
Abstract
Four hitherto unknown aristolane-type sesquiterpenes, including one novel 8,9-secoaristolane, namely secoaristolenedioic acid (1), two aristolone derivatives, namely 1α,2β-dihydroxyaristolone (2), 9-epidebilon (3), and one rare aristolane-chalcone hybrid, namely 3′-hydroxynardoaristolone A (4) were isolated from the ethanol extract of the roots and rhizomes of Nardostachys chinensis. Their structures were elucidated on the basis of extensive spectroscopic analysis. In addition, the structure of aristolanhydride, recently isolated from the same species, was corrected by reanalysis of the published NMR data.
Graphical Abstract

Similar content being viewed by others

1 Introduction
Nardostachys chinensis is a perennial herb belonging to the genus Nardostachys (Valerianaceae family), and mainly distributed in the Himalayan mountain areas [1]. The roots and rhizomes of N. chinensis, known as “Gansong”, is a traditional Chinese medicine, which has been used as analgesic and sedative agents [2, 3]. Previous phytochemical studies led to the isolation of a series of aristolane, nardosinane, and guaiane sesquiterpenoids [3]. Aristolane is a rare class of natural sesquiterpenoid featuring a gem-dimethylcyclopropane moiety, and mainly isolated from the genus Nardostachys (terrestrial plants), Laurencia (marine organisms), Axinyssa (sponges), and Russula (fungi) [4].
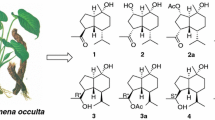
As part of a BioBioPha [http://www.chemlib.cn] objective to assemble a large-scale natural product library valuable in the discovery of new drug leads from nature, the phytochemical investigation on the roots and rhizomes of N. chinensis resulted in the isolation of four new aristolane-type sesquiterpenoids, including one novel 8,9-secoaristolane, namely secoaristolenedioic acid (1), two aristolone derivatives, namely 1α,2β-dihydroxyaristolone (2), 9-epidebilon (3), and one rare aristolane-chalcone hybrid, namely 3′-hydroxynardoaristolone A (4) (Fig. 1), together with eight known aristolanes (aristolone [5], debilon [5], kanshone H [6], anthracophyllone [7], axinysone A [8], axinysone B [8], 1(10)-aristolen-2-one [9], and aristola-1(10),8-dien-2-one [10]), six know nardosinanes (nardosinone [11], nardosinonediol [11], desoxo-narchinol A [11], kanshone A [12], kanshone B [12], and narchinol B [13]), three known guaianes (isonardoperoxide [14], 10-epiteuclatriol [15], and aromadendrane-4β,10α-diol [16]), and three known eudesmanes (eudesma-3,11-dien-2-one [17], 7-epi-5-eudesmene-1β,11-diol [18], and nardoeudesmol A [6]). Herein, we describe the isolation and structure elucidation of these new compounds.

Structures of compounds 1–4
2 Results and Discussion
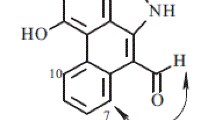
Compound 1 was obtained as a white amorphous powder, and its molecular formula was determined to be C15H22O4 based on HRESIMS (pos.): m/z 289.1410 [M + Na]+ (calcd. for C15H22O4Na, 289.1410) with five degrees of unsaturation. The IR spectrum showed characteristic absorption bands for carbonyl functionalities (1687 cm−1). The 1H NMR spectrum (Table 1) in pyridine-d5 showed the signals for three tertiary methyls [δH 1.32, 1.81, and 1.91 (each 3H, s)], one secondary methyl [δH 0.95 (3H, d, J = 6.9 Hz)], two mutually coupled methine protons [δH 1.65 (1H, d, J = 9.8 Hz) and 1.92 (1H, d, J = 9.8 Hz)], and one olefinic proton [δH 6.96 (1H, t, J = 3.7 Hz)]. The 13C and DEPT spectra (Table 1) displayed a total of 15 carbon resonances, including four methyls, two methylenes, four methines (one olefinic at δC 135.1), and five quaternary carbons (two carbonyls at δC 170.8 and 175.3, one olefinic at δC 141.7). The cross-peaks of H-1/H-2, H-2/H-3, H-3/H-4, and H-6/H-7 in the 1H-1H COSY spectrum disclosed the following connectivities: C-1–C-2–C-3–C-4 and C-6–C-7. A gem-dimethylcyclopropane unit was deduced from the HMBC correlations (Fig. 2) from H3-12 (δH 1.91) to C-6 (δC 45.3), C-7 (δC 30.8), C-11 (δC 29.1), and C-13 (δC 32.4), from H3-13 (δH 1.32) to C-6, C-7, C-11, and C-12 (δC 16.9). Together with the HMBCs from H3-14 (δH 1.81) to C-4 (δC 37.0), C-5 (δC 40.8), and C-6, and from H3-15 (δH 0.95) to C-3 (δC 25.8), C-4, and C-5, an aristolane-type sesquiterpene skeleton was speculated for compound 1 [5]. A double bond located at C-1(10) was confirmed by the HMBCs from H3-14 (δH 1.81) to C-5 and C-10 (δC 141.7), and from H-1 (δH 6.96) to C-3 (δC 25.8) and C-5. Then, the presence of two newly formed carbonyl groups at C-8 and C-9 was determined by the HMBC correlations of H-1 (δH 6.96) to C-9 (δC 170.8) and H-6 (δH 1.65) to C-8 (δC 175.3), which indicated an oxidative cleavage had happened at C-8–C-9 in the ring B. The stereochemistry was defined by observation of the ROESY correlations between H-4/H-6 and H-6/H-7 (Fig. 3), consistent with that of the co-occurring aristolanes. Therefore, the structure of 1 was established as 8,9-seco-1(10)-aristolene-8,9-dioic acid and given the trivial name secoaristolenedioic acid. To our knowledge, this compound represents the first example of a secoaristolane.

Key HMBC
 correlations of 1–4
correlations of 1–4

Key ROESY
 correlations of 1–3
correlations of 1–3
It was worth mentioning that the NMR data of compound 1 were almost identical to those of aristolanhydride (an unprecedented 8,9-cleavage anhydride derivative of aristolane-type sesquiterpene), recently isolated by Chen and co-workers from the same species [19]. Since the NMR spectra of aristolanhydride were initially measured in methanol-d4, we then re-tested the 1H and 13C-NMR of 1 using methanol-d4 and found that their NMR spectra were exactly the same. The structural difference between them was whether the dicarboxylic acid dehydrated to form a cyclic anhydride or not. Then we carefully examined the HRESIMS spectra of 1 and aristolanhydride. For compound 1, a positive-ion HRESIMS exhibited an [M + Na]+ ion peak at m/z 289.1410, suggesting a molecular formula of C15H22O4 (calcd. for C15H22O4Na, 289.1410), while for aristolanhydride, a negative mode HRESIMS gave an [M − H]− ion peak at m/z 265.1441, but Chen and co-workers mistakenly believed that it was an [M + OH]− ion peak, and then led to a wrong dehydrated molecular formula C15H20O3. Furthermore, HPLC and TLC analysis were performed on an Agilent 1200 series HPLC system and silica gel F254 TLC plates, respectively, and a broad chromatogram peak and a long trailing spot (chloroform/methanol = 10/1) were observed, which also hinted the existence of carboxyl group not an anhydride. Above all, the structure of aristolanhydride should be revised as secoaristolenedioic acid (1) (Fig. 4).

Structural revision of aristolanhydride
Compound 2 was isolated as a white amorphous powder and possessed a molecular formula of C15H22O3 determined by positive HRESIMS: m/z 273.1462 [M + Na]+ (calcd. for C15H22O3Na, 273.1461) with five degrees of unsaturation. The IR spectrum showed characteristic absorption bands for hydroxy group (3445 cm−1) and conjugated carbonyl functionalities (1642 cm−1). The 1H NMR spectrum (Table 1) in methanol-d4 displayed a series of characteristic proton signals of an aristolane sesquiterpene: three tertiary methyls [δH 1.23, 1.25, and 1.35 (each 3H, s)], one secondary methyl [δH 1.08 (3H, d, J = 7.0 Hz)], two mutually coupled methine protons [δH 1.56 (1H, d, J = 7.8 Hz) and 1.74 (1H, dd, J = 7.8, 1.2 Hz)], and one olefinic protons [δH 5.87 (1H, d, J = 1.2 Hz)]. The 13C NMR and DEPT spectra (Table 1) showed a total of 15 carbon signals, characterized by four methyls, one methylene, six methines (including one olefinic at δC 130.0, two oxygenated at δC 71.9 and 77.3), and four quaternary carbons (including one keto carbonyl at δC 199.7 and one olefinic at δC 168.3). The above-mentioned NMR data revealed its structural features related to those of aristolone [5], with the major difference being the presence of two oxymethine groups at δH 3.91 (1H, q-like, J = 2.9 Hz)/δC 71.9 and δH 4.07 (1H, dd, J = 3.0, 1.3 Hz)/δC 77.3, indicative of the presence of two hydroxy groups. One of the hydroxy group located at C-1 was supported by the HMBC correlations (Fig. 2) from H-9 (δH 5.87) to C-1 (δC 77.3) and C-5 (δC 40.2), and from H-1 (δH 4.07) to C-5, C-9 (δC 130.0), and C-10 (δC 168.3), and the other was assigned to C-2 on the basis of the HMBCs from H-2 (δH 3.91) to C-4 (δC 32.8) and C-10. α-Axial orientation of the hydroxy at C-1 was deduced from the strong ROESY correlation of H-1/H-9 as well as the absence of correlation of H-1/H3-14 in the ROESY spectrum (Fig. 3). Moreover, the small di-equatorial couplings (Jee = 3.0 Hz) of Heq-1/Heq-2, Heq-2/Heq-3 and the small axial-equatorial coupling (Jea = 3.0 Hz) of Heq-2/Hax-3 revealed a β-axial orientation of the hydroxy at C-2. Hence, the structure of 2 was established as 1α,2β-dihydroxyaristolone.
Compound 3, colorless oil, gave a molecular formula of C15H22O2 as determined by positive HRESIMS: m/z 257.1511 [M + Na]+ (calcd. for C15H22O2Na, 257.1512) with five degrees of unsaturation. The IR spectrum revealed the absorptions for hydroxy (3432 cm−1) and conjugated carbonyl groups (1646 cm−1). The NMR spectra (Table 1) in chloroform-d also exhibited a series of characteristic signals due to an aristolane sesquiterpene: three tertiary methyls [δH 0.95, 1.02, and 1.25 (each 3H, s)], one secondary methyl [δH 1.06 (3H, d, J = 6.6 Hz)], two mutually coupled up-field methine protons [δH 0.67 (1H, d, J = 9.1 Hz) and 0.93 (1H, ddd, J = 9.6, 9.1, 4.0 Hz)], and one olefinic proton [δH 6.12 (1H, d, J = 1.8 Hz)] in the 1H NMR spectrum; a total of 15 carbon resonances classified as four methyls, two methylenes, five methines (including one olefinic at δC 120.8 and one oxygenated at δC 67.2), and four quaternary carbons (including one keto carbonyl at δC 199.2 and one olefinic at δC 174.7) in the 13C and DEPT spectra. The above NMR spectroscopic features were highly similar to those of debilon [9α-hydroxy-1(10)-aristolen-2-one], with the obvious difference being the chemical shift of the oxymethine [5]. The hydroxy was assigned to C-9 according to the HMBC correlations (Fig. 2) from H-1 (δH 6.12) to C-9 (δC 67.2), from H-9 (δH 4.46) to C-1 (δC 120.8), C-8 (δC 29.9), and C-10 (δC 174.7), which suggested that compound 3 shared the same planar structure with debilon. The strong ROESY correlation of H-9/H3-14 (Fig. 3), together with the di-axial coupling (J = 12.0 Hz) of Hax-9/Hax-8 and axial-equatorial coupling (J = 7.6 Hz) of Hax-9/Heq-8, suggested a β-equatorial orientation of the hydroxy group at C-9. Therefore, compound 3 was established as 9β-hydroxy-1(10)-aristolen-2-one and named as 9-epidebilon.
Compound 4 was isolated as an orange amorphous powder, with a molecular formula of C32H34O8 according to the positive HRESIMS: m/z 569.2147 [M + Na]+ (calcd. for C32H34O8Na, 569.2146). In the NMR spectra (Table 2), three tertiary methyls [δH 1.30, 1.31, and 1.37 (each 3H, s)], one secondary methyl [δH 1.08 (3H, d, J = 6.5 Hz)], two mutually coupled methine doublets [δH 1.49 (1H, d, J = 7.8 Hz) and 2.51 (1H, d, J = 7.8 Hz)], together with 15 carbon resonances (four methyls, two methylenes, four methines, and five quaternary carbons) were recognized as an aristolane structural unit. Moreover, a trans-α,β-unsaturated ketone group [δH 7.78 (1H, d, J = 15.4 Hz)/δC 123.9; δH 7.73 (1H, d, J = 15.4 Hz)/δC 143.4; δC 190.8], a chelated hydroxy [δH 13.99 (1H, s)] and one aromatic proton [δH 6.05 (1H, s)] due to a pentasubstituted phenyl ring, as well as one typical ABX system [δH 7.37 (1H, d, J = 2.0 Hz), 6.88 (1H, d, J = 8.3 Hz), and 7.09 (1H, dd, J = 8.3, 2.0 Hz)] were observed, which could be recognized as a chalcone structural unit. The above-mentioned NMR data were very close to those of nardoaristolone A, an unprecedented aristolane-chalcone hybrid recently isolated from the same species [20], and were almost identical in the aristolane part. The prominent difference between the two compounds came from the substituted pattern of ring B in chalcone part. The presence of a 1,2,4-trisubtituted ring B in 4, instead of p-substituted in nardoaristolone A, was confirmed from the HMBC correlations (Fig. 2) from H-2′ (δH 7.37) to C-4′ (δC 149.0) and C-6′ (δC 123.7), from H-5′ (δH 6.88) to C-1′ (δC 129.1) and C-3′ (δC 146.0), and from H-6′ (δH 7.09) to C-2′ (δC 112.1) and C-4′. Also a newly emerged hydroxy [δH 5.85 (1H, s)] and a methoxy group [δH 3.94 (3H, s)] located at C-3′ and C-4′, respectively, were deduced from the HMBC correlations of the hydroxy signal (δH 5.85) to C-3′ and the methoxy signal (δH 3.94) to C-4′, together with the ROESY correlations of H-2′↔OH-3′ and H-5′↔OCH3-4′. Hence, the structure of 4 was established and named as 3′-hydroxynardoaristolone A. To our knowledge, only the two aristolane-chalcone hybrids (nardoaristolone A and 3′-hydroxynardoaristolone A) were reported until now.
3 Experimental Section
3.1 General Experimental Procedures
Optical rotations were measured on a Jasco P-1020 automatic digital polarimeter. UV data were obtained from HPLC online analysis. IR spectra (KBr) were obtained on a Bruker Tensor-27 infrared spectrophotometer. NMR spectra were carried out on a Bruker Avance III 600 or Bruker DRX-500 spectrometer with deuterated solvent signals used as internal standards. ESIMS and HRESIMS were measured using an Agilent G6230 time-of-flight mass spectrometer. MPLC was performed on a Büchi apparatus equipped with Büchi fraction collector C-660, Büchi pump module C-605 and manager C-615. Preparative HPLC separation was performed using an Agilent 1260 series HPLC system equipped with a Zorbax SB-C18 column (5 μm, 21.2 × 150 mm). Silica gel (200–300 mesh, Qingdao Marine Chemical Inc., China), MCI gel CHP-20P (75–150 μm, Mitsubishi Chemical Corporation, Japan), Chromatorex C18 (40–75 μm, Fuji Silysia Chemical Ltd., Japan) and Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) were used for column chromatography. Fractions were monitored and analyzed using TLC, in combination with Agilent 1200 series HPLC system equipped by an Extend-C18 column (5 μm, 4.6 × 150 mm).
3.2 Plant Material
The roots and rhizomes of Nardostachys chinensis were purchased from Luosiwan Chinese herbal medicine market in Kunming, People’s Republic of China, in August 2015 and identified by Mr. Yu Chen of Kunming Institute of Botany, Chinese Academy of Sciences. A voucher specimen (No. BBP0656026NC) was deposited at BioBioPha Co., Ltd.
3.3 Extraction and Isolation
The air-dried and powdered roots and rhizomes of N. chinensis (12.0 kg) were extracted with 95% aqueous EtOH (3 × 18 L) at room temperature to obtain a crude extract (900 g) after evaporation of the solvent. The crude extract was fractionated by silica gel column chromatography successively eluted with a gradient of increasing acetone in petroleum ether (PE) (10:1, 8:1, 6:1, 5:1, 2:1, 1:1, 0:1; v/v) and then methanol to afford fractions A–H, respectively. Fraction E was separated by Sephadex LH-20 (CHCl3/MeOH, 1:1; v/v), MPLC (MeOH/H2O; 20% → 30%), silica gel (CHCl3/acetone, 10:1; v/v), and prep-HPLC (MeOH/H2O; 51%) to yield compound 3 (70 mg). Fraction F was separated by silica gel (CHCl3/acetone; 100:1 → 50:1), Sephadex LH-20 (MeOH), and prep-HPLC (MeOH/H2O; 48%) to yield compounds 1 (47 mg) and 4 (31 mg). Fraction G was further isolated sequentially using Sephadex LH-20 (MeOH), prep-MPLC (MeOH/H2O; 0 → 100%), and prep-HPLC (MeOH/H2O; 55%) to afford compound 2 (14 mg). The retention times (tR) of 1–4 on an analytical HPLC Thermo Hypersil BDS C18 column (20% → 100% MeOH in H2O over 8.0 min followed by 100% MeOH to 13.0 min, 1.0 ml/min, 22 °C) were 8.51, 7.25, 8.68, and 10.37 min, respectively.
3.4 Secoaristolenedioic Acid (1)
White amorphous powder; [α] 25D −104.0 (c 0.20, MeOH); UV (MeOH) λmax: 215 nm; IR (KBr) νmax: 3443, 3428, 2921, 1687, 1676, 1638, 1446, 1417, 1384, 1294 cm−1; 1H and 13C NMR data: see Table 1; ESIMS (pos.): m/z 289 [M + Na]+; HRESIMS (pos.): m/z 289.1410 [M + Na]+ (calcd. for C15H22O4Na, 289.1410).
3.5 1α,2β-Dihydroxyaristolone (2)
White amorphous powder; [α] 25D −245.3 (c 0.20, MeOH); UV (MeOH) λmax: 235 nm; IR (KBr) νmax: 3445, 2966, 2922, 1642, 1449, 1383, 1054 cm−1; 1H and 13C NMR data: see Table 1; ESIMS (pos.): m/z 273 [M + Na]+; HRESIMS (pos.): m/z 273.1462 [M + Na]+ (calcd. for C15H22O3Na, 273.1461).
3.6 9-Epidebilon (3)
Colorless oil; [α]25 D +14.0 (c 0.20, MeOH); UV (MeOH) λmax: 242 nm; IR (KBr) νmax: 3432, 2967, 2942, 2882, 1710, 1646, 1455, 1383, 1193, 1053 cm−1; 1H and 13C NMR data: see Table 1; ESIMS (pos.): m/z 257 [M + Na]+; HRESIMS (pos.): m/z 257.1511 [M + Na]+ (calcd. for C15H22O2Na, 257.1512).
3.7 3′-Hydroxynardoaristolone A (4)
Orange amorphous powder; [α] 25D +223.7 (c 0.20, MeOH); UV (MeOH) λmax: 262, 315 (sh), 379 nm; IR (KBr) νmax: 3443, 2960, 2930, 1632, 1589, 1560, 1509, 1441, 1270, 1223, 1030 cm−1; 1H and 13C NMR data: see Table 2; ESIMS (pos.): m/z 569 [M + Na]+; HRESIMS (pos.): m/z 569.2147 [M + Na]+ (calcd. for C32H34O8Na, 569.2146).
References
Editorial Committee of Flora Reipublicae Popularis Sinicae, Flora Reipublicae Popularis Sinicae, vol. 75 (Academic Press, Beijing, 1986), p. 25
P. Zhang, Y.G. Cheng, W.P. Cheng, Acta Chin. Med. Pharm. 43, 99–100 (2015)
J.B. Zhang, M.L. Liu, C. Li, Y. Zhang, Y. Dai, X.S. Yao, Fitoterapia 100, 195–200 (2015)
M.J. Durán-Peña, J.M.B. Ares, J.R. Hanson, I.G. Collado, R. Hernández-Galán, Nat. Prod. Rep. 32, 1236–1248 (2015)
M. Furusawa, T. Hashimoto, Y. Noma, Y. Asakawa, Chem. Pharm. Bull. 54, 861–868 (2006)
M.L. Liu, Y.H. Duan, J.B. Zhang, Y. Yu, Y. Dai, X.S. Yao, Tetrahedron 69, 6574–6578 (2013)
C. Intaraudom, N. Boonyuen, S. Supothina, P. Tobwor, S. Prabpai, P. Kongsaeree, P. Pittayakhajonwui, Phytochem. Lett. 6, 345–349 (2013)
E. Zubia, M.J. Ortega, J.L. Carballo, J. Nat. Prod. 71, 2004–2010 (2008)
Z.B. Cheng, W.W. Shao, Y.N. Liu, Q. Liao, T.T. Lin, X.Y. Shen, S. Yin, J. Nat. Prod. 76, 664–671 (2013)
H.H. Wu, Y.T. Xu, X.M. Gao, Y.P. Chen, S.S. Ying, Y.T. Liu, P.Z. Dong, Y. Zhu, Faming Zhuanli Shenqing, CN105152894 A 20151206 (2015)
H. Itokawa, K. Masuyama, H. Morita, K. Takeya, Chem. Pharm. Bull. 41, 1183–1184 (1993)
A. Bagchi, Y. Oshima, H. Hikino, Phytochemistry 27, 1199–1201 (1988)
J.S. Hwang, S.A. Lee, S.S. Hong, X.H. Han, C. Lee, D. Lee, C.K. Lee, J.T. Hong, Y. Kim, M.K. Lee, B.Y. Hwang, Bioorg. Med. Chem. Lett. 22, 706–708 (2012)
Y. Takaya, K. Kuramada, Y. Takeuji, H.S. Kim, Y. Shibata, N. Ikemoto, Y. Wataya, Y. Oshima, Tetrahedron Lett. 39, 1361–1364 (1998)
M. Bruno, M.C. de la Torre, B. Rodriguez, A.A. Omar, Phytochemistry 34, 245–247 (1993)
G. Goldsby, B.A. Burke, Phytochemistry 26, 1059–1063 (1987)
E. Demole, P. Enggist, Helv. Chim. Acta 66, 1381–1391 (1983)
B. Bläs, J. Zapp, H. Becker, Phytochemistry 65, 127–137 (2004)
Y.P. Chen, S.S. Ying, H.H. Zheng, Y.T. Liu, Z.P. Wang, H. Zhang, X. Deng, Y.J. Wu, X.M. Gao, T.X. Li, Y. Zhu, Y.T. Xu, H.H. Wu, Sci. Rep. 7, 15114 (2017)
M.L. Liu, Y.H. Duan, Y.L. Hou, C. Li, H. Gao, Y. Dai, X.S. Yao, Org. Lett. 15, 1000–1003 (2013)
Acknowledgements
This work was financially supported by the “Large-scale Compound Library” project of National Development and Reform Commission of China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, LX., Jiang, XJ., Li, XM. et al. Aristolane-type Sesquiterpenoids from Nardostachys chinensis and Revised Structure of Aristolanhydride. Nat. Prod. Bioprospect. 9, 149–155 (2019). https://doi.org/10.1007/s13659-019-0200-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-019-0200-7