
Abstract
Two new sesquiterpenoids, artemlavanins A (1) and B (3), together with fifteen known compounds (2 and 4−17) were isolated from the EtOH extract of Artemisia lavandulaefolia. The structures of new compounds were elucidated by extensive spectroscopic analyses (HRESIMS, 1D and 2D NMR) and ECD calculations. Compound 1 was a sesquiterpenoid lactone possessing a rearranged eudesmane skeleton; compounds 2–5, 6–8, 9 and 10–12 belonged to the eudesmane, guaiane, oppositane and farnesane sesquiterpenoids, respectively; compounds 13–17 were the phenyl derivatives with a 4-hydroxyacetophenone moiety. Twelve compounds (1–3, 5–7, 10–12, 14, 15 and 17) displayed cytotoxicity against hepatic stellate cell line LX2 (HSC-LX2) with IC50 values ranging from 35.1 to 370.3 μM. Compounds 2, 7, 10–12 and 17 exhibited the stronger cytotoxicity than silybin (IC50, 169.6 μM) with IC50 values of 82.1, 35.1, 95.0, 83.8, 81.6 and 90.1 μM. Compound 7 as the most active one showed significant inhibition on the deposition of human collagen type I (Col I), human hyaluronic acid (HA) and human laminin (HL) with IC50 values of 10.7, 24.5 and 13.3 μM.
Graphic Abstract

Similar content being viewed by others

1 Introduction
Hepatic fibrosis is characterized by the abnormal accumulation of extracellular matrix (ECM), which results from the liver diseases such as viral hepatitis and alcoholic or nonalcoholic steatohepatitis. The global prevalence of alcohol-use disorders, diabetes and hepatitis B virus were approximately 75 million, 422 million and 257 million people [1], thus increasing the risks for liver cirrhosis or cancer and leading to a significant burden for liver diseases and medical costs. Although over 20 candidate molecules for hepatic fibrosis involving the inhibition of inflammatory response, ECM production and HSCs activation have achieved positive progress in recent years, most of which are being assessed in clinical trials and none of them have been commercialized as antifibrotic drugs [2,3,4,5]. Therefore, the discovery of antifibrotic agents with high efficacy and low side effect are urgently required. Recently, a review of 60 natural products with antihepatic fibrosis activity has been reported, indicating alkaloids, polysaccharides, flavonoids, polypeptides, terpenoids and polyphenols as the active ingredients [6].
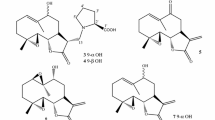
Artemisia is a large genus with over 300 species and belongs to the family Asteraceae, many of which were found to be effective for the liver diseases, such as A. capillaris [7, 8], A. scoparia [9], A. sacrorum [10], A. absinthium [11], A. vulgaris [12], A. iwayomogi [13] and A. campestris [14]. A. lavandulaefolia as a substitute of the traditional Chinese medicine A. argyi was used for the treatment of hemorrhage, menstruation-related symptoms, diarrhea, skin diseases, stomatitis, bronchitis and liver ailments in diverse cultures [15,16,17]. Phytochemical research of A. lavandulaefolia has led to reports on monoterpenoids [18, 19], sesquiterpenoids [19,20,21], triterpenoids [18, 22], steroids [18, 22], phenyl derivatives [18, 19], phenylpropanoids [18, 19, 23] and flavonoids [22, 24], as well as essential oils [17]. The present investigation on the EtOH extract of A. lavandulaefolia resulted artemlavanins A (1) and B (3) together with fifteen known compounds (2 and 4–17), which were evaluated on the hepatic stellate cell line LX2 (HSC-LX2). Herein, the isolation, structural elucidation and cytotoxicity of these compounds were reported (Fig. 1).

Chemical structures of compounds 1–17
2 Results and Discussion
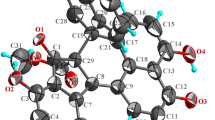
Artemlavanin A (1) was assigned a molecular formula of C15H18O2 by the (+)-HRESIMS ion at m/z 231.1383 [M+H]+ (calcd for C15H19O2, 231.1380), implying seven degrees of unsaturation. Its IR spectrum indicated the absorption bands for carbonyl (1752 cm−1) and aromatic ring (1488 cm−1 and 1454 cm−1) functionalities. The 1H NMR data (Table 1) displayed the presence of five methines at δH 6.97 (H-2 and H-3), 4.92 (H-8), 2.93 (H-11) and 2.63 (H-7), two methylenes at δH 3.22 (H-9a), 2.87 (H-9b), 2.73 (H-6a) and 2.38 (H-6b), and three methyl protons at δH 2.30 (H-15), 2.29 (H-14) and 1.28 (H-13). The 13C NMR (DEPT) data (Table 1) showed 15 signals for a carbonyl at δC 179.3, an aromatic ring at δC 134.8, 133.4, 132.1, 132.0 and 128.0 (× 2), three methines at δC 77.1, 38.3 and 38.0, two methylenes at δC 29.9 and 23.6, and three methyls at δC 19.4, 19.3 and 10.6. Two methyls were attached to C-1 and C-4 based on the HMBC correlations (Fig. 2) from H3-14 (δH 2.29) to C-2 (δC 128.0) and C-10 (δC 132.1) and from H3-15 (δH 2.30) to C-3 (δC 128.0) and C-5 (δC 134.8). The 1H–1H COSY cross-peaks of H-6/H-7/H-8/H-9 and H-7/H-11/H-13 together with the HMBC correlations from H2-6 (δH 2.73 and 2.38) to C-4 (δC 132.0) and C-10 (δC 132.1) and from H2-9 (δH 3.22 and 2.87) to C-1 (δH 133.4) and C-5 (δC 134.8) verified that the aromatic ring was fused with a hexatomic ring via C-10 and C-5 in compound 1. In addition, the lactone ring was established by the HMBC correlations of H-7 (δH 2.63), H-11 (δH 2.93) and H-13 (δH 1.28) with C-12 (δC 179.3). The planar structure of compound 1 was therefore determined to be consistent with α-(1,4-dimethyl-7-hydroxy-5,6,7,8-tetrahydro-6-naphthyl)-propionic acid lactone as previously reported [25, 26], but the stereochemistry of this compound remained unclear. The coupling constant (J = 6.4 Hz) between H-7 and H-8 specified a cis-relationship [27, 28]. Furthermore, the ROESY correlation of H-8 with H-11 indicated that they were on the same side. Comparison of the experimental and calculated ECD spectra (Fig. 3) revealed that compound 1 was elucidated as 7α,11αH-14(10 → 1)abeo-eudesma-1,3,5(10)-trien-12,8β-olide.

Key HMBC (red), 1H–1H COSY (blue) and ROESY (black) correlations of compounds 1 and 3

Experimental and calculated ECD spectra for compound 1 in MeOH (σ = 0.25, shift = ‒3 nm)
Artemlavanin B (3) possessed a molecular formula of C16H26O3 by the (+)-HRESIMS ion at m/z 289.1754 [M+Na]+ (calcd for C16H26O3Na, 289.1774), indicating four indices of hydrogen deficiency. The IR absorptions at 3455, 1713 and 1625 cm‒1 suggested the existence of hydroxy, carbonyl and double bond groups. The 1H NMR data (Table 1) exhibited characteristic signals for a methoxy at δH 3.70 (MeO-15) and three methyls at δH 1.10 (H3-14) and 1.19 (H3-12 and H3-13). The 13C NMR (DEPT) data (Table 1) showed 16 carbon resonances including a carbonyl at δC 171.0, a tetrasubstituted double bond at δC 150.5 and 124.3, two quaternary carbons at δC 72.7 and 35.6, a methine at δC 50.9, six methylenes at δC 41.9, 39.4, 28.9, 28.1, 23.0 and 18.6, three methyls at δC 27.1, 27.0 and 24.9, and a methoxy at δC 51.5. Its 1H NMR and 13C NMR spectra were similar with those of (+)-γ-eudesmol [29, 30], except that the methyl at C-4 in (+)-γ-eudesmol was replaced by a methoxycarbonyl group in compound 3. The location of methoxycarbonyl was verified at C-4 by the HMBC correlations (Fig. 2) from MeO-15 (δC 3.70) and H2-3 (δH 2.31 and 2.16) to C-15 (δC 171.0). The relative configuration of compound 3 was identical with (+)-γ-eudesmol based on the ROESY correlation of H-8β with H-6β, H3-14 and H3-12(13). Combined with the positive optical rotation, compound 3 was established as methyl 11-hydroxyeudesm-4-en-7αH-15-oate.
The known compounds were identified as 11α,13-dihydroyomogin (2) [31], 1β,6α-dihydroxyeudesm-4(15)-ene (4) [32], 4-epi-vulgarin (5) [33], ludartin (6) [34], dehydroleucodin (7) [35], matricarin (8) [36], (7R*)-5-epi-opposit-4(15)-ene-1β,7-diol (9) [37], cis-hydroxydavanone (10) [38], (6S,7S,l0R)-2-hydroperoxy-2,6,10-trimethyl-7,10-epoxydodeca-3,11-dien-5-one (11) [39], 11-hydroxy-8-oxo-9,10-dehydro-10,11-dihydronerolidol (12) [40], 4-hydroxyacetophenone (13) [41], espeletone (14) [42], (S)-3-(1-hydroxy-3-methylbutyl)-4-methoxyacetophenone (15) [43], (R)-1-[5-(1-hydroxyethyl)-2-methoxyphenyl]-3-methyl-1-butanone (16) [44], and 6-acetyl-3,7-dihydroxy-2,2-dimethyl-chromene (17) [45] by comparison of their spectroscopic data with those previously reported.
Compounds 1–17 were evaluated for their cytotoxicity against HSC-LX2. Twelve compounds showed activities with inhibitory ratios higher than 50% at 400 μM (Table 2) and IC50 values ranging from 35.1 to 370.3 μM (Table 3). Compounds 2, 7, 10–12 and 17 displayed potent cytotoxicity with IC50 values of 82.1, 35.1, 95.0, 83.8, 81.6 and 90.1 μM, indicating more potent cytotoxicity than the positive control silybin (IC50, 169.6 μM); compound 14 exhibited moderate cytotoxicity with the IC50 value of 151.7 μM; compounds 1, 3, 5, 6 and 15 demonstrated less effective than silybin with the IC50 values in the range of 204.9–370.3 μM. In order to better comprehend the cytotoxic mechanism, the effects of active compounds on the deposition of Col I, HA and HL in HSC-LX2 were assayed by ELISA analysis. Notably, compound 7 as the most active one against HSC-LX2 showed obvious inhibitory effects on the secretion of Col I, HA and HL with IC50 values of 10.7, 24.5 and 13.3 μM, which were significantly superior to silybin with IC50 values of 54.3, 228.4 and 89.9 μM (Table 4).
3 Conclusion
In conclusion, the phytochemical investigations on A. lavandulaefolia revealed two new sesquiterpenoids and fifteen known compounds, of which artemlavanin A (1) was a rearranged eudesmanolide with an aromatic ring and artemlavanin B (3) was a methyl ester derivative of (+)-γ-eudesmol. In addition, the chemical constituents isolated from A. lavandulaefolia were composed of eudesmane (2, 4 and 5), guaiane (6–8), oppositane (9) and farnesane (10–12) sesquiterpenoids, together with the phenyl derivatives with a 4-hydroxyacetophenone moiety (13–17). Compounds 2, 7, 10–12 and 17 showed obvious cytotoxicity against HSC-LX2 with IC50 values of 82.1, 35.1, 95.0, 83.8, 81.6 and 90.1 μM. Interestingly, compound 7 possessed inhibitory activity on not only HSC-LX2 but also the accumulation of Col I, HA and HL with IC50 values of 10.7, 24.5 and 13.3 μM. These results provided the evidence for the antihepatic fibrosis potential of A. lavandulaefolia.
4 Experimental Section
4.1 General Experimental Procedures
Optical rotations were measured on an Autopol VI automatic polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA). UV spectra were recorded on a Shimadzu UV2401PC spectrophotometer (Shimadzu, Kyoto, Japan). IR spectra were determined on a NICOLET iS10 (Thermo scientific, Madison, USA) spectrometers using ATR ITX-DIAMOND mode. HRESIMS data were acquired on an LCMS-IT-TOF mass spectrometer (Shimadzu, Kyoto, Japan). 1D and 2D NMR data were obtained on Avance III-600 spectrometers (Bruker, Bremerhaven, Germany). ECD spectra were collected on an Applied Photophysics Chirascan spectrometer (Applied Photophysics, Surrey, UK). Silica gel (200–300 mesh, Qingdao Makall group Co., Ltd., Qingdao, China) and Sephadex LH-20 (GE Healthcare Amersham Biosciences, Uppsala, Sweden) were used for column chromatography. MPLC separations was conducted on a Dr-Flash II apparatus (Lisure Science Co., Ltd., Suzhou, China) using MCI gel CHP 20P column (75–150 μm, Mitsubishi Chemical Corporation, Tokyo, Japan). Semipreparative HPLC separations were performed on a Shimadzu LC-CBM-20 system (Shimadzu, Kyoto, Japan) with an Agilent Eclipse XDB-C18 column (5 μm, 9.4 × 250 mm, Agilent Technologies, Santa Clara, USA) unless otherwise specified. TLC analyses were carried out using silica gel GF254 plates (Jiangyou Silica Gel Development Co., Ltd., Yantai, China), and compounds were detected by heating after spraying with 10% H2SO4 in EtOH. Hepatic stellate cell line LX2 (Jining Industrial Co., Ltd., Shanghai, China) were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (Gibco BRL, NY, USA). The cytotoxicity was assessed by MTT (BioFROXX, Guangzhou, China) method. The ELISA kits (Cusabio Biotech Co., Ltd., Wuhan, China) was used for the detection of the human collagen type I (Col I) and human hyaluronic acid (HA), and the ELISA kit (Boster Biological Technology Co., Ltd., California, USA) was applied to determine the human laminin (HL). The absorbance was measured on a microplate reader (Bio-Rad, Hercules, CA, USA).
4.2 Plant Materials
The aerial parts of Artemisia lavandulaefolia DC. were collected in October, 2018, from Dali, Yunnan Province, China, and authenticated by Prof. Dr. Li-Gong Lei (Key Laboratory of Biodiversity and Biogeography, Kunming Institute of Botany, Chinese Academy of Sciences). A voucher specimen (No. 20181104) was deposited at the Laboratory of Anti-virus and Natural Medicinal Chemistry, Kunming Institute of Botany, Chinese Academy of Sciences.
4.3 Extraction and Isolation
The air-dried aerial parts of Artemisia lavandulaefolia (20 kg) were powdered and extracted with 95% EtOH for two times (2 × 100 L × 4 day) at room temperature. The combined extract was concentrated under reduced pressure to yield the residue, which was suspended in H2O and subsequently partitioned with EtOAc. The EtOAc fraction (814 g) was subjected to silica gel column chromatography (CC) using EtOAc–petroleum ether (PE) gradient (0:100, 5:95, 10:90, 20:80, 40:60 and 100:0, v/v) to give fractions A–F (122, 75, 87, 34, 91 and 363 g).
Fraction C (87 g) was separated by silica gel CC with Me2CO–PE gradient (5:95, 10:90 and 20:80) as the eluent to give fractions C1–C3 (36, 35 and 10 g). Fraction C1 was submitted on MPLC eluted with H2O–MeOH gradient (10:90 and 0:100) to give two fractions (Frs. C1.1 and C1.2). Compounds 1 (12 mg, tR = 31 min) and 14 (37 mg, tR = 26 min) was obtained from fraction C1.1 by repeated silica gel CC (EtOAc–PE, 10:90) and semipreparative HPLC (H2O–MeCN, 56:44). Fraction C2 was purified by MPLC using H2O–MeOH gradient (50:50, 30:70, 10:90 and 0:100) to give four fractions (Frs. C2.1–C2.4). Fraction C2.2 was loaded on a silica gel CC and eluted with EtOAc–PE gradient (5:95, 10:90 and 20:80) to yield three fractions (Frs. C2.2.1–C2.2.3). Fraction C2.2.1 was separated by silica gel CC (EtOAc–CHCl3, 2:98) to afford compounds 10 (296 mg) and 11 (753 mg), and subsequently purified by semipreparative HPLC (H2O–MeCN, 67:33) to give compounds 4 (85 mg, tR = 26 min) and 12 (10 mg, tR = 29 min). Fraction C2.3 was chromatographed on a silica gel CC and eluted with Me2CO–PE (5:95 and 10:90) to provide three fractions (Frs. C2.3.1–C2.3.3). Fraction C2.3.1 was carried out on silica gel CC and eluted with PE–CHCl3 gradient (1:99 and 2:98) to produce two fractions (Frs. C2.3.1.1 and C2.3.1.2). Compounds 15 (8 mg, tR = 50 min) and 16 (9 mg, tR = 54 min) were obtained from fraction C2.3.1.1 by semipreparative HPLC (H2O–MeCN, 73:27) using Opti-Chiral® C1-5 (RP) column (5 μm, 10 × 250 mm, ColumnTek, LLC, 200 Innovation Blvd., Suite 258A, State College, PA 16803 USA). Fraction C2.3.1.2 was chromatographed on a Sephadex LH-20 CC (MeOH–CHCl3, 50:50) to provide compound 13 (10 mg) and subsequently purified by semipreparative HPLC (H2O–MeCN, 53:47) to yield compound 17 (189 mg, tR = 14 min). Compounds 3 (25 mg), 6 (57 mg) and 7 (16 mg) were obtained from fraction C2.3.2 by repeated silica gel CC (EtOAc–PE, 3:97 and 10:90) and Sephadex LH-20 CC (MeOH–CHCl3, 50:50). Fraction C3 was separated by MPLC using H2O–MeOH gradient (30:70, 10:90 and 0:100) to generate three fractions (Frs. C3.1–C3.3). Compound 9 (10 mg) was isolated from fraction C3.1 by repeated silica gel CC (EtOAc–PE, 10:90) and Sephadex LH-20 CC (MeOH–CHCl3, 50:50).
Fraction D (34 g) was subjected to MPLC and eluted with H2O–MeOH gradient (40:60, 60:40, 20:80, and 0:100) to yield fractions D1–D4 (0.9, 6.8, 9.0 and 11 g). Fraction D3 was separated by silica gel CC eluted with EtOAc–PE (20:80) and semipreparative HPLC (H2O–MeCN, 70:30) to give compounds 5 (57 mg, tR = 18 min) and 8 (31 mg, tR = 34 min). Fraction D4 was chromatographed on a silica gel CC using EtOAc–CHCl3 as eluent (5:95) to afford two fractions (Frs. D4.1 and D4.2). Compound 2 (46 mg, tR = 18 min) was obtained from fraction D4.1 by semipreparative HPLC (H2O–MeCN, 70:30).
Artemlavanin A (1): colorless oil; [α]22D + 15.2 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 220 (3.91) nm; ECD (MeOH) λmax (Δε) 205 (+ 43.2) nm; IR vmax 1752, 1488, 1454, 1189 cm−1; 1H NMR and 13C NMR data see Table 1; (+)-HRESIMS m/z 231.1383 [M+H]+ (calcd for C15H19O2, 231.1380).
Artemlavanin B (3): colorless oil; [α]20D + 68.9 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 225 (3.77) nm; IR vmax 3455, 1713, 1625, 1241, 1218 cm−1; 1H NMR and 13C NMR data see Table 1; (+)-HRESIMS m/z 289.1754 [M+Na]+ (calcd for C16H26O3Na, 289.1774).
4.4 Cytotoxicity Assay
The cytotoxicity of compounds was determined using the MTT method. Briefly, HSC-LX2 were cultured in 96-well plates at a density of 1 × 104 cells/well in RPMI 1640 medium supplemented with 10% fetal bovine serum at 37 ℃ with 5% CO2. After overnight incubation, cells were treated with compounds at different concentrations for 48 h. Subsequently, the culture medium was exchanged by 100 μL of MTT reagent (1 mg/mL). After co-incubation for 4 h at 37 °C, the solution was removed and 100 μL of DMSO was added to dissolve the formazan crystals. The absorbance was recorded on a microplate reader at 490 nm. All the experiments were carried out in triplicate. The inhibitory ratios were calculated as [A(control)−A(sample)]/A(control) × 100%, and IC50 values were calculated using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA).
4.5 Col I, HA and HL Secretion Assay
HSC-LX2 were seeded at 8 × 104 cells/well in 24-well plates overnight and then treated with the compounds at different concentrations. After 72 h incubation, Col I, HA and HL levels of culture media were collected and determined using the commercial ELISA kits according to the manufacturer's instructions.
References
S.K. Asrani, H. Devarbhavi, J. Eaton, P.S. Kamath, J. Hepatol. 70, 151–171 (2019)
L. Chen, D.A. Brenner, T. Kisseleva, Hepatol. Commun. 3, 180–192 (2019)
D. Schuppan, M. Ashfaq-Khan, A.T. Yang, Y.O. Kim, Matrix Biol. 68–69, 435–451 (2018)
C. Trautwein, S.L. Friedman, D. Schuppan, M. Pinzani, J. Hepatol. 62, S15–S24 (2015)
W.D. Zhang, R.F. Wang, H.M. Wu, H. Yang, G.C. Wang, Acta Pharm. Sin. 53, 667–675 (2018)
L. Shan, Z.N. Liu, L.L. Ci, C. Shuai, X.W. Lv, J. Li, Int. Immunopharmacol. 75, 105765 (2019)
J.M. Han, H.G. Kim, M.K. Choi, J.S. Lee, J.S. Lee, J.H. Wang, H.J. Park, S.W. Son, S.Y. Hwang, C.G. Son, Exp. Toxicol. Pathol. 65, 837–844 (2013)
Y. Zhao, C.A. Geng, C.L. Sun, Y.B. Ma, X.Y. Huang, T.W. Cao, K. He, H. Wang, X.M. Zhang, J.J. Chen, Fitoterapia 95, 187–193 (2014)
Z.Q. Wang, X.H. Zhang, Y.M. Yu, R.C. Tipton, I. Raskin, D. Ribnicky, W. Johnson, W.T. Cefalu, Metabolism 62, 1239–1249 (2013)
H.D. Yuan, G.Z. Jin, G.C. Piao, J. Ethnopharmacol. 127, 528–533 (2010)
N. Amat, H. Upur, B. Blažeković, J. Ethnopharmacol. 131, 478–484 (2010)
M.L. Corrêa-Ferreira, M.H. Verdan, F.A. dos Reis Lívero, L.F. Galuppo, J.E.Q. Telles, M.É.A. Stefanello, A. Acco, C.L. de Oliveira Petkowicz, Phytomedicine 24, 68–76 (2017)
J.M. Han, H.G. Kim, M.K. Choi, J.S. Lee, H.J. Park, J.H. Wang, J.S. Lee, S.W. Son, S.Y. Hwang, C.G. Son, Food Chem. Toxicol. 50, 3505–3513 (2012)
M. Sefi, H. Bouaziz, N. Soudani, T. Boudawara, N. Zeghal, Pestic. Biochem. Physiol. 101, 71–79 (2011)
S. Wang, J. Li, J. Sun, K.W. Zeng, J.R. Cui, Y. Jiang, P.F. Tu, Fitoterapia 85, 169–175 (2013)
J.D. Cha, Y.H. Kim, J.Y. Kim, Food Sci. Biotechnol. 19, 185–191 (2010)
J.D. Cha, M.R. Jeong, H.J. Choi, S.I. Jeong, S.E. Moon, S.I. Yun, Y.H. Kim, B.S. Kil, Y.H. Song, Planta Med. 71, 575–577 (2005)
L. Ma, B.H. Wei, L. Hu, J. Guangzhou Univ. Tradit. Chin. Med. 29, 450–453 (2012)
L.F. Ding, G.M. Yang, Y.D. Guo, L.D. Song, X.D. Wu, Chin. Tradit. Herb. Drugs 49, 1995–1999 (2018)
L.F. Ding, L.Y. Peng, H.F. Zhou, L.D. Song, X.D. Wu, Q.S. Zhao, Tetrahedron Lett. 61, 151872 (2020)
J.L. Lv, Z. Li, L.M. Guo, L.B. Zhang, Chem. Biodiversity 15, e1700548 (2018)
X.Q. Wang, C.J. Zhou, N. Zhang, G. Wu, M.H. Li, J. Chin. Med. Mater. 34, 234–236 (2011)
J.Y. Xie, G.L. Zhang, Z.G. Yu, Chin. J. Pestic. Sci. 21, 383–388 (2019)
L. Ma, H.L. Zhou, Y.M. Gong, L. Hu, Q. Duan, Tradit. Chin. Drug Res. Clin. Pharmacol. 23, 555–578 (2012)
W.G. Dauben, P.D. Hance, W.K. Hayes, J. Am. Chem. Soc. 77, 4609–4612 (1955)
M. Sumi, W.G. Dauben, W.K. Hayes, J. Am. Chem. Soc. 80, 5704–5705 (1958)
Y. Peng, J. Xiao, X.B. Xu, S.M. Duan, L. Ren, Y.L. Shao, Y.W. Wang, Org. Lett. 18, 5170–5173 (2016)
Y.S. Cai, Z. Wu, X.Q. Zheng, C. Wang, J.R. Wang, X.X. Zhang, G.F. Qiu, K.K. Zhu, S.G. Cao, J.Q. Yu, Org. Chem. Front. 7, 303–309 (2020)
T.A. Van Beek, R. Kleis, M.A. Posthumus, A. Van Veldhuizen, Phytochemistry 28, 1909–1911 (1989)
X.L. Gao, Z.M. Xiong, G. Zhou, Y.L. Li, Synthesis 1, 37–39 (2001)
J. Jakupovic, A. Schuster, F. Bohlmann, M.O. Dillon, Phytochemistry 27, 1113–1120 (1988)
T. Ohmoto, K. Ikeda, S. Nomura, M. Shimizu, S. Saito, Chem. Pharm. Bull. 35, 2272–2279 (1987)
M.A. Metwally, J. Jakupovic, M.I. Youns, F. Bohlmann, Phytochemistry 24, 1103–1104 (1985)
V.E. Sosa, J.C. Oberti, R.R. Gil, E.A. Rúveda, V.L. Goedken, A.B. Gutiérrez, W. Herz, Phytochemistry 28, 1925–1929 (1989)
H.F. Wong, G.D. Brown, J. Nat. Prod. 65, 481–486 (2002)
A.A. Ahmed, T. Gáti, T.A. Hussein, A.T. Ali, O.A. Tzakou, M.A. Couladis, T.J. Mabry, G. Tóth, Tetrahedron 59, 3729–3735 (2003)
L.R. Soares, Quim. Nova. 35, 323–326 (2012)
K.K. Wan, C.D. Evans-Klock, B.C. Fielder, D.A. Vosburg, Synthesis 45, 1541–1545 (2013)
G. Appendino, P. Gariboldi, G.M. Nano, P. Tétényi, Phytochemistry 23, 2545–2551 (1984)
F. Bohlmann, N. Ates, J. Jakupovic, R.M. King, H. Robinson, Phytochemistry 21, 2691–2697 (1982)
H.Y. Ding, H.C. Lin, C.M. Teng, Y.C. Wu, J. Chin. Chem. Soc. 47, 381–388 (2000)
X.N. Fan, S. Lin, C.G. Zhu, Y. Liu, J.F. Hu, X.G. Chen, W.J. Wang, N.H. Chen, J.G. Shi, China J. Chin. Mater. Med. 36, 48–56 (2011)
M.D.R. Cuenca, S. Borkosky, C.A.N. Catalan, V.L. Goedken, J.G. Diáz, W. Herz, Phytochemistry 32, 1509–1513 (1993)
F. Bohlmann, N. Rao, Chem. Ber. 106, 3035–3038 (1973)
A. Zana, Z. Hajdú, N. Jedlinszki, I. Máthé, G. Dombi, J. Hohmann, Tetrahedron 71, 4817–4820 (2015)
Acknowledgements
This work was supported by the Yunnan Wanren Project (YNWR-KJLJ-2019-002), the Program of Yunling Scholarship, the Reserve Talents of Young and Middle-aged Academic and Technical Leaders in Yunnan Province, and the Youth Innovation Promotion Association, CAS (2013252).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shen, C., Huang, XY., Geng, CA. et al. Artemlavanins A and B from Artemisia lavandulaefolia and Their Cytotoxicity Against Hepatic Stellate Cell Line LX2. Nat. Prod. Bioprospect. 10, 243–250 (2020). https://doi.org/10.1007/s13659-020-00254-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-020-00254-0