthalmology cystinosis forum. Ophthalmol Ther. 2018;7(2):307–322.
Gahl WA, Kuehl EM, Iwata F, Lindblad A, Kaiser-Kupfer MI. Corneal crystals in nephropathic cystinosis: natural history and treatment with cysteamine eyedrops. Mol Genet Metab. 2000;71:100–20.
Liang H, Baudouin C, Hassani RTJ, Brignole-Baudouin F, Labbe A. Photophobia and corneal crystal density in nephropathic cystinosis: an in vivo confocal microscopy and anterior-segment optical coherence tomography study. Invest Ophthalmol Vis Sci. 2015;56:3218–25.
Dureau P, Broyer M, Dufier JL. Evolution of ocular manifestations in nephropathic cystinosis: a long-term study of a population treated with cysteamine. J Pediatr Ophthalmol Strabismus. 2003;40:142–6.
Dufier JL, Dhermy P, Gubler MC, Gagnadoux MF, Broyer M. Ocular changes in long-term evolution of infantile cystinosis. Ophthalmic Paediatr Genet. 1987;8:131–7.
Pinxten AM, Hua MT, Simpson J, Hohenfellner K, Levtchenko E, Casteels I. Clinical Practice: a proposed standardized ophthalmological assessment for patients with cystinosis. Ophthalmol Ther. 2017;6:93–104.
Tsilou E, Zhou M, Gahl W, Sieving PC, Chan CC. Ophthalmic manifestations and histopathology of infantile nephropathic cystinosis: report of a case and review of the literature. Surv Ophthalmol. 2007;52:97–105.
Jones NP, Postlethwaite RJ, Noble JL. Clearance of corneal crystals in nephropathic cystinosis by topical cysteamine 0.5%. Br J Ophthalmol. 1991;75:311–2.
McKenzie B, Kay G, Matthews KH, Knott R, Cairns D. Preformulation of cysteamine gels for treatment of the ophthalmic complications in cystinosis. Int J Pharm. 2016;515(1–2):575–82.
European Medicines Agency: Cystadrops 3.8 mg/mL eye drops solution. Summary of product characteristics. http://www.ema.europa.eu/documents/product-information/cystadrops-epar-product-information_en.pdf (2017). Accessed 3 Oct 2018.
Labbe A, Baudouin C, Deschenes G, et al. A new gel formulation of topical cysteamine for the treatment of corneal cystine crystals in cystinosis: the Cystadrops OCT-1 study. Mol Genet Metab. 2014;111:314–20.
Liang H, Labbe A, Le Mouhaer J, Plisson C, Baudouin C. A new viscous cysteamine eye drops treatment for ophthalmic cystinosis: an open-label randomized comparative phase III pivotal study. Invest Ophthalmol Vis Sci. 2017;58:2275–83.
Royal College of Ophthalmologists, UK Ophthalmic Pharmacy Group: Ophthalmic special order products. http://www.rcophth.ac.uk/wp-content/uploads/2016/09/Ophthalmic-Special-Order-Products-updated-August-2016.pdf (2016). Accessed 2018.
Dixon P, Fentzke RC, Bhattacharya A, Konar A, Hazra S, Chauhan A. In vitro drug release and in vivo safety of vitamin E and cysteamine loaded contact lenses. Int J Pharm. 2018;544(2):380–91.
Marcano DC, Shin CS, Lee B, et al. Synergistic cysteamine delivery nanowafer as an efficacious treatment modality for corneal cystinosis. Mol Pharm. 2016;13:3468–77.
Acknowledgements
The authors would like to thank the INC patients, and their families, who attended the Manchester Royal Eye Hospital Paediatric Ophthalmology clinic for allowing us to report on their condition and treatment. The authors would also like to thank Ms Donna Warburton, Lead Paediatric Orthoptist, Manchester Royal Eye Hospital, Jaya Chidarambaram BSc Hons, MB BS, MRCOphth, PhD, Consultant Ophthalmologist, Manchester Royal Eye Hospital, Manchester Foundation Trust, for their participation to this work.
Funding
Article processing charges were funded through Manchester Royal Eye Hospital. No funding or sponsorship was received for this study. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Editorial Assistance
Editorial assistance in the preparation of this article was provided by Catherine Rees of Springer Healthcare Communications. Support for this assistance was funded by Orphan Europe Ltd.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Susmito Biswas has received honorarium and travel awards from Orphan Europe. Krishanthy Sornalingam has nothing to disclose.
Compliance with Ethics Guidelines
As this was a retrospective review of anonymised patient records, no ethics approval was required and the study was approved by the Manchester University Hospital NHS Foundation Trust Review Board.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced digital features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.7387448.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Biswas, S., Sornalingam, K. The Ocular Status of Cystinosis Patients Receiving a Hospital Pharmacy-Made Preparation of Cysteamine Eye Drops: A Case Series. Ophthalmol Ther 8, 125–136 (2019). https://doi.org/10.1007/s40123-018-0156-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40123-018-0156-4