
Abstract
In animals, voltinism is a result of evolutionary adaptations to environmental conditions. These evolutionary adaptations may profoundly affect the population structure and social organization level. To study the bivoltinism of the solitary bee Tetrapedia diversipes, we performed comparative transcriptomics analyses of foundresses and larvae from the two reproductive generations (G1 and G2) produced per year by this bee. Most of the differentially expressed genes (DEGs) were found between foundresses: 52 DEGs between adults, but only one between the larvae. Among the DEGs in foundresses, 46 were higher expressed in G1 and most of them (38) have no functional annotation defined in the database. Interestingly, mitochondrial genes and long non-coding RNAs were the only type of identified transcripts in the set of upregulated genes. These results highlight the importance of developing studies on non-model species and suggest that maternal genes may be of importance for determining larval diapause in T. diversipes.
Similar content being viewed by others


1 Introduction
The number of reproductive generations presented by a species in a year determines its pattern of voltinism (Corbet et al. 2006). In insects, the voltine trait is considered the result of evolutionary adaptations to environmental conditions, such as temperature, humidity, photoperiod, latitude, and food resources (Corbet et al. 2006; Altermatt 2010; Cardoso and Silveira 2012; Hunt 2012). And it may have had profound consequences at the levels of population structure and social organization (Hunt and Amdam 2005). This is especially noticeable in species where sociality has evolved recently, such as in Halictinae bees (Brady et al. 2006). Within the Halictinae, it has been demonstrated that social behavior may be facultative and directly related to voltine generations (Yanega 1988). Halictus rubicundus females, for example, present solitary behavior when individuals are univoltine, but they may become social when individuals are bivoltine (two generations per year) and enter diapause in the second generation (Soucy 2002). Indeed, bivoltinism associated with diapause in one offspring generation is the biological system on which Hunt’s bivoltine ground plan hypothesis on the evolution of insect sociality in temperate climates is based (Hunt and Amdam 2005). According to this hypothesis, social behavior could evolve from solitary species if females from the first reproductive generation remain in the nest and changes in environmental conditions suppress the entry into prepupal diapause of the second generation (Hunt and Amdam 2005; Hunt 2012). Thus, studies of ecological and molecular mechanisms involved in voltinism can be important pieces to understand the puzzle of social behavior in bees and its evolution.
In the tropics, bivoltinism has been observed in different solitary bees (Silveira et al. 2002; Alves-dos-Santos et al. 2007) including Tetrapedia diversipes Klug 1810 (subfamily Apinae, tribe Tetrapediini; Michener 2007). This oil-collecting bee, native of the Neotropical region, has two main reproductive generations during the year, each presenting different developmental times (Alves-dos-Santos et al. 2002). Individuals of the first generation (G1) show direct development from egg to adults, and they emerge within a few weeks during the hot and wet months. In contrast, individuals from the second generation (G2) halt their development in the fifth larval instar during the cold and dry season and emerge as adults only after a diapause period (Camillo 2005; Alves-dos-Santos et al. 2006). Due to this diapause, the developmental time of G2 individuals may be four times longer in comparison to G1 (Alves-dos-Santos et al. 2002). In addition to this interesting developmental aspect, T. diversipes is an attractive species to study because (i) it easily nidifies in trap nests (Camillo 2005; Alves-dos-Santos et al. 2006; Menezes et al. 2012; Neves et al. 2012; Rocha-Filho and Garófalo 2015), (ii) it has already characterized molecular markers (Arias et al. 2016), and (iii) its genome is currently being sequenced. In combination, all these characteristics make T. diversipes a promising emerging model for developmental and evolutionary studies on bees in tropical climates.
In this exploratory study, we report the complete transcriptomes of foundress females and non-diapause larvae of T. diversipes using next-generation sequencing (RNA-Seq) and these data were used to search for major differences in the expression profiles of the two reproductive generations. This sequencing approach has become widely used in gene expression studies, especially for non-model species, as it provides a non-directional and unbiased way of obtaining data on the relative frequencies of messenger RNAs (mRNAs) with no need for species-specific probes or a reference genome (Wang et al. 2009).
2 Material and methods
2.1 Sample collection
Wooden trap nests, as described in Alves-dos-Santos et al. (2002), were placed at the Universidade de São Paulo campus in São Paulo city, Brazil. Foundresses were collected using an entomological net between 10:00 and 12:00 in front of their nests while constructing. Larvae were collected directly from inside the nests that had been completed and closed by the foundresses. All instars (first to fifth) in the non-diapause (G1) or prediapause state (G2) were sampled. Individuals were immediately frozen in liquid nitrogen. G1 samples were collected from November to December (mean temperature 26.4 °C; mean humidity 62.2%) and G2’s from March to beginning of July (mean temperature 24 °C; mean humidity 59.2%). The larvae were not sexed prior to pooling for RNA extraction.
2.2 RNA extraction and sequencing
Total RNA was extracted from the whole body using the RNeasy® Kit (Qiagen) and following the manufacturer’s protocol. RNA quality was verified by the sequencing facility (Macrogen) using a Bionalyzer® system, and results were interpreted as discussed in Winnebeck et al. (2010). Nine foundresses and nine larvae from each generation were selected for RNA sequencing. Samples were divided into three replicates, each containing the RNA of three individuals from the same developmental stage. This sampling approach was adopted to improve gene identification and differential expression analyses (Hart et al. 2013; Lin et al. 2016). Although the larval samples contained all developmental stages, the larvae in each replication were pooled according to their instar—one pool of larvae from the first to fourth instar; one from second to fifth; and one consisting of fifth instar larvae only. This approach was adopted to focus on the identification of major differences between the two-generation groups without confounding effects from the larval developmental instars. Altogether, 12 samples were sequenced, 6 from G1 (3 pools of adults and 3 of larvae) and 6 from G2 (3 pools of adults and 3 of larvae), using a HiSeq2000® sequencer (Illumina). Paired-end reads of 100 bp were sequenced, and about 50 million paired reads were obtained per sample. Sequencing and library preparation were done by Macrogen (South Korea).
2.3 Transcriptome assembly
Quality assessment of the reads was performed using the FastQC program (version 0.11.2; Andrews 2010) before and after cleaning. The FASTX Toolkit (version 0.0.14; Hannon Lab 2009) was used to trim the first 14 bp of all reads because of the initial GC bias (Hansen et al. 2010). Low-quality bases (phred score below 30) and small reads (less than 31 bp) were removed by the SeqyClean program (version 1.9.3; Zhbannikov 2013).
Prior to assembly, the data from each developmental stage and reproductive generation were digitally normalized (20× coverage) to increase assembly efficiency using the Brown et al. (2012) protocol incorporated in Trinity (version 2.0.6, Grabherr et al. 2013). Normalized data were independently assembled de novo using Trinity with default parameters. Assemblies from different reproductive generations (G1 and G2) were then concatenated with the CD-Hit program (version 4.6; Huang et al. 2010) at 95% similarity, generating one assembly for adults and another one for larvae. Next, cleaned libraries were aligned to the respective assemblies by the TopHat2 program (version 2.1.0; Kim et al. 2013), i.e., replicate libraries from larva were aligned to the larval transcriptome and adult libraries to the adult transcriptome. These realignments were used as input in Corset (version 1.03; Davidson and Oshlack 2014) with minimum coverage of 50× to improve transcript assemblies.
Final transcriptomes were then annotated with Annocript (version 1.2, Musacchia et al. 2015) using the UniProt Reference Clusters (UniRef) database (version February 2016; Suzek et al. 2015). Transcripts with significant blast hits (E value < 1e−5) against possible contaminants (plants, fungus, mites, and bacteria) in UniRef were removed from the final dataset and were also used to identify other contaminants based on cluster analysis from Corset, as described in Araujo et al. (2016). Quality assessments of the final assembly were performed with QUAST (version 4.0; Gurevich et al. 2013), BUSCO (version 2; Simão et al. 2015), and Qualimap (version 2.2; García-Alcalde et al. 2012).
2.4 Differential expression analysis
The Trinity script was used to automate the differential expression analyses using the Bowtie2 program (version 2.2.5; Langmead and Salzberg 2012) to realign all cleaned libraries to the final transcriptome; the RSEM program (version 1.2.22; Li and Dewey 2011) to count the realigned reads; and the edgeR package (version 3.14.0; Robinson et al. 2009) for the statistical analyses (minimum FDR p value < 1e−5). Differential expression analyses were performed independently for each developmental stage (adult and larva) by comparing the two reproductive generations (G1 × G2).
3 Results
3.1 Transcriptome assembly and annotation
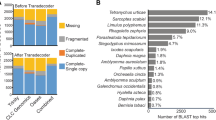
The main parameters from the final transcriptome assemblies of foundresses and larvae are listed in Table I. The final adult transcriptome (after removal of contaminants) contains 44,486 transcripts, of which 27,098 are reported by at least one blast hit. The Annocript pipeline also reported that from all the transcripts, 709 are probably long non-coding RNAs (lncRNAs) and 32,037 have coding potential based on their open reading frame (ORF) or annotation. The larval transcriptome had similar results, presenting a total of 41,354 transcripts, of which 27,393 had at least one blast hit; 30,975 transcripts are potentially coding and 539 were identified as potential lncRNAs. The final transcriptome assemblies and annotation tables, containing all the statistics for coding and lncRNAs, are available at GitHub (https://github.com/nat2bee/bivoltine_apidologie) and will eventually be deposited in public databases.
The database for Hymenoptera orthologs available via BUSCO software has a total of 4,415 genes of which 3,645 (82.6%) were identified as complete in the T. diversipes adult transcriptome and 3,432 (77.7%) in the larval one. A small portion was identified as fragmented genes, 268 (6.1%) and 358 (8.1%) in the adult and larval transcriptome, respectively. Thus, only 502 (11.3%) and 625 (14.2%) of all the Hymenoptera orthologs were missing in the adult and larval final dataset, respectively.
3.2 Differential expression analyses
Gene expression analyses with the edgeR program reported 52 differentially expressed genes (DEGs) between the two adult generations (Online Resource 1). Among these, 46 genes were highly expressed in adult G1 females and 6 had elevated levels of expression in females from G2 (Figure 1). Four G1 upregulated genes (cytochrome b; COII; NADH4 and one encoding a predicted uncharacterized protein) and three upregulated genes from G2 (defensin-2; DHRS11 and one for an uncharacterized protein) had a significant blast hit against the UniRef database (Figure 1; Online Resource 2). Another five of the higher represented transcripts in G1 females were reported as possible lncRNAs by Annocript according to their small ORF size (≤ 100 bp), their absence in databases and their sequence length (≥ 200 bp) (Figure 1; Online Resource 2). When comparing the larval generations, only one DEG was found, and this was higher expressed in individuals from G2 (Online Resource 1). This gene was annotated as a replicase polyprotein according to the UniRef database (Online Resource 2).

Heatmap of the differentially expressed genes among T. diversipes adult females of generations G1 and G2. Replicate samples for G1 foundresses are fem1, fem2 and fem3; fem4, fem5 and fem6 are replicates for G2 foundresses. Indicated in bold on the y-axis are the names of the annotated transcripts and lncRNAs. Uncharacterized refers to transcripts that were retrieved as uncharacterized proteins by blast queries against the UniRef database. Expression scale is log2. FDR p value < 10e−5.
4 Discussion
We report here for the first time a transcriptome assembly of female foundresses and non-diapause larvae of the solitary bee T. diversipes, which we consider an emerging model for voltinism and its possible consequences for the evolution of sociality in bees. Quality analyses of the transcripts indicate that the assembly is comparable to other bee transcriptomes (Colgan et al. 2011; Kocher et al. 2013; Rehan et al. 2014; Harrison et al. 2015) and has a high sequencing coverage (about 150 x per transcriptome). In both life cycle stages, more than 60% (60.9% in adult females and 66.2% in larvae) of the transcripts had a significant blast hit against a sequence in the UniRef database, especially against bees and other Hymenoptera sequences (Online Resource 3). Moreover, it is expected that an additional 8% of the non-annotated transcripts are protein coding according to their ORF characteristics, but since they did not have a significant blast hit, they may represent genes that are either novel or significantly divergent from any of the database sequences. This leaves about 30% of the sequences as possible miss-assemblies, non-coding RNAs, or transcriptional noise (retained introns and similar) in the T. diversipes transcriptomes.
Differential expression analyses revealed that between non-diapause larvae from G1 and G2 only, the gene encoding a replicase polyprotein was upregulated in G2. Genes for replicase polyproteins or RNA-dependent RNA polymerases (RdRps) are well-known genes that encode for proteins essential for genome replication of RNA viruses (Ahlquist 2002; Iyer et al. 2003). Recently, however, it has been demonstrated that many eukaryotes may actually have one or more RdRp copies in their genomes (Nishikura 2001; Ahlquist 2002; Anantharaman et al. 2002; Chapman and Carrington 2007) leading to the assumption that RdRps may have a different biological function acting directly in post-transcriptional regulatory mechanisms (especially in RNA silencing) and host response (Ahlquist 2002; Anantharaman et al. 2002; Chapman and Carrington 2007). Thus, the higher expression of this gene in G2 larvae may indicate either a season-related viral infection or an aspect of gene expression regulation when compared to larvae in G1. It is possible, for example, that once G2 larvae will enter diapause, the higher RdRp expression may be involved with regulatory functions in the gene expression cascade necessary for diapause onset. However, in several arthropod genomes, including Drosophila, no homologs of an RdRp gene have been detected (Zong et al. 2009). Thus, although post-transcriptional regulatory mechanisms related to RdRps are likely to be functional in insects, it is not clear if other proteins are involved in the process instead (Ahlquist 2002; Zong et al. 2009). Therefore, identifying the actual true origin of the reported RdRp will only be possible after completion of the T. diversipes genome assembly.
Most of the DEGs between the two reproductive generations were reported in foundresses. In G2 foundresses, whose offspring will enter diapause, six transcripts were higher represented. One of these, the defensin-2 gene, is known to be expressed in honey bee fat body and is responsible for immune defense against a number of parasites (as discussed in Klaudiny et al. 2005). For Apis mellifera, it has been reported that the level of defensin-2 expression only increases in response to infections (reviewed by Ilyasov et al. 2012). Thus, our finding suggests that G2 females are more likely to be exposed to pathogens than G1 females. Another higher expressed gene in G2 foundresses that also may be involved in some type of immune response is dehydrogenase/reductase SDR family member 11 (DHRS11), a gene encoding an NAD(P)(H)-dependent oxidoreductase that belongs to the large superfamily of short-chain dehydrogenase/reductases (Persson et al. 2009; Endo et al. 2016). Genes from this superfamily are known to be involved in the metabolism of lipids, carbohydrates, vitamins, drugs, and xenobiotics (Endo et al. 2016). Specifically in honeybees, DHRS11 has been found to play a role in host resistance against the varroa mite (Parker et al. 2012), and in larval caste differentiation, it appears as one of the genes possibly necessary for worker ovary differentiation (Guidugli et al. 2004; Lago et al. 2016). Furthermore, the DHRS11 product is one of the proteins present in bee venom (Li et al. 2013).
Among the higher expressed genes identified in G1 foundresses are COII, NADH4, and cytochrome b. These genes are involved in energy production through oxidative phosphorylation (Cooper 2000), suggesting that adult females from G1 have higher energy demands than G2 females, which may be associated with environmental factors such as food availability and temperature. It is also worthy of note that other five higher expressed genes in females from G1 are putative lncRNAs. LncRNAs are poly(A) RNAs which, different from other mRNAs, are not translated into functional proteins. Presently, lncRNAs are defined as probable non-coding RNAs longer than 200 bp (as discussed in Rinn and Chang 2012). These transcripts have recently emerged as critical elements in many genetic regulatory mechanisms including epigenetic regulation DNA imprinting, control of cell pluripotency, transcriptional silencing, and co-activation, among others (Rinn and Chang 2012; Kung et al. 2013). In honey bees, for instance, one specific lncRNA, lncov-1, was shown to be associated with programmed cell death in the larval worker ovary (Humann et al. 2013).
Taken together, two important aspects related to bivoltinism in T. diversipes emerged from our gene expression analyses. First, the RdRp gene, if not related to viral infection, may be one of the first genes to have its expression pattern specifically regulated in prediapause larvae from G2. Second, most of the observed expression differences between G1 and G2 individuals were seen in adult females and not in larvae. Thus, DEGs of foundresses might be important for triggering diapause in the subsequent offspring generation. Nevertheless, it is important to mention that the experimental design used in this study for larval comparisons was directed towards retrieving only major differences between larvae from both generations, without accounting for differences related to sex or the respective developmental larval phases. In effect, we believe that the pooling approach used here permitted to reveal the possible main players between the two generations. Further, more in-depth studies can now be devised to reveal such sex- or stage-specific differences.
With respect to the adult stages, our data showed a strong bias in DEGs towards immunity and energy-related processes. Diapause is known to be also induced by changes in the mother’s metabolism in many insects (Denlinger 2002). This induction may be direct, as in the case of the silkworm Bombyx mori that produces a specific diapause hormone (Fukuda 1951), or indirect, where the correlation between progenitor gene expression and brood diapause is not so clearly established, because even changes experienced during the mother’s development may affect the offspring (Denlinger 1998, 2002). It is possible that in T. diversipes, the overexpression of metabolic genes and of lncRNAs (or any other of the as yet non-identified genes) in the foundresses provides a molecular or epigenetic signal that prevents the larvae from entering diapause. Similar mechanisms of maternal epigenetic suppression to larval diapause have already been described in the fly Sarcophaga bullata and in the GABAergic circuit of B. mori (Çabej 2013; Reynolds et al. 2013, 2016). Additionally, lncRNAs have been reported as paramount in glucose and lipid metabolism, which are important energetic pathways for diapause (Denlinger 2002; Lang-Ouellette et al. 2014).
Another important outcome of this study is that most of the DEGs represent genes that are unknown with respect to their function. Thus, the 42 non-characterized DEGs reported in foundresses may also play an important role in inducing or suppressing larval diapause, but we were unable to infer on their biological relevance based on the information currently available in genome databases. Although there has been an expressive improvement in databases content over the last years (O’Leary et al. 2016), they are still highly biased towards model species. Therefore, studies on non-model species are clearly desirable to improve our understanding of complex biological systems.
5 Conclusions
Herein we report the complete transcriptome assembly from two different life stages of T. diversipes. Using these assemblies, we compared gene expression differences between the two reproductive generations of T. diversipes. These analyses have shown that an RdRp gene is the only one that is highly expressed in larvae from G2 before entering diapause and that genes expressed in the mothers are likely to influence the developmental timing of the larval offspring. This can be inferred based on the fact that most of the observed gene expression differences occurred in the adult life stage. Among the higher expressed genes in G1 adult females, the only identified transcripts are the ones involved in oxidative phosphorylation and lncRNAs, while in G2 adult females, higher expressed genes are possibly related to immune responses.
References
Ahlquist, P. (2002) RNA-dependent RNA polymerases, viruses, and RNA silencing. Science 296 (5571), 1270–1273
Altermatt, F. (2010) Climatic warming increases voltinism in European butterflies and moths. Proc. R. Soc. B Biol. Sci. 277 (1685), 1281–1287
Alves-dos-Santos, I., Melo, G.A.R., Rozen Jr, J.G. (2002) Biology and immature stages of the bee tribe Tetrapediini (Hymenoptera: Apidae). Am. Museum Nat. Hist. 3377, 1–45
Alves-dos-Santos, I., Naxara, S.R.C., Patrício, E.F.L.R.A. (2006) Notes on the morphology of Tetrapedia diversipes Klug 1810 (Tetrapediini, Apidae), an oil-collecting bee. Braz. J. Morphol. Sci. 23 (3–4), 425–430
Alves-dos-Santos, I., Machado, I.C., Gaglianone, M.C. (2007) História natural das abelhas coletoras de óleo. Oecol. Bras. 11 (4), 544–557
Anantharaman, V., Koonin, E. V, Aravind, L. (2002) Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 30 (7), 1427–64
Andrews, S. (2010) FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics [online] http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 05 Feb 2016)
Araujo, N. de S., Zuntini, A.R., Arias, M.C. (2016) Getting useful information from RNA-Seq contaminants: A case of study in the oil-collecting bee Tetrapedia diversipes transcriptome. Omi. A J. Integr. Biol. 20 (8), 491–492
Arias, M.C., Aulagnier, S., Baerwald, E.F., Barclay, R.M.R., Batista, J.S. et al. (2016) Microsatellite records for volume 8, issue 1. Conserv. Genet. Resour. 8 (1), 43–81
Brady, S.G., Sipes, S., Pearson, A., Danforth, B.N. (2006) Recent and simultaneous origins of eusociality in Halictid bees. Proc. R. Soc. B 273, 1643–1649
Brown, C.T., Howe, A., Zhang, Q., Pyrkosz, A.B., Brom, T.H. (2012) A reference-free algorithm for computational normalization of shotgun sequencing data. Genome Announc. 2 (4), 1–18
Çabej, N. (2013) Living and adapting to its own habitat. In: Building the most complex structure on Earth: an epigenetic narrative of development and evolution of animals, 1st edn. Elsevier, Oxford, pp. 193–238
Camillo, E. (2005) Nesting biology of four Tetrapedia species in trap nests (Hymenoptera: Apidae: Tetrapediini). Rev. Biol. Trop. 53, 175–186
Cardoso, C.F., Silveira, F.A. (2012) Nesting biology of two species of Megachile (Moureapis) (Hymenoptera: Megachilidae) in a semideciduous forest reserve in southeastern Brazil. Apidologie 43, 71–81
Chapman, E.J., Carrington, J.C. (2007) Specialization and evolution of endogenous small RNA pathways. Nat. Rev. Genet. 8 (11), 884–896
Colgan, T.J., Carolan, J.C., Bridgett, S.J., Sumner, S., Blaxter, M.L., Brown, M.J. (2011) Polyphenism in social insects: insights from a transcriptome-wide analysis of gene expression in the life stages of the key pollinator, Bombus terrestris. BMC Genomics 12 (623), 1–20
Cooper, G. (2000) The mechanism of oxidative phosphorylation. In: The Cell: A Molecular Approach (ed Hausman RE). Sinauer Associates, Sunderland. [online] https://www.ncbi.nlm.nih.gov/books/NBK9885/
Corbet, P.S., Suhling, F., Soendgerath, D. (2006) Voltinism of Odontada: a review. Int. J. Odonatol. 9 (1), 1–44
Davidson, N.M., Oshlack, A. (2014) Corset: enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 15 (410), 1–14
Denlinger, D.L. (1998) Maternal control of fly diapause. In: Maternal effects as adaptations (eds Mousseau TA, Fox CW), pp. 275–286. Oxford University Press Oxford.
Denlinger, D.L. (2002) Regulation of diapause. Annu. Rev. Entomol. 47, 93–122
Endo, S., Miyagi, N., Matsunaga, T., Hara, A., Ikari, A. (2016) Human dehydrogenase/reductase (SDR family) member 11 is a novel type of 17B-hydroxysteroid dehydrogenase. Biochem. Biophys. Res. Commun. 472 (1), 231–236
FASTX-Toolkit (2009) by Hannon Lab [online] http://hannonlab.cshl.edu/fastx_toolkit/index.html (accessed on 05 Feb 2016)
Fukuda, S. (1951) The production of the diapause eggs by transplanting the suboesophageal ganglion in the silkworm. Proc. Jpn. Acad. 27, 672–677
García-Alcalde, F., Okonechnikov, K., Carbonell, J., Cruz, L.M., Götz, S., Tarazona, S., Dopazo, J., Meyer, T.F., Conesa, A. (2012) Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 28 (20), 2678–2679
Grabherr, M.G., Haas, B.J., Yassour, M., Levin, J.Z., Thompson, D.A. et al. (2013) Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29 (7), 644–652
Guidugli, K.R., Hepperle, C., Hartfelder, K. (2004) A member of the short-chain dehydrogenase/reductase (SDR) superfamily is a target of the ecdysone response in honey bee (Apis mellifera) caste development. Apidologie 35 (1), 37–47
Gurevich, A., Saveliev, V., Vyahhi, N., Tesler, G. (2013) QUAST: quality assessment tool for genome assemblies. Bioinformatics 29 (8), 1072–1075
Hansen, K.D., Brenner, S.E., Dudoit, S. (2010) Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 38 (12), 1–7
Harrison, M., Hammond, R., Mallon, E. (2015) Reproductive workers show queen-like gene expression in an intermediately eusocial insect, the buff-tailed bumble bee Bombus terrestris. Mol. Ecol. 24, 3043–3063
Hart, S.N., Therneau, T.M., Zhang, Y., Poland, G.A., Kocher, J.P. (2013) Calculating sample size estimates for RNA sequencing data. J. Comput. Biol. 20, 1–9
Huang, Y., Niu, B., Gao, Y., Fu, L., Li, W. (2010) CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 26 (5), 680–682
Humann, F.C., Tiberio, G.J., Hartfelder, K. (2013) Sequence and expression characteristics of long noncoding RNAs in honey bee caste development-potential novel regulators for transgressive ovary size. PLoS One 8 (10), e78915
Hunt, J.H. (2012) A conceptual model for the origin of worker behaviour and adaptation of eusociality. J. Evol. Biol. 25 (1), 1–19
Hunt, J.H., Amdam, G. V (2005) Bivoltinism as an antecedent to eusociality in the paper wasp genus Polistes. Science 308, 264–267
Ilyasov, R., Gaifullina, L., Saltykova, E., Poskryakov, A., Nikolenko, A. (2012) Review of the expression of antimicrobial peptide defensin in honey bees Apis mellifera L.. J. Apic. Sci. 56 (1), 115–123
Iyer, L.M., Koonin, E. V, Aravind, L. (2003) Evolutionary connection between the catalytic subunits of DNA-dependent RNA polymerases and eukaryotic RNA-dependent RNA polymerases and the origin of RNA polymerases. BMC Struct. Biol. 3, 1–23
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., Salzberg, S.L. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14 (4R36), 1–13
Klaudiny, J., Albert, S., Bachanova, K., Kopernicky, J., Simuth, J. (2005) Two structurally different defensin genes, one of them encoding a novel defensin isoform, are expressed in honeybee Apis mellifera. Insect Biochem. Mol. Biol. 35 (1), 11–22
Kocher, S.D., Li, C., Yang, W., Tan, H., Yi, S. V et al. (2013) The draft genome of a socially polymorphic halictid bee, Lasioglossum albipes. Genome Biol. 14 (12), R142
Kung, J.T.Y., Colognori, D., Lee, J.T. (2013) Long noncoding RNAs: past, present, and future. Genetics 193 (3), 651–69
Lago, D.C., Humann, F.C., Barchuk, A.R., Abraham, K.J., Hartfelder, K. (2016) Differential gene expression underlying ovarian phenotype determination in honey bee, Apis mellifera L, caste development. Insect Biochem. Mol. Biol. 79, 1–12
Langmead, B., Salzberg, S.L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods 9 (4), 357–359
Lang-Ouellette, D., Richard, T.G., Morin, P. (2014) Mammalian hibernation and regulation of lipid metabolism: A focus on non-coding RNAs. Biochemistry 79 (11), 1161–1171
Li, B., Dewey, C.N. (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12 (323), 1–16
Li, R., Zhang, L., Fang, Y., Han, B., Lu, X., Zhou, T., Feng, M., Li, J. (2013) Proteome and phosphoproteome analysis of honeybee (Apis mellifera) venom collected from electrical stimulation and manual extraction of the venom gland. BMC Genomics 14 (766), 1–12
Lin, Y., Golovnina, K., Chen, Z.X., Lee, H.N., Negron, Y.L.S., Sultana, H., Oliver, B., Harbison, S.T. (2016) Comparison of normalization and differential expression analyses using RNA-Seq data from 726 individual Drosophila melanogaster. BMC Genomics 17 (28), 1–20
Menezes, G.B., Gonçalves-Esteves, V., Bastos, E.M. A. F., Augusto, S.C., Gaglianone, M.C. (2012) Nesting and use of pollen resources by Tetrapedia diversipes Klug (Apidae) in Atlantic Forest areas (Rio de Janeiro, Brazil) in different stages of regeneration. Rev. Bras. Entomol. 56, 86–94
Michener, C.D. (2007) The bees of the world. Johns Hopkins University Press, Baltimore
Musacchia, F., Basu, S., Petrosino, G., Salvemini, M., Sanges, R. (2015) Annocript: A flexible pipeline for the annotation of transcriptomes able to identify putative long noncoding RNAs. Bioinformatics 31 (13), 2199–2201
Neves, C.M. de L., de Carvalho, C.A.L., Souza, V.A., de Lima Junior, C.A. (2012) Morphometric characterization of a population of Tetrapedia diversipes in restricted areas in Bahia, Brazil ( Hymenoptera: Apidae ). Sociobiology 59 (3), 767–782
Nishikura, K. (2001) A short primer on RNAi: RNA-directed RNA polymerase acts as a key catalyst. Cell 107 (4), 415–418
O’Leary, N.A., Wright, M.W., Brister, J.R., Ciufo, S., Haddad, D. et al. (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44 (D1), D733–D745
Parker, R., Guarna, M.M., Melathopoulos, A.P., Moon, K.M., White, R., Huxter, E., Pernal, S.F., Foster, L.J. (2012) Correlation of proteome-wide changes with social immunity behaviors provides insight into resistance to the parasitic mite, Varroa destructor, in the honey bee (Apis mellifera). Genome Biol. 13 (9R81), 1–15
Persson, B., Kallberg, Y., Bray, J.E., Bruford, E., Dellaporta, S.L. et al. (2009) The SDR (short-chain dehydrogenase/reductase and related enzymes) nomenclature initiative. Chem. Biol. Interact. 178 (1–3), 94–98
Rehan, S.M., Berens, A.J., Toth, A.L. (2014) At the brink of eusociality: transcriptomic correlates of worker behaviour in a small carpenter bee. BMC Evol. Biol. 14 (1), 260
Reynolds, J.A., Clark, J., Diakoff, S.J., Denlinger, D.L. (2013) Transcriptional evidence for small RNA regulation of pupal diapause in the flesh fly, Sarcophaga bullata. Insect Biochem. Mol. Biol. 43 (10), 982–989
Reynolds, J.A., Bautista-Jimenez, R., Denlinger, D.L. (2016) Changes in histone acetylation as potential mediators of pupal diapause in the flesh fly, Sarcophaga bullata. Insect Biochem. Mol. Biol. 76, 29–37
Rinn, J.L., Chang, H.Y. (2012) Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81, 145–66
Robinson, M.D., McCarthy, D.J., Smyth, G.K. (2009) edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26 (1), 139–140
Rocha-Filho, L.C., Garófalo, C.A. (2015) Natural history of Tetrapedia diversipes (Hymenoptera: Apidae) in an Atlantic Semideciduous forest remnant surrounded by coffee crops, Coffea arabica (Rubiaceae). Ann. Entomol. Soc. Am. 109 (2), 183–197
Silveira, F.A., Melo, G.A.R., Almeida, E.A.B. (2002) Abelhas brasileiras: Sistemática e Identificação. Fundação Araucária, Belo Horizonte.
Simão, F.A., Waterhouse, R.M., Ioannidis, P., Kriventseva, E.V., Zdobnov, E.M. (2015) BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31 (19), 3210–3212
Soucy, S.L. (2002) Nesting biology and socially polymorphic behavior of the sweat bee Halictus rubicundus (Hymenoptera: Halictidae). Entomol. Soc. Am. 95 (1), 57–65
Suzek, B.E., Wang, Y., Huang, H., McGarvey, P.B., Wu, C.H., UniProt Consortium (2015) UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31 (6), 926–932
Wang, Z., Gerstein, M., Snyder, M. (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10 (1), 57–63
Winnebeck, E.C., Millar, C.D., Warman, G.R. (2010) Why does insect RNA look degraded?. J. Insect Sci. 10 (159), 1–7
Yanega, D. (1988) Social plasticity and early-diapausing females in a primitively social bee. Proc. Natl. Acad. Sci. U. S. A. 85, 4374–4377
Zhbannikov, I. (2013) SeqyClean [online] https://bitbucket.org/izhbannikov/seqyclean.git (accessed on 05 Feb 16)
Zong, J., Yao, X., Yin, J., Zhang, D., Ma, H. (2009) Evolution of the RNA-dependent RNA polymerase (RdRP) genes: Duplications and possible losses before and after the divergence of major eukaryotic groups. Gene 447 (1), 29–39
Acknowledgements
The authors would like to thank Isabel Alves-dos-Santos and Guraci D. Cordeiro for the support during bee collection, to Susy Coelho for technical assistance, and to the reviewers and editor for their contributions to the final version of this manuscript. Financial support was provided by FAPESP (São Paulo Research Foundation, process numbers 2013/12530-4 and 2012/18531-0). PKFS received a CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) scholarship. MCA received research fellowship from CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico). This work was developed in the Research Centre on Biodiversity and Computing (BioComp) of the Universidade de São Paulo (USP), supported by the USP Provost’s Office for Research. Part of the bioinformatics analyses was performed using the cloud computing service provided by USP.
Author information
Authors and Affiliations
Contributions
NSA participated in the design and interpretation of the data; NSA and PKFS performed experiments and analysis; NSA and MCA wrote the paper and participated in the revisions of it. All authors read and approved the final manuscript.
Corresponding author
Additional information
Manuscript editor: Klaus Hartfelder
Le séquençage de l’ARN révèle que les gènes mitochondriaux et les ARN longs non-codants peuvent jouer des rôles importants dans les générations bivoltines de l’abeille néotropicale non sociale, Tetrapedia diversipes
transcriptome / bivoltinisme / diapause / abeille solitaire / ARN-Seq
RNA-Seq Ergebnisse zeigen, dass mitochondriale Gene und lange, nichtkodierende RNAs eine wichtige Rolle spielen in der Abfolge der bivoltinen Generationen der nicht sozialen neotropishcen Biene Tetrapedia diversipes
Transkriptom / Bivoltinismus / Diapause / solitäre Biene / RNA-Seq
Electronic supplementary material
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Araujo, N.S., Santos, P.K.F. & Arias, M.C. RNA-Seq reveals that mitochondrial genes and long non-coding RNAs may play important roles in the bivoltine generations of the non-social Neotropical bee Tetrapedia diversipes . Apidologie 49, 3–12 (2018). https://doi.org/10.1007/s13592-017-0542-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13592-017-0542-2