
Abstract
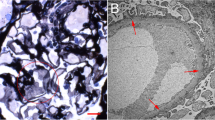
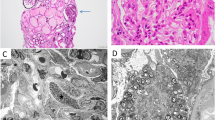
A 52-year-old woman had been found to have hematuria at her annual checkup 5 years in a row. She hoped to donate her kidney to her husband, so we performed a percutaneous kidney biopsy at our department. It was difficult for us to detect apparent abnormalities under a light microscopic examination, and she was determined to meet the eligibility criteria for living kidney transplantation. However, the sample for electron microscopy was not evaluated before kidney donation. She subsequently underwent living kidney transplantation as a donor. A 1-h biopsy revealed swelling and obvious vacuolation of the glomerular podocytes, which were characteristic of Fabry disease. Her medical history and examinations were reviewed. No findings or episodes were observed. Pre-donation electronmicroscopy revealed numerous zebra bodies in the podocytes. A definite diagnosis of heterozygous Fabry disease was made based on the GLA gene mutation despite the normal range of leukocyte α-Gal A activity. Based on the pathological deposition of GL-3, chaperone therapy was initiated to suppress the progression of organ damage. In this case, we could not confirm a diagnosis of Fabry disease despite performing a renal biopsy prior to kidney donation. Kidney donor candidates may sometimes have factors that cannot be assumed based on medical or family history. Thus, it is important to perform a renal biopsy before kidney donation when necessary, and to always conduct a detailed evaluation including electron microscopy.

Similar content being viewed by others

References
Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry’s disease. N Engl J Med. 1967;27:1163–7.
Kint JA. Fabry’s disease: alpha-galactosidase deficiency. Science. 1970;167:1268–9.
Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. https://doi.org/10.1186/1750-1172-5-30.
MacDermot KD, Holmes A, Miners AH, Rowland HS. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. 2001;38(11):750–60.
MacDermot KD, Holmes A, Miners AH, Rowland HS. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001;38(11):769–75.
Linthorst GE, Vedder AC, Aerts JMFG, Hollak CEM. Screening for Fabry disease using whole blood spots fails to identify one-third of female carriers. Clin Chim Acta. 2005;353(1–2):201–3.
Linthorst GE, Poorthuis BJHM, Hollak CEM. Letters to the editor enzyme activity for determination of presence of Fabry disease in women results in 40 % false-negative results. J Am Coll Cardiol. 2008;51(21):2082–3.
Hoshino Y, Kaga T, Abe Y. Renal biopsy findings and clinical indicators of patients with hematuria without overt proteinuria. Clin Exp Nephrol. 2015;19(5):918–24.
Pierides A, Voskarides K, Athanasiou Y, Ioannou K, Damianou L, Arsali M, et al. Clinico-pathological correlations in 127 patients in 11 large pedigrees, segregating one of three heterozygous mutations in the COL4A3 COL4A4 genes associated with famikial hematuria and significant late progression to proteinuria and chronic kidney disease from focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2009;24(9):2721–9.
Muzaale AD, Massie AB, Wan MC, Montgomery RA, McBride MA, Wainright JL, et al. Risk of end-stage renal disease following live kidney donation. JAMA. 2014;311(6):579–86.
Grünfeld JP, Le Porrier M, Droz D, Bensaude I, Hinglais N, Crosnier J. Renal transplantation in patients suffering from Fabry's disease. Kidney transplantation from an heterozygote subject to a subject without Fabry's disease. NouvPresse Med. 1975;4(29):2081–5.
Popli S, Molnar ZV, Leehey DJ, Daugirdas JT, Roth DA, Adams MB, et al. Involvement of renal allograft by Fabry's disease. Am J Nephrol. 1987;7(4):316–8.
Puliyanda DP, Wilcox WR, Bunnapradist S, Nast CC, Jordan SC. Case report Fabry disease in a renal allograft. Am J Transplant. 2003;3:1030–2.
Paull LS, Lipinski MJ, Wilson WG, Lipinski SE. Female with Fabry disease unknowingly donates affected kidney to sister: a call for pre-transplant genetic testing. JIMD Rep. 2012;4:1–4. https://doi.org/10.1007/8904_2011_108.
Taneda S, Honda K, Nakajima I, Huchinoue S, Oda H. Renal transplantation between siblings with unrecognized Fabry disease. Transplant Proc. 2013;45(1):115–8.
Nishioka R, Sofue T, Moritoki M, Nishijima Y, Nishioka S, Hara T, et al. Case report: a case of living-donor kidney transplantation from a heterozygote mother to a hemizygote son of Fabry disease diagnosed by donated allograft biopsy. Nihon NaikaGakkaiZasshi. 2015;104(4):775–80.
Odani K, Okumi M, Honda K, Ishida H, Tanabe K. Kidney transplantation from a mother with unrecognized Fabry disease to her son with low α-galactosidase A activity: a 14-year follow-up without enzyme replacement therapy. Nephrology. 2016;21(Suppl. 1):57–9.
Ortiz A, Oliveira J, Waldek S, Warnock DG, Cianciaruso B, Wanner C. Nephropathy in males and females with Fabry disease: cross-sectional description of patients before treatment with enzyme replacement therapy. Nephrol Dial Transplant. 2008;23(5):1600–7.
Biegstraaten M, Arngrímsson R, Barbey F, Boks L, Cecchi F, Deegan PB, et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: the European Fabry Working Group consensus document. Orphanet J Rare Diseases. 2015;10:36. https://doi.org/10.1186/s13023-015-0253-6.
Wanner C, Arad M, Baron R, Burlina A, Elliott PM, Feldt-Rasmussen U, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018;124(3):189–203.
Tøndel C, Bostad L, Larsen KK, Hirth A, Vikse BE, Houge G, et al. Agalsidase benefits renal histology in young patients with Fabry disease. J Am Soc Nephrol. 2013;24:137–48.
Tanaka A, Takeda T, Hoshina T, Fukai K, Yamano T. Enzyme replacement therapy in a patient with Fabry disease and the development of IgE antibodies against agalsidase beta but not agalsidase alpha. J Inherit Metab Dis. 2010;33(Suppl 3):249–52.
Hughes DA, Nicholls K, Shankar SP, Sunder-Plassmann G, Koeller D, Nedd K. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet. 2017;54:288–96.
Fan JQ, Ishii S, Asano N, Suzuki Y. Accelelated transport and maturation of lysosomal α–galactosidase A in Fabrylymphoblasts by an enzyme inhibitor. Nat Med. 1999;5(1):112–5.
Yam GH, Zuber C, Roth J. A synthetic chaperone corrects the trafficking defect and disease phenotype in a protein misfolding disorder. FASEB J. 2005;19(1):12–8.
Asano N, Kizu H, Ikeda K, Yasuda K, Ishii S, Martin OR, et al. In vitro inhibition and intracellular enhancement of lysosomal α-galactosidasea activity in fabrylymphoblasts by 1-deoxygalactonojirimycin and its derivatives. Eur J Biochem. 2000;267(13):4179–86.
Kobayashi M, Ohashi T, Kaneshiro E, Higuchi T, Ida H. Mutation spectrum of α-galactosidase gene in Japanese patients with Fabry disease. J Hum Genet. 2019;64(7):695–9.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Minami, M., Mizuma, E., Nakahara, M. et al. A case of latent heterozygous Fabry disease in a female living kidney donor candidate. CEN Case Rep 10, 30–34 (2021). https://doi.org/10.1007/s13730-020-00510-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13730-020-00510-9