
Abstract
This article reports a successful nanostructure formation from block copolymer having broad distribution of molecular weight. The block copolymer synthesis and the nanosphere formation are facile; therefore, it is promising for fabrication of nanostructure materials in large-scale manufactory. The polydisperse diblock copolymer of polystyrene-block-poly(4-vinyl pyridine) (PSVP) was prepared by the nitroxide-mediated radical polymerization that contains the fraction of poly(4-vinyl pyridine) block of 45 mol% and the overall polydispersity index of 2.08. The phase separation of PSVP was induced by the simple evaporation of co-solvent DMF:THF (70:30 v/v) of the PSVP solution. The SEM images of the self-assembled polydisperse PSVP display the spherical morphology with the diameter of ~ 50 nm, which is larger than that of block copolymer having narrow molecular weight distribution. By simply immersing the self-assembled film into iron chloride solution, the transformation from the spherical structure to the porous structure occurred directly without sacrificing the block copolymer component indicating the advantage of stimuli-response properties of the self-assembled PSVP. The results demonstrated that the polydisperse block copolymer could be used for the nanostructure formation by simple synthesis and evaporation procedures and, therefore, it is suitable for industrial applications.
Graphical Abstract

Similar content being viewed by others


Introduction
Microphase separation of the amphiphillic block copolymer (BCP) of PSVP, which is driven by the strong repulsive interaction between the non-polar polystyrene (PS) block and the polar poly(4-vinyl pyridine) (P4VP) block, generates the various morphologies at nanometer scale such as sphere, cylinder, lamellar, and gyroid. The diversity of the nanometer morphologies has broadened their applications [1] from electronics devices [2], (nano)lithography [3, 4], nanoporous structure [5], self-cleaning surface, membrane filtration [6,7,8] to biomedical materials [9, 10]. Therefore, the studies on the microphase separation of BCP have been attracted many attentions in academia and industry. However, in the large-scale or in the industrial synthesis, the self-assembly process for nanostructure morphologies is difficult because the molecular weight distribution (MWD) of the polymer is broader than that synthesized in the ideal laboratory conditions (polydispersity index, PDI < 1.2) [11]. Thereby, more attention has been focused on the nanostructure formation from polydisperse BCPs, which are synthesized by controlled/”living” radical polymerization (CRP) [12].
Strategies to approach the polydisperse BCP are either by blending of block copolymers/homopolymer having different molecular weight [13,14,15] or by adjusting the polymerization conditions [16,17,18] of CRP. The latter is considered closer to the nature of BCP structure in the large-scale industrial process [12]. Previous reports demonstrated that the morphologies such as disordered microstructure [19,20,21], ordered lamellar or cylinder [17, 19,20,21] were observed depending on the fraction of the individual components. The coexistence of two morphologies [19] and the shift of the phase boundaries [17, 18] were assigned to the broad PDI of BCPs. In addition, the sizes [13, 17] and the size distribution [20] of the microdomain increased as the PDI increased. However, there is a disagreement issue on the presence of macrophase domain [13, 18, 19, 22], which is due to the phase separation of low molecular weight homopolymer incorporated to polydisperse BCPs. The macrophase separation was observed in the self-assembly of polydisperse BCPs having high concentration of homopolymer [19] and high PDI (above 1.8) [13].
In this research, we synthesized polystyrene via TEMPO-mediated radical polymerization which is the simplest CRP method with adequate polydispersity. It is well known that thermal self-initiation of styrene and other side reactions occurred at high temperature, such as 130 and 145 °C, leading to gradual decrease in the controllability of polymerization [23, 24]. Thereby, the extension of PS with 4VP monomer produces the polydisperse PSVP. The polydisperse PSVP has been subsequently self-assembly to afford the nanostructure materials by simple evaporation of DMF/THF cosolvents (Fig. 1). The chemical characterizations of PSVP and the morphology of the nanostructure materials were revealed.

The polydisperse polystyrene-block-poly(4-vinyl pyridine) structure and their self-assembly structures at nanometer scale
Materials and experimentals
Materials
Styrene (St, Acros, 99%) was freshly purified by passing through neutral aluminium oxide column under nitrogen gas before being used. 4-Vinyl Pyridine (4VP, Acros, 99%) was freshly distilled before being used. 2,2,6,6-Tetramethylpiperidin-1-yl oxy (TEMPO, Acros, 98%), benzoyl peroxide (BPO, China, 99%), FeCl2·4H2O, FeCl3·6H2O (Xilong Scientific Co. Ltd, China), and solvents (Xilong Scientific Co, Ltd., China) were used as received.
Preparation of polystyrene capped with TEMPO (PS)
The synthesis of polystyrene (PS) was carried out in a 250-mL schlenk flask under nitrogen. The reactor was charged with St (50 mL), BPO (0.36 g, 1.50 × 10−3 mol), and TEMPO (0.31 g, 1.95 × 10−3 mol), degassed by bubbling nitrogen gas for 20 min. The polymerization was carried out at 95 °C for 60 min, raised to 130 °C for 7.5 h. The viscous solution was diluted with THF, precipitated in methanol, filtered, and dried at 70 °C for 12 h. The white powder of PS having TEMPO end functional group was obtained.
Preparation of polystyrene-block-poly(4-vinyl pyridine) (PSVP)
The synthesis of polystyrene-block-poly(4-vinyl pryridine) (PSVP) was carried out in a 250-mL schlenk flask under nitrogen gas. The synthesis procedure of PSVP is the same as PS with the reactor ratio of [4VP]:[BPO]:[PS] = 290:1:1.3. The reaction was proceeded for 2 h at 130 °C under inert gas. Then, the solution was exposed to air to stop the reaction. PSVP was precipitated from the THF solution in cold hexane, filtered, washed several times, and dried at 70 °C for 12 h.
Preparation of self-assembly film
The glass substrates were cleaned from the organic contaminants by immersing in piranha solution (H2O2 30%:H2SO4 98%—1:3 v/v) at room temperature for 60 min, cleaning with distilled water, acetone and ethanol, and drying under nitrogen flow. Then, the 20 wt% solution of PSVP in co-solvents of DMF:THF (70:30) was casted on slide glass, dried at 50 °C for 20 min, immersed in distilled water overnight, and dried under reduced atmosphere for 24 h. The film was subsequently immersed in 13 wt% iron chloride solution ([FeCl2]:[FeCl3] = 1:2) for 24 h at room temperature to afford the porous structure. The porous film was dried at ambient condition.
Characterization
Molecular weight and molecular weight distribution of polymer were determined by gel permeation chromatography (PL GPC 50Plus—Varian) equipped with Mesopore columns (7.5 × 300 mm), THF eluent at the flow rate 1 mL/min. Linear polystyrene standards were used for calibration. The polymer structure was characterized by Fourier transform infrared spectroscopy (FT-IR, EQUINOX 55 Bruker, Germany) and proton nuclear magnetic resonance (1H-NMR, 500 MHz, Bruker Avance, Germany). The morphology of nanostructure was observed from FE-SEM images (FE-SEM S4800, Hitachi, Japan).
Results and discussion
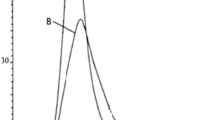
In CRP, TEMPO is used to reversibly terminate the growing polymer chain. As a result, the polymerization is controlled in a “living” fashion and the polymer structures such as the architecture, the molecular weight, and the MWD can be controlled. Since TEMPO is a stable free radical, high temperature is required for the activation/deactivation of the growing radical chains during the TEMPO-mediated radical polymerization, but most of polymerization temperature was induced at 125–135 °C to avoid the homolysis of alkoxyamine [25]. TEMPO-mediated polymerization of St with BPO as initiator at high temperature of 145 °C has not been reported yet although the synthesis at high temperature is of interest in the industrial polymerization [26]. In our experiment, the polymerizations of PS were induced at temperature of 130–145 °C with and without the initial decomposition of BPO at 95 °C. The kinetic plots and the evolution of the molecular weight are presented in Fig. 2. After the decomposition period of BPO (~ 60 min), the PS 95–130 exhibits the slow polymerization rate (St conversion ~ 0.1 for 6 h, Fig. 2a) because the excess concentration of TEMPO predominantly capped the initiator radical [27,28,29]. This stage is reduced to 2 h when the temperature increases to 145 °C (PS 95–145, Fig. 2a). That is because the large initiator radical concentration sufficiently generates from the decomposition of BPO and the thermal self-initiation of St [26]. After the slow stage, polymerization rates abruptly increase in proportion to temperature. Without BPO decomposition, the polymerization rate of PS 145 is slightly higher than PS 95–145 which may result from the additional radical stemmed from the thermal self-initiated of St at high temperature. Clearly, temperature strongly affects to the kinetic of PS polymerization. The evolution of the number-average molecular weight (Mn) and the molecular weight distribution (Mw/Mn) have been presented in Fig. 2b. It can be seen that after the initial stage, the Mn of PS increases linearly with the conversion which is the evidence of the “living” polymerization. However, the evolution of Mn of PS 145 deviates from the linearity after the St conversion of 40% (Fig. 2b). It is to be noted that the PDIs of PS 95–145 and PS 95–130 decrease to 1.26 with the conversion but the PDI of PS 145 start to increase at the conversion of 40% to the value of 1.51 (Fig. 2b). Interestingly, the same behavior of TEMPO-mediated PS polymerization at 145 °C (PS 145) was reported at 130 °C for the system without BPO initiator [30]. These authors stated that the Mn deviation from linearity is due to the termination reactions, which are degenerative transfer and bimolecular termination reactions, and the loss of functional end group. These results suggested that the polymerization of PS 145 involves the predominant thermal self-polymerization of St; hence, the termination reactions and the chain end loss cause the deviation. This assumption will be further discussed in terms of MWD (Figs. 2b, 3).

Kinetic plots (a) and evolution of Mn and Mw/Mn versus conversion (b) for the PS polymerization at 145 °C (PS 145); 95 °C—60 min—145°C (PS 95–145) and 95°C—60 min—130°C (PS 95–130). Polymerization condition: [St]:[BPO]:[TEMPO] = 290:1:1.3

GPC chromatograms of the evolution of molecular weight versus conversion for PS polymerization conditions of a 145 °C, b 95 °C—60 min—145 °C and c 95 °C—60 min—130 °C and PS-b-P4VP (PSVP)
The chromatograms (Fig. 3) gradually shift to lower retention time with increasing St conversion, indicating the increase of PS molecular weight. In addition, the unimodal curves of PS 95-130, PS 95-140 chromatograms are the evidence for the “living” behavior of PS polymerization. On the contrary, the bimodal curve of PS 145 (St conversion ~ 0.57) indicates the loss chain-end. This result supports the deviation from linearity of the evolution of Mn as discussed above. Although PS 95–145 and PS 95–130 possess the narrow PDIs, the appearance of the tails on GPC curves may indicate that there are polymer chains that lose their functional end groups. These terminated chains are confirmed through the bimodal curves of the chain extension of PS 95–130 with 4VP (PSVP on Fig. 3c). It can be observed on GPC curve that there is the similar peak at evolution time (~ 450 s) of PSVP to the peak of PS 95–130 at conversion of ~ 3%. Hence, it can be concluded that PSVP contains a portion of PS low molecular weight ~ 3500 g/mol. The overall number-average molecular weight and PDI of PSVP are 24,000 g/mol and 2.08, respectively.
The chemical structure and composition of PS and PSVP were determined through FT-IR (Fig. 4) and 1H-NMR (Fig. 5). The typical vibrations of PS are observed around 3070–3030, 2930–2850, and 1600 cm−1 attributing for the aromatic, aliphatic C–H stretching and C=C ring stretching vibrations, respectively. Aromatic overtone vibration peaks are also observed around 1900–1700 cm−1 [31] (PS in Fig. 4). The more prominent peaks at 1600, 1560, 1500, 1460, and 1410 cm−1 on FT-IR are assigned for the vibration of C=C and C=N stretching of pyridine rings [31] which are the characteristic vibrations of pyridine groups in PSVP. Further evidence of 4VP incorporated to PS is observed on 1H-NMR (Fig. 5) with the representative chemical shifts of proton on pyridine ring and phenyl ring at 8–8.5 ppm and 6–7.5 ppm, respectively. Based on the integration ratio of these peaks, we can calculate the PS:P4VP ratio approximately 1.2, yielding the fraction of P4VP of ~ 45 mol% of PSVP. Hence, the roughly estimated composition of PSVP composes of PS13k-b-P4VP11k and PS3.5k.

Fourier transform infrared spectra of PS and PS-b-P4VP (PSVP)

1H-NMR spectrum of PS-b-P4VP (PSVP)
Previous reports [32, 33] stated that the solution of PSVP having narrow PDI in DMF:THF existed in the form of the spherical to thread-like micelles with P4VP core and PS shell. In our experiment, after fast evaporation of solvent at 50 °C for 20 min, the morphology of thick films appears as bumps with the size of ~ 50 nm and partly regular (Fig. 6). At the defect edge (black arrow in Fig. 6a), the spherical morphology of the self-assembled PSVP can be clearly observed. The defect surface is possibly due to the dewetting of PSVP film from the substrate surface during the immersion. In addition, the separation distances between the bumps of ~ 14 nm can be measured. Since, the loose packing spheres were reported for the blending system of PS90k homopolymer with PS133k-b-P2VP132k [34], it is reasonable to state that the PS3.5k homopolymer in PSVP probably locates in the PS phase due to the preferred interactions (as illustrated in Fig. 1). The dispersion of low molecular weight PS3.5 k causes the stretching and relaxing of PS block in PSVP leading to increase the corona region, and thereby an increase in the interspace between the spheres. At the similar composition, the spherical diameter of current polydisperse PSVP (PS13k-b-P4VP11k, PDI 2.08) micelle is higher than that of the PS20k-b-P4VP17k (PDI 1.08) [35], which is 50 nm and 34 nm, respectively. This result clearly shows that the broad MWD of the polydisperse PSVP increases the sphere domain. The low molecular weight PSVP chains are probably located between the other high molecular weight PSVP chains leading to decrease in the packing density and increase in the PS/P4VP interfacial curvature. Therefore, the spherical size of polydisperse PSVP is higher than that of narrow MWD PSVP. The white arrow regions are unresolved region by FE-SEM (white arrow in Fig. 6a) and it may generate the macrophase separation of low molecular weight of terminated chains of PS.

Scanning electron microscopy (SEM) images (a) and the magnification of the circle area (b) of PSVP film. White scale bar is 200 nm
Owing to the possibility of the coordination with metal ion of pyridine group, we exposed the partly dried film of PSVP in iron chloride solution. Interestingly, the open pores at various pore sizes from ~ 30 to 100 nm are observed on the surface (Fig. 7a) and the cross-section images of the film (Fig. 7b, c). The pore size is approximately similar to the micelle size as presented in Fig. 6. This result proves the fact that P4VP assembles in the core, surrounded by the PS corona (Fig. 1). This result is consistent with the self-assembly structures of narrow PSVP suggested by Peneimann et al. [8, 33], indicating the possibility of organizing polydisperse BCP into ordered structure. The larger pore may be formed as the collapse of unstable structure during the evaporation. This is because PS chain in corona is “freeze” in water, the P4VP active part in the core, which can be partly swollen and coordinated with iron ion, is isolated. Consequently, the evaporation of the solvent to afford pores can damage the structure and create larger pores. In addition, the voids (Fig. 7c) can be formed due to the removal of the trapped solvent by the insufficient mass transfer during drying process [8, 32, 33].

SEM images of surfaces (a, b) and cross-sections (c, d) of self-assembled PSVP in iron chloride solution
These results showed that the polydisperse BCP could be employed for microphase separation for many applications such as nano-coating layer for self-cleaning surface, as nanoporous materials for membrane fabrication. The incorporation of PSVP with metal ion also promises for the development of the hydrid inorganic–organic nanomaterials for electronic or smart devices. The interesting stimuli-responsive properties of PSVP are suitable for the application in drug-delivery system or in other biomedical applications.
Conclusion
In this paper, the simple self-assembly from BCP to nanostructure has been reported. BCP composed of PS and P4VP has been synthesized at high polymerization temperature in the presence of TEMPO as the capping agent. The “living” fashion of PS polymerization has been observed. However, at high temperatures, the spontaneous self-initiation of St and the termination reactions have occurred that caused the low molecular weight PS homopolymer as the observed tails in chromatogram curves. Hence, the extension of PS with 4VP monomer produced block copolymer with broad breadth (PDI 2.08) and bimodal on GPC curves. The self-organization of BCP is driven by the differences in polarity between PS block and P4VP showed the loose packing of spheres on thick film fabricated from high-concentration solution in DMF/THF co-solvents. The spherical morphology is speculated to compose of P4VP block core and the PS block corona. Low molecular weight of terminated PS interacts with these corona relaxing PS chains that lead to increase the corona domains. Additionally, the short chains of PSVP distributed among the long chains of PSVP cause the chains’ packing density decrease and change the interfacial curvature. As a result, the micelle domains is large with the average diameter of 50 nm. Furthermore, this speculated structure is indirectly proved by the immersion of the film into the iron chloride solution to perform the swelling and coordinating of metal with P4VP core. The subsequent solvent evaporation generated the opened pores with the similar pore sizes to original P4VP spherical core. Our results are the experimental evidences for the self-assemblable nanoscale structure of high polydispersity of BCP by the simple polymerization and self-organization which is expected to be able to the scale-up to industrial production for many applications.
References
Schacher, F.H., Rupar, P.A., Manners, I.: Functional block copolymers: nanostructured materials with emerging Applications. Angew. Chem. Int. Ed. 51, 7898–7921 (2012)
Kosonen, H., Valkama, S., Hartikainen, J., Eerikäinen, H., Torkkeli, M., Jokela, K., Serimaa, R., Sundholm, F., ten Brinke, G., Ikkala, O.: Mesomorphic structure of poly(styrene)-block-poly(4-vinylpyridine) with oligo(ethylene oxide)sulfonic acid side chains as a model for molecularly reinforced polymer electrolyte. Macromolecules 35, 10149–10154 (2002)
Cummins, C., Borah, D., Rasappa, S., Chaudhari, A., Ghoshal, T., O’Driscoll, B.M.D., Carolan, P., Petkov, N., Holmes, J.D., Morris, M.A.: Self-assembly of polystyrene-block-poly(4-vinylpyridine) block copolymer on molecularly functionalized silicon substrates: fabrication of inorganic nanostructured etchmask for lithographic use. J. Mater. Chem. C 1, 7941–7951 (2013)
Roman, G., Martin, M., Joachim, P.S.: Block copolymer micelle nanolithography. Nanotechnology 14, 1153–1160 (2003)
Qiu, X., Yu, H., Karunakaran, M., Pradeep, N., Nunes, S.P., Peinemann, K.-V.: Selective separation of similarly sized proteins with tunable nanoporous block copolymer membranes. ACS Nano 7, 768–776 (2013)
Nunes, S.P.: Block copolymer membranes for aqueous solution applications. Macromolecules 49, 2905–2916 (2016)
Darling, S.B.: Directing the self-assembly of block copolymers. Prog. Polym. Sci. 32, 1152–1204 (2007)
Peinemann, K.-V., Abetz, V., Simon, P.F.W.: Asymmetric superstructure formed in a block copolymer via phase separation. Nat. Mater. 6, 992–996 (2007)
Nabeela, K., Thomas, R.T., Nair, J.B., Maiti, K.K., Warrier, K.G.K., Pillai, S.: TEMPO-oxidized nanocellulose fiber-directed stable aqueous suspension of plasmonic flower-like silver nanoconstructs for ultra-trace detection of analytes. ACS Appl. Mater. Interfaces. 8, 29242–29251 (2016)
Yu, C.-H., Chuang, Y.-H., Tung, S.-H.: Self-assembly of polystyrene-b-poly(4-vinylpyridine) in deoxycholic acid melt. Polymer 52, 3994–4000 (2011)
Bates, F.S., Fredrickson, G.H.: Block copolymers—designer soft materials. Phys. Today 52, 32–38 (1999)
Lynd, N.A., Meuler, A.J., Hillmyer, M.A.: Polydispersity and block copolymer self-assembly. Prog. Polym. Sci. 33, 875–893 (2008)
Matsushita, Y., Noro, A., Iinuma, M., Suzuki, J., Ohtani, H., Takano, A.: Effect of composition distribution on microphase-separated structure from diblock copolymers. Macromolecules 36, 8074–8077 (2003)
Noro, A., Cho, D., Takano, A., Matsushita, Y.: Effect of molecular weight distribution on microphase-separated structures from block copolymers. Macromolecules 38, 4371–4376 (2005)
Phillip, W.A., Dorin, R.M., Werner, J., Hoek, E.M.V., Wiesner, U., Elimelech, M.: Tuning structure and properties of graded triblock terpolymer-based mesoporous and hybrid films. Nano Lett. 11, 2892–2900 (2011)
Gromadzki, D., Lokaj, J., Šlouf, M., Štĕpánek, P.: Dilute solutions and phase behavior of polydisperse A-b-(A-co-B) diblock copolymers. Polymer 50, 2451–2459 (2009)
Widin, J.M., Kim, M., Schmitt, A.K., Han, E., Gopalan, P., Mahanthappa, M.K.: Bulk and thin film morphological behavior of broad dispersity poly(styrene-b-methyl methacrylate) diblock copolymers. Macromolecules 46, 4472–4480 (2013)
Ruzette, A.-V., Tencé-Girault, S., Leibler, L., Chauvin, F., Bertin, D., Guerret, O., Gérard, P.: Molecular disorder and mesoscopic order in polydisperse acrylic block copolymers prepared by controlled radical polymerization. Macromolecules 39, 5804–5814 (2006)
Bendejacq, D., Ponsinet, V., Joanicot, M., Loo, Y.L., Register, R.A.: Well-ordered microdomain structures in polydisperse poly(styrene)–poly(acrylic acid) diblock copolymers from controlled radical polymerization. Macromolecules 35, 6645–6649 (2002)
Zaremski, M.Y., Chen, X., Orlova, A.P., Blagodatskikh, I.V., Nikonorova, N.I.: Block copolymers of styrene and 4-vinylpyridine: synthesis and structure. Polym. Sci. Ser. B 57, 181–196 (2015)
Shoji, M., Eguchi, M., Layman, J.M., Cashion, M.P., Long, T.E., Nishide, H.: Microphase-separated poly(vinylpyridine) block copolymer prepared with a novel bifunctional initiator. Macromol. Chem. Phys. 210, 579–584 (2009)
Nguyen, D., Zhong, X.-F., Williams, C.E., Eisenberg, A.: Effect of ionic chain polydispersity on the size of spherical ionic microdomains in diblock ionomers. Macromolecules 27, 5173–5181 (1994)
Grubbs, R.B.: Nitroxide-mediated radical polymerization: limitations and versatility. Polym. Rev. 51, 104–137 (2011)
Nicolas, J., Guillaneuf, Y., Lefay, C., Bertin, D., Gigmes, D., Charleux, B.: Nitroxide-mediated polymerization. Prog. Polym. Sci. 38, 63–235 (2013)
Goto, A., Kwak, Y., Yoshikawa, C., Tsujii, Y., Sugiura, Y., Fukuda, T.: Comparative study on decomposition rate constants for some alkoxyamines. Macromolecules 35, 3520–3525 (2002)
Payne, K.A., Nesvadba, P., Debling, J., Cunningham, M.F., Hutchinson, R.A.: Nitroxide-mediated polymerization at elevated temperatures. ACS Macro. Lett. 4, 280–283 (2015)
Veregin, R.P.N., Odell, P.G., Michalak, L.M., Georges, M.K.: The pivotal role of excess nitroxide radical in living free radical polymerizations with narrow polydispersity. Macromolecules 29, 2746–2754 (1996)
Roa-Luna, M., Nabifar, A., Díaz-Barber, M.P., McManus, N.T., Vivaldo-Lima, E., Lona, L.M.F., Penlidis, A.: Another perspective on the nitroxide mediated radical polymerization (nmrp) of styrene using 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and dibenzoyl peroxide (BPO). J. Macromol. Sci. A 44, 337–349 (2007)
Baumann, M., Schmidt-Naake, G.: Controlled radical copolymerization of styrene and 4-vinylpyridine. Macromol. Chem. Phys. 201, 2751–2755 (2000)
Pan, G., Sudol, E.D., Dimonie, V.L., El-Aasser, M.S.: Thermal self-initiation of styrene in the presence of TEMPO radicals: bulk and miniemulsion. J. Polym. Sci. A 42, 4921–4932 (2004)
Silverstein, R.M., Webster, F.X., Kiemle, D.J.: Spectrometric Identification of Organic Compounds. Hoboken, NJ (2005)
Radjabian, M., Abetz, C., Fischer, B., Meyer, A., Abetz, V.: Influence of solvent on the structure of an amphiphilic block copolymer in solution and in formation of an integral asymmetric membrane. ACS Appl. Mater. Interfaces. 9, 31224–31234 (2017)
Oss-Ronen, L., Schmidt, J., Abetz, V., Radulescu, A., Cohen, Y., Talmon, Y.: Characterization of block copolymer self-assembly: from solution to nanoporous membranes. Macromolecules 45, 9631–9642 (2012)
Park, H., Kim, J.-U., Park, S.: High-throughput preparation of complex multi-scale patterns from block copolymer/homopolymer blend films. Nanoscale 4, 1362–1367 (2012)
Ghoshal, T., Chaudhari, A., Cummins, C., Shaw, M.T., Holmes, J.D., Morris, M.A.: Morphological evolution of lamellar forming polystyrene-block-poly(4-vinylpyridine) copolymers under solvent annealing. Soft Matter 12, 5429–5437 (2016)
Acknowledgements
This research is funded by the Vietnam National University Ho Chi Minh City (VNU-HCM) under Grant Number C2015-18-15.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to urisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nguyen, H.T., Tran, T.T.T. & Nguyen-Thai, N.U. Preparation of polydisperse polystyrene-block-poly(4-vinyl pyridine) synthesized by TEMPO-mediated radical polymerization and the facile nanostructure formation by self-assembly. J Nanostruct Chem 8, 61–69 (2018). https://doi.org/10.1007/s40097-018-0254-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40097-018-0254-9