
Abstract
Proteins physiologically exist as “proteoforms” that arise from one gene and acquire additional function by post-translational modifications (PTM). When multiple PTMs coexist on single protein molecules, top-down proteomics becomes the only feasible method of characterization; however, most top-down methods have limited quantitative capacity and insufficient throughput to truly address proteoform biology. Here we demonstrate that top-down proteomics can be quantitative, reproducible, sensitive, and high throughput. The proteoforms of histone H4 are well studied both as a challenging proteoform identification problem and due to their essential role in the regulation of all eukaryotic DNA-templated processes. Much of histone H4’s function is obfuscated from prevailing methods due to combinatorial mechanisms. Starting from cells or tissues, after an optimized protein purification process, the H4 proteoforms are physically separated by on-line C3 chromatography, narrowly isolated in MS1 and sequenced with ETD fragmentation. We achieve more than 30 replicates from a single 35-mm tissue culture dish by loading 55 ng of H4 on column. Parallelization and automation yield a sustained throughput of 12 replicates per day. We achieve reproducible quantitation (average biological Pearson correlations of 0.89) of hundreds of proteoforms (about 200–300) over almost six orders of magnitude and an estimated LLoQ of 0.001% abundance. We demonstrate the capacity of the method to precisely measure well-established changes with sodium butyrate treatment of SUM159 cells. We show that the data produced by a quantitative top-down method can be amenable to parametric statistical comparisons and is capable of delineating relevant biological changes at the full proteoform level.

Similar content being viewed by others


Introduction
Top-down proteomics is capable of capturing full proteoform information; however, previous approaches have mostly been non-quantitative, semi-quantitative, or have suffered from moderate to poor reproducibility. High sample requirements and low throughput have mostly limited the scope of top-down proteomics to technological demonstrations on limited biological questions with a few data points [1]. Recent efforts in top-down proteomics have largely focused on identification of proteoforms, with only a few recent studies being quantitative [2]. This seemingly easier problem of identification is also replete with substantial largely unsolved challenges. A recent inter-laboratory comparison of the identification of histone H4 proteoforms from a single pooled sample showed relatively low concordance and wide variation in the number of proteoforms identified; however, a high similarity in the proteoforms identified, with minor differences in the exact construction of the major proteoforms [3]. The variation in the number of proteoforms was largely a function of the methods used and the effort applied. As the number of observations increased, the concordance improved despite the presence of minor errors. It is fallacious to expect the elimination of error in either identity or quantity. In fact, a core foundation of science is the measurement of error. Without error, we do not understand precision. The gold standard for proteoform identity remains a single annotated spectrum (as we provide in supplemental data). This dramatically improves the reliability of such confounding identifications; however, it is nonetheless anecdotal evidence and is not immune to anecdotal false identification. Reproducible observation, reproducible quantitation, functional changes in abundance, informed biochemical manipulation, and the positing and confirmation of proteoform-level hypotheses provide much stronger validation of proteoform biology. Thus, quantitative top-down proteomics promises not only deeper functional insights into proteoform biology but also complementary support for the identification of proteoforms because bona fide proteoforms should also be reproducibly observed, yield low quantitative variance, and respond to relevant biological cues.
We demonstrate a complete top-down HPLC-MS/MS method, starting from cells or tissue to analyze histone H4 proteoforms that is robust, high-throughput, reproducible, and exceptionally quantitative using reversed-phased techniques and electron transfer dissociation on a commercially available Orbitrap Fusion Lumos instrument. To our knowledge, the quantitative precision exceeds previous efforts at all levels of analysis: from the full proteoform to binary combinations to the fully marginalized discrete PTM level. With a sustained throughput of weeks of continuous operation and almost no consumable cost, this method is economically competitive with western blot analysis of histone H4 PTMs. An estimate of the true cost of western blot approaches is difficult, especially when accounting for salaries and overhead. However, commercial services cost about $25/lane ($200/8 lanes), including salaries, overhead, and most materials (excluding the primary antibody and the prepared samples). The cost to perform western blots plus salary and overhead can reasonably be estimated to be 20–50% of this in an exceptionally efficiently run academic laboratory, giving a $5–$12 per lane estimate. Our comprehensive costs are approximately 47 cents per proteoform and $8 per discrete PTM. Thus, despite the steep capital investment required, our approach is comparable in net cost per analysis, while also providing much deeper molecular characterization and greatly enhanced quantitation. Cost and sustained throughput are rarely addressed in top-down proteomics, despite being crucial metrics of utility that should be addressed, especially as they approach or exceed prevailing technologies. The method far exceeds western blots in precision while unbiasedly quantitating individual PTMs, allowing for quantitative cross-comparison between relative abundances of PTMs and of course revealing the rich single-molecule combinations of PTMs inherent to proteoform-level measurements that are otherwise inaccessible.
Most chromatin studies focused on histone PTMs have used antibodies for quantification [4]. Antibodies can suffer from epitope occlusion and often have only moderate specificity for a specific histone PTM. There are three general mass spectrometry-based approaches for the analysis of histones: bottom-up, middle-down, and top-down. The development of bottom-up mass spectrometry methods for the quantitation of histone PTMs has contributed greatly to our understanding of histones and the chromatin-based regulation eukaryotic gene transcription [5]. PTMs affect the ionization efficiency of both peptides and proteins by modulating the chemical characteristics of the molecule; however, the relative effect on the net ionization efficiency of molecules is approximately inversely proportional to the size of the molecule or to the total number of ionizable residues [6]. Thus, bottom-up methods are inherently more prone to ionization bias, and top-down methods may be more accurate in their quantitation of PTMs. Bottom-up approaches have a sensitivity advantage by disposing of most combinations by proteolysis; as with all enzymatic digestions, losses are also incurred during processing. Most importantly, both bottom-up and middle-down approaches critically suffer from the loss of connectivity between at least some of the PTMs on the same molecule. Middle-down approaches have successfully quantitated histone H4 PTMs while capturing many combinations of PTMs [7,8,9]. Middle-down approaches most often do not fully characterize proteoforms: in particular, almost all efforts do not include K31ac. They have also been most effectively performed with advanced and challenging separation techniques that offer unique selectivity but can also lead to decreased reproducibility and throughput. Additionally, the digestion and re-purification steps require additional time, labor, and cost as well as decreasing sensitivity due to substantial losses during these steps.
In this work, we focus on the quantitative capacity of top-down proteomics generally and the efficacy of the method presented specifically; however, science enabled by these approaches is furthering a mechanistic understanding of the regulation of eukaryotic transcription. Briefly, the physiological state of the human genome is chromatin, a nucleo-protein complex which is approximately 50% protein. A histone octamer and approximately 147 base pairs of DNA form the basic subunit of chromatin: the nucleosome [10, 11]. Each histone octamer is composed of two copies of H2A, H2B, H3, and H4 [12]. These histone proteins have highly basic tail domains that interact with multiple regulatory proteins [13]. Histone tails can be post-translationally modified at multiple residues and in a staggering number of combinatorial patterns, each defining a unique proteoform with the potential to transduce specific information [14, 15]. In addition to modifications, several variants of histones H2A, H2B, and H3 exist [16, 17]. Many of these have unique vital functions such as DNA damage response and development [18, 19]. Histone modifications on specific variants play unique roles in these processes [20]. The diversity, malleability, and capacity to interact with multiple regulatory proteins have firmly established the role of histones as key regulators of transcription and genome maintenance. The post-translational modifications (PTM) added to histone tails regulate transcription primarily by being “read” by other proteins [21]. Methylation (me), acetylation (ac), and phosphorylation (ph) are common PTMs found on histone H4 tails [22, 23]. These modifications are thought to either increase transcription, such as H4K16ac, or inhibit transcription and promote heterochromatin, such as H4K20me3 [24,25,26]. Acetylations are “written” by histone acetyl transferases (HATs) and removed by histone deacetylases (HDACs) [27, 28]. Methylation and phosphorylation are similarly written and erased [29]. PTMs are recognized by proteins containing specific domains, such as chromodomains for methylation and bromodomains for acetylation [30]. Many of these “reader” proteins contain tandem or multiple PTM recognition domains, enabling the direct recognition of combinatorial patterns of PTMs. Proteins that primarily site-specifically recognize only one modification, interact with the entire tail region and the full set of modifications present that can modulate such events through many other mechanisms, such as epitope occlusion. There is significant synergy between modifications, and between histones of the same nucleosome or proximal nucleosomes [31]. To fully understand, this complex and dynamic environment full proteoform characterization and quantitation is required.
We have previously used precursor methods to this one to investigate chromatin biochemical mechanisms [32, 33]. We have also shown quantitation of middle-down H4 [9]. Here we aim to demonstrate the capacity of our mature method to precisely and reproducibly measure change in the abundances of proteoforms. We use SUM159 cells, a triple-negative breast cancer cell line, and measure the difference between asynchronous basal conditions and butyrate treatment [34, 35]. Sodium butyrate as an HDAC inhibitor that inhibits class I, class IIa, and class IV HDACs [36]. Butyrate has been used as an anti-cancer treatment and can kill cells in tissue culture at sufficient doses and time [37,38,39]. In 2 h, butyrate can sensitize cells to additional treatments, while longer treatments kill cells by oxidative stress [40, 41]. The effects of the treatment of cells with sodium butyrate on histones have been studied previously and thus serve to validate the method we present here on a well-characterized cellular perturbation. However, the results of our analysis substantially exceed previous efforts in both dynamic range and quantitative confidence and enable new insights into the proteoforms that change in response to biological perturbations.
Experimental Procedures
Cell Lines
Sum 159 cells were cultured with F-12 Nutrient Media (Life Technologies), with 10% newborn calf serum, 100 U/mL penicillin and streptomycin, 10 mM HEPES, and 1 μg/mL hydrocortisone. Cells were passaged at 80% confluency and maintained at 37 °C and 5% CO2. Sum159 cells were treated with 5 mM sodium butyrate for 4 h. Cells were harvested by scraping and centrifugation at 500 RCF for 3 min, aspirating the supernatant media and washing with 4 °C PBS twice. For this study, one 10-cm tissue culture dish was used per biological replicate, roughly 7.5 million cells.
High-Throughput Histone Purification
Histones were purified according to previous work [32, 33]. Briefly, cells were lysed and nuclei isolated with 4 °C NIB (15 mM Tris-HCL (pH 7.5), 60 mM KCl, 15 mM NaCl, 5 mM MgCl, 21 mM CaCl2, 250 mM sucrose, 1 mM DTT, 0.5 mM AEBSF, 5 nM microcystin, 10 mM sodium butyrate, 0.3% NP-40 alternative) for 5 min. Nuclei were centrifuged at 600 RCF for 5 min and washed with detergent-free NIB. Non-histone proteins were precipitated with 0.4 N H2SO4 for 2 h at 4 °C. To the supernatant, a saturated solution of TCA equal to 25% of the starting was added and incubated for 45 min at 4 °C after which precipitated histones were centrifuged for 5 min at 30,000 RCF.
Off-line HPLC Histone Purification
Histones were dissolved in buffer A (5% acetonitrile, 0.2% trifluoroacetic acid) and loaded on a C-18 column (HiCHROM 218TP54: Vydac 218TP 5 μm C18, 250 × 4.6 mm) pre-equilibrated at 30% buffer B (95% acetonitrile, 0.188% trifluoroacetic acid). Histones were separated by a linear gradient from 30%B to 60%B over 100 min. Fractions were collected every minute and H4-containing fractions were combined and evaporated to dryness in a speedvac (Thermo Scientific, model SPD131DDA) without added heat. Off-line separation is recommended given that histone H3 is not feasibly analyzed by top-down methods, and that H2A can interfere with H4 analysis.
LC-MS/MS Analysis
A Thermo Scientific DIONEX UltiMATE 3000 was used for on-line liquid chromatography with buffer A (98% H2O, 2% ACN, 0.1% FA) and buffer B (2% H2O, 98% ACN, 0.1% FA). A linear gradient from 27%B to 30%B over 70 min at 200 nL/min was used to separate proteoforms of histone H4. A lab-made column of 15 cm, 100 μm ID and ZORBAX C3-300SB, 3.5 μm, and 300-Å pore beads was used. Histone H4 samples were dissolved in buffer A, and 1 μL of sample was injected per experiment; this results in around 55 ng of histone H4 loaded onto the column. For each analysis, we inject 55 ng on column (based on an external UV-HPLC calibration curve at 214 nm of pure recombinant histone H4, analytically weighed, and serially diluted). Ionization was achieved by a Nanospray Flex source with 1700 V in positive ion mode. A Thermo Scientific Orbitrap Fusion Lumos was used for all data acquisition. The front-end ion transfer tube was set to 320 °C and the ion funnel an RF of 80%. The Orbitrap was used for both MS1 and MS2 analyses. For MS1, a scan window of 700–1400 m/z, a resolution setting of 60,000, 5.0e5 AGC, 200-ms maximum injection time, and 5 microscans were used. The resolution is sufficient for isotopic resolution of histone H4 precursors. An H4 proteoform specific targeted mass list was used to pick MS1 peaks (Supp. Table 1). All major H4 peaks are captured by this list. A data-dependent top-20 decision method was used to select precursors for MS2. Isolation was performed by the quadrupole with a 1 m/z isolation window. ETD fragmentation was used with a 14-ms reaction time, 5.0e5 ETD reagent target, and 200-ms maximum ETD reagent injection time. MS2 spectra were acquired with a resolution setting of 60,000, AGC of 5.0e5, 3 microscans, and profile data type.
Data Analysis
Data processing is performed by a custom analysis suite adapted from [42]. Briefly, a 3.4-Da window for precursor mass error constrains the proteoforms considered for each spectrum. This precursor mass tolerance resolves methylation states but intermingles acetylation and trimethylation. We compute isotopic distributions for all theoretical MS2 fragments, for all possible chosen PTM combinations. Spectra are not deconvoluted; instead, for each fragment, the full isotopic distribution of multiple charge states is computed. MS2 spectra are matched to the most abundant isotope in the theoretical spectra within 10-ppm error and checked for an unambiguous isotope pattern. A mixed integer linear optimization model is used to solve the optimal proportions of a simple mixture of a limited number of proteoforms to account for the spectrum. This model maximizes the MS2 area matched by the theoretical fragment ions of multiple proteoforms. Our model has been optimized such that few proteoforms (2–4) are matched to a given scan, and multiple scans have no mixture. In practice, if three proteoforms explain 80% of the fragment peaks, and 2 different proteoforms also explain the same peaks, the two proteoforms will be selected. The proportion of the spectrum that a proteoform represents is multiplied by the precursor ion intensity to compute an incremental proteoform abundance. The final abundance of each proteoform is the integral of incremental proteoform abundance across the entire LC-MS analysis. Values are normalized to total percent of histone H4 and discrete and combinatorial values are obtained through marginalization of proteoform quantities. Proteoforms are denoted by angel brackets “<>,” while marginalized discrete PTMs and binary combinations are denoted by braces “{}.” For example, the relative abundance of {K16ac} is simply the sum of the relative abundances of all proteoforms containing K16ac. The relative abundance of the single-molecule binary combination of K16ac and K20me2, {K16acK20me2}, is simply the sum of the relative abundance of all proteoforms containing both K16ac and K20me2, irrespective of the presence or absence of other PTMs. This is different from the singular proteoform containing only K16ac and K20me2, <K16acK20me2>, which is definitively not modified at all other sites. Statistical analysis and figure generation is achieved by use of R and Python programming languages and packages (matplotlib). Statistical significance is defined as p < 0.05 derived from Welch’s two-tail student’s t test and Pearson correlations are used to determine reproducibility. Multiple hypothesis corrections are done with the FDR method, with q values of 0.10 unless otherwise specified.
Results
Throughput
The method provides sufficient throughput for the biological study of proteoforms. The analysis time per sample is 90 min. With the use of an additional 35-min wash step, the carryover is less than 1%. The minimal elapsed time between the harvesting of cells and the final data results is about 12 h for a single technical replicate. The acid extraction and off-line HPLC purification stage require about 8 h of total time and data analysis requires another 20 min per run. These steps are automated and parallelized, allowing for the analysis of many samples with minimal labor and at low overall cost. The resulting throughput is 12 analyses per day and allows for more than sufficient technical and biological replicates to reach firm and robust biological conclusions from extensive datasets in a few days. At the time of writing, this method and earlier versions of the method have been used for 1826 runs over the past 2 years, nearly always with productive results at the demonstrated throughput. We have used the method for sustained periods and intermittently with many other methods and samples with robust and reproducible results. The majority of the cost of our method is mass spectrometer depreciation. Including time and labor, we approximate that we measure the abundance of a proteoform for about 47 cents and the abundance of a discrete PTM for about 8 dollars. This makes top-down mass spectrometry of histone H4 with this method an economically viable alternative to antibody-based methods, while providing much deeper functional and mechanistic insight.
Sample Requirements
Sample requirements to achieve these results are significantly below minimum feasible levels for most applications. From one 10-cm tissue culture plate, about 5–8 million cells, we obtain sufficient histone H4 for around 150 technical replicates. From one 35-mm tissue culture plate, we achieve about 30 technical replicates. This is approximately 1 million cells. The method is readily adapted to other eukaryotes due to identical or near identical histone H4 sequences. Minor optimization of acid extraction is required for other organisms and typically yields similar sample requirements on a genome-sized normalized basis. These sample requirements greatly enable many experiments, such as cell sorting-based experiments and the analysis of subsections of in vivo organs.
Chromatography
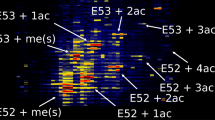
The chromatography implemented here is sufficiently selective toward proteoforms to enable top-down identification of proteoforms and sufficiently reproducible for precise quantitation of proteoforms (Figure 1). Current middle-down methods separate H4 N-terminal tails primarily by acetylation state [7]. We sought to develop a complementary chromatographic approach that resolves full proteoforms by leveraging modifications that are not used in most H4 middle-down approaches. The method fully separates H4 proteoforms that contain M84 oxidation, in accordance with previous results [43], and moderately separates the degree and site of acetylation on the histone tails (Figure 1a). M84ox, which is not present in the 1-23aa peptide used in middle-down analyses, is highly variable and its separation is essential for high reproducibility in top-down quantitation of H4 (Supp. Figure 1). The methylation state of K20, although not well chromatographically separated, is readily separated in the mass spectrometer. This is complementary to HILIC approaches and also allows for effective interrogation of distal PTM relationships over longer distances. There are strong proteoform ordering relationships in the chromatography. Individual proteoforms elute with a near-Gaussian peak shape, enabling precise quantitation. As with all proteoform analyses, not all proteoforms are baseline resolved from each other; however, there is sufficient separation to distinguish and accurately quantitate structural isomers in conjunction with of a mixture model for data analysis.

Overview of the method. (a) Elution profile of histone H4 from reverse phase C3 chromatography. Black lines are technical replicates of the total ion current. Separation is primarily by oxidation on M84 and secondarily by lysine acetylations. Near-Gaussian elution is observed for specific proteoforms (red, blue, yellow, and purple dots). (b) Orbitrap MS1 spectra with a precursor selection window of 750–765 m/z containing the + 15 charge state. A 1 m/z isolation window is shown which is selective for one family of isomeric or isobaric proteoforms. (c) Partial separation of proteoforms by C3 chromatography and mixture model-based data analysis allows for accurate quantitation of isomers. (d) A single MS2 spectra from ETD fragmentation, and ion map with coverage for a full proteoform identifies <Nα-acK8acK16acK20me2>
Chromatographic Reproducibility
High chromatographic reproducibility, multiple technical and biological replicates and low sample requirements are essential to reproducible and robust quantitation of the proteoforms in a biological context. The method has a technical retention time coefficients of variance (CV) of 0.71%, and a biological retention time CV of 8.37% in an abundance and proteoform-dependent manner (Supp. Table 2). Significant changes in the abundance a given proteoform between samples only slightly skews the retention time for that given proteoform. The reproducibility is highly consistent over time. Although the chromatography is relatively robust with respect to loading amounts (10–250 ng of total H4, sub-ng for each proteoform), overloading or underloading the column shifts the chromatography and affects the quantitation as separation between proteoforms is lost (data not shown). Minor shifts in the chromatography preserve the relationship between proteoforms and do not affect relative quantitation. These effects are also observed in other proteoform selective chromatographies, such as WCX-HILIC; however, they are significantly less problematic and more easily controlled here. Although the method is notably robust with respect to sample loading, we typically inject 55 ng on column (based on a UV-HPLC calibration curve at 214 nm) for each analysis for maximum reproducibility.
MS2 Spectra
Both the quality and quantity of MS2 spectra generated in a single run are important for the accurate identification and quantitation of proteoforms. Over our 90-min analysis, we acquire around 1800 MS2 spectra of which around 1300 MS2 spectra are identified and quantified. This is performed by isolating a 1 m/z precursor mass window, which results in spectra that are almost entirely composed of structural isomers or isobaric near structural isomers (Figure 1b). On average, each MS2 contains 30 matched ions, with more intense precursor ions resulting in larger numbers of MS2 matched ions (up to 260 ions matched) (Figure 1d). We require at least 10 matched ions for an identification. Typically, this allows for the ready localization of modifications among the first 20+ amino acids of H4. Although sometimes inferred for weaker proteoforms, we have sufficient sequence depth to directly localize K31ac in multiple scans. Multiple MS2s are acquired of each precursor across each chromatographic peak. We do not use any dynamic exclusion to maximize identification information of moderately resolved and less abundant proteoforms and achieve consistent cadence and extensive sampling of chromatographic peaks for quantitation. The method uses a resolution setting of 60,000 which is sufficient to isotopically separate all fragment ions while not significantly reducing sampling rate. The high sampling rate yields better resolution of isobaric proteoforms that are distinguished by only a few MS2 ions and only slightly time resolved (Figure 1c). For each peak, we will detect some proteoforms multiple times in excess of the needs for identification but also minor proteoforms that may otherwise be missed. The abundant spectra and the near constant sampling cadence are essential for high quantitative reproducibility.
Number of Proteoforms Quantitated
The combination of our chromatography, mass spectrometry, and data analysis allows for the identification and reproducible quantitation of hundreds of proteoforms per analysis. We preliminarily identify over 600 proteoforms without filtering with all runs combined. A large portion of these proteoforms have insignificant abundances and are also of limited identity confidence. Thus, analogous to standard practices in bottom-up proteomics, we filter our data for proteoforms that reproducibly appear on multiple technical (2/3) and biological replicates (2/4). This results in an average of 226 (control) and 338 (butyrate) proteoforms per run (minimum of 211 and a maximum of 345). Although correlated with spectral quality metrics, the reproducible observation of proteoforms provides a complementary line of evidence and this filter removes spurious identifications of proteoforms that appear superficially accurate at the single-spectrum level. The number of reliably identified proteoforms increases in butyrate-treated samples to an average of 338 proteoforms due to a greater diversity of acetylation state.
Technical Proteoform Quantitation
The method exhibits high technical reproducibility for the quantitation of proteoforms. Absolute quantitation is achievable through our highly reproducible off-line separation and quantitation. We have generated a standard curve using purified H4 that is linear (R2 = 0.99) from at least 1.375 to 22 ug (Sup. Figure 2). This allows us to accurately and consistently load precise amounts (55 ng per replicate) for our final chromatography and determine absolute proteoform amounts detected. We run each biological sample in interleaved technical triplicates. Replicates are thus analyzed distantly from each other to mitigate order bias. We observe technical replicates with a CV of 8% for the abundant proteoforms (between 35 and 1% abundance) (Sup. Figure 3 A). Average variance in the measured abundance of proteoforms increases as abundance decreases, reaching CVs of 42% for proteoforms of 0.1% abundance. Proteoforms that are 0.01 to 0.001% abundant have a large range of CVs and an average approaching 100% CV (i.e., mean = standard deviation) and are considered on average weakly quantitated. However, not all proteoforms within this range exhibit high variance. Most importantly, we rigorously test the statistical value of this data below and these proteoforms are not more likely to present a false-positive change in abundance than more abundant proteoforms. Type 1 errors (false-positive changes in abundance) are not abundance dependent. Within this range of low abundances and high CVs, we do not expect to detect vanishingly small absolute changes in quantity; however, we readily observe increases greater than twofold from these reproducibly measured low levels and are able to apply rigorous two-tailed statistics with confidence. When proteoforms increase in abundance, they become easier to detect and are more reproducible relative to their abundance, although the absolute size of the error bars concomitantly increase.
Biological Proteoform Quantitation
The biological reproducibility is comparable with the technical reproducibility of the method. In this study, 4 biological replicates for each condition were used for SUM159 cells along with 3 technical replicates. We achieve an average Pearson R value of 0.92 for technical replicates of filtered data (data points that have quantities on all 3 technical replicates). We achieve exceptional reproducibility with Pearson correlation values of 0.89 for control samples and 0.86 for butyrate-treated samples for biological replicates when the data is filtered by removing low-abundance and non-reproducible proteoforms (a proteoform must be found in 2/3 technical and 2/4 biological runs) (Figure 2). Further, our overall data structure is normally distributed when log normalized which is suitable for metrics such as Pearson correlation. We observe the same unbiased, linear quantitation with biological replicates as with technical replicates. The biological variance is substantially greater than our technical variance (Sup. Figure 3 B). This has been consistently observed across multiple projects and sample types (e.g., various cell types, organisms, organs, treatments) over the past 2 years with progenitor versions of this method. Thus, the precision of the method exceeds the inherent biological variance and is more than sufficient to enable confident statistical comparisons between different biological conditions. Biological variance typically differs between experimental conditions and over time in predictable ways. For example, measurements taken on cells after perturbation exhibit greater variance than steady-state samples. This is likely reflective of bona fide dispersion of cellular physiology within the sample.

Correlation plot of full proteoforms. Correlation matrix of full proteoforms from four biological (Ctrl_1, Ctrl_2 …) and four sets of three technical replicates (Ctrl_1_1, Ctrl_1_2 …) of SUM159 control cell lines with log10 normalized percent abundances. Graphs below the diagonal are scatter plots of filtered data. On the diagonal are histograms with density curves for each technical replicate exhibiting normal-like distributions for all proteoforms. Top boxes above the diagonal contain Pearson correlation values. Average Pearson R = 0.94 for technical and average Pearson R = 0.86 for biological replicates
Statistical Assumptions
Proteoform quantities are appropriate for statistical analysis. Commonly used parametrical tests for determining statistical significance require specific assumptions to be true: normally distributed data, independent variables, and sufficient replicates to establish a distribution. Histone H4 proteoforms are normally distributed. While normality is challenging to establish, it is possible to show that proteoforms are likely normally distributed to a point that enables parametric tests. Scaled top 20 proteoforms measured in the control samples (240 measurements) achieve a near-normal distribution (Sup. Figure 4 A). When observing all proteoforms, the majority of them qualitatively approach a normal distribution (Sup. Figure 4 B and C). Overall, we find that proteoforms are not only reproducibly detected and quantitated, they are also distributed normally and thus standard parametric tests are readily applicable.
Clustering
Unsupervised hierarchical clustering readily delineates technical and biological replicates as well as butyrate treatment (Figure 3). All technical replicates clustered together and the major separation between the samples is the addition of butyrate. Lowly acetylated proteoforms are more abundant in the control samples as observed in cluster 1. Highly acetylated proteoforms are increased in butyrate-treated samples as observed in clusters 3 and 4. These clusters include proteoforms that were not identified in the control samples. All proteoforms observed in the control samples were observed in the combined butyrate samples. A large proportion of proteoforms are significantly changed by butyrate treatment (Sup. Figure 5). These results are in agreement with previous butyrate data: the inhibitor acts broadly and the majority of proteoforms are affected (Figure 4).

Heatmap of full proteoforms. Log10 normalized full proteoform filtered data was clustered with Euclidean distance and average linkage. On the right hand side, the presence of N-terminal tail acetylations are indicated (Red: K5ac, Blue: K8ac, Orange: K12ac, Green: K16ac). Technical replicates cluster together without exception. Biological replicates also perfectly cluster together. K-means clustering of proteoforms separates proteoforms into different groups depending on the change in abundance of proteoforms upon butyrate treatment. The biggest source of variance is the butyrate treatment. Cluster 1 is enriched in the hypo-acetylated proteoforms that are abundant in the control samples. Cluster 2 contains mostly low-abundance proteoforms that are not substantially changed by butyrate treatment. Clusters 3 and 4 contain the, mostly hyper-acetylated, proteoforms that greatly increase upon butyrate treatment. Many of these were not observed in the control group. Cluster 5 contains proteoforms that are not significantly different between the two

Discrete acetylations increase upon butyrate treatment. Butyrate treatment increases acetylations on H4 at the discrete PTM level. Acetylation is significantly increased on K5, K8, K12, K16, and K31 after 4 h of treatment (p < 0.001, K31ac p < 0.01). S1 phosphorylation and R3 methylation are also significantly increased (p < 0.01). K20 methylations and M84 oxidation are unchanged by butyrate treatment
Butyrate Treatment
Hyper-acetylated proteoforms are significantly increased, while hypo-acetylated proteoforms are decreased (Figure 5). Upon butyrate treatment, multiple proteoform level changes are observed: the total number of identified proteoforms increase in the butyrate-treated samples increase by 79 proteoforms and acetylated proteoforms increase in a highly reproducible and consistent way, while hypo-acetylated proteoforms decrease in abundance. The 79 proteoforms that are found exclusively in the butyrate-treated samples all contain 2 or more acetylations. By fold change, the motif K5acK8acK12acK16ac is the most increased (Supp. Table 3). This represents a fully acetylated N-terminal tail. Multiple other acetylation combinations are observed increasing in response to butyrate, establishing the method as capable of quantitating proteoforms that respond to biological perturbations.

Volcano plot of proteoform differences between control- and butyrate-treated SUM159. Several acetylated proteoforms (red) are increased over 10-fold. Conversely, the primary substrate for acetylation <Nα-acK20me2> is decreased slightly in fold change along with other unacetylated proteoforms (black). The grey box contains proteoforms that are only identified in the butyrate samples. The black horizontal dashed line represents a p value of 0.05, the blue line an FDR-corrected q value of 0.10, and the red line a q value of 0.05. Vertical dashed lines are 2-fold change
Marginalized PTMs
We obtain quantitative information for marginalized PTMs with accuracies comparable with those of bottom-up and middle-down methods [43,44,45]. At the discrete modification level, we achieve CVs of 8.10% on average (CV of 5.62% excluding R39 sulfate and M84ox) with a low of 0.23%, and an average standard error of 0.008 for our control- and butyrate-treated cells (Figure 4). Our statistical power easily detects the expected increases in acetylation from butyrate treatment and does so with previously unachieved quantitative precision and depth. There is a significant increase of acetylation on K5, K8, K12, and K16 of H4 (p < 0.001). To a much lesser extent, K31ac is increased (p < 0.01), suggesting an alternative regulatory mechanism for K31ac. This differential behavior of K31ac has not been previously observed, but few studies have aimed to quantitatively study K31ac much less its functional significance or regulation. Additionally, K31 is located in a relatively structurally inaccessible part of the nucleosome structure, suggesting that it may be less prone to rapid change. The sensitivity is such that it is possible to detect sub 2% changes (< 1.02-fold) for the most reproducible PTMs.
Low-Abundance PTMs
The sensitivity of the method is sufficient to study low-abundance PTMs. R3 monomethylation is a low-abundance PTM that, according to our data, is significantly increased by butyrate treatment, likely due to downstream effects, or misidentifications. R3me1 exists at around 0.27% of total histone H4 at basal conditions in SUM159 cells. Butyrate treatment increases R3me1 methylation by 1.7-fold to 0.46%. This is a relatively small difference caused by butyrate treatment on a fold change basis and absolute change basis. S1 phosphorylation is also significantly increased by 1.3-fold, from 0.69 to 0.88%. Both of these PTMs are very low in abundance, and their differences are minor between groups, yet the method quantitates the abundance of these PTMs with a precision sufficient to detect these clearly indirect and downstream changes.
Non-Enzymatic PTMs
The method separates, localizes, and quantitates M84 oxidation and a sulfate adduct. M84 oxidation is localized by several fragment ions unambiguously. At the technical level, M84 oxidation abundance is highly reproducible with an average CV of 4.8%. Additionally, chromatographic separation and abundance for M84ox is highly reproducible (RSD of ~ 0.2 min, AUC CV of 11.2%). At the biological level, M84ox has substantial variance: on average, it has an abundance of 24.34% for control, but with a standard deviation of 6.48 percentage points (CV of 26% for the control, and 36% for the butyrate treated). Given that the technical reproducibility of M84ox is significantly better than the biological reducibility, and the biological variance of M84ox is extremely high, and that M84ox increases with sample exposure to air and time, the variation is most likely due to processing artifacts prior to HPLC-MS/MS. We do not observe any proteoform bias for M84ox and find similar number of ox proteoforms as non-oxidized. Similarly, we often detect what appears to be a sulfate adduct, which we consider when searching for PTMs. In general, adducts and non-enzymatic PTMs can obfuscate the true signal unless they are properly identified and addressed. While it is likely that other adducts and modifications exist, we find no evidence for additional adducts, by exhaustive automated and manual searching.
Novel PTMs
Enabled by chromatography, the method detects previously unreported modifications on histone H4. Two early eluting peaks contain approximate H4 sized precursors that had similar MS1 patterns as the predominant species of H4 (Supp. Figure 6 A). Manual analysis has determined the first peak (Supp. Figure 6 B and D) to have a − 1.027-Da mass shift. This corresponds closest to conversion of lysine to allysine (< 10 ppm). However, the precursor signal intensity is weak, and the modification is in a poorly fragmented area of the protein. Thus, proper identification and confident localization was not achieved. The second peak, which eluted later, contained over 10× the signal by AUC, which enabled the identification and localization of aspartic acid 68 conversion to succinimide (− 18 Da, < 10 ppm) (Supp. Figure 6 C and E). These modifications are difficult to observe without good chromatographic separation: both off-line and on-line separations are necessary to effectively capture these minor H4 peaks. In this study, they are not reproducibly found at the biological level. The biological significance of these modifications has not been established, and it is possible they are both artifacts of processing; however, the method establishes the capacity to test this. Identification of these potential PTMs is impossible if only the N-terminal tail of H4 is targeted.
Discussion
Here we present a method for the quantitative top-down analysis of histone H4. The method is enabled by innovations in chromatography, mass spectrometry, and data analysis: extremely shallow and reproducible gradients, fast and efficient ETD fragmentation and high-resolution spectra, and mixture model optimization analysis. We demonstrate the capabilities of this method by analyzing SUM159 cells, a triple-negative breast cancer cell line, and the effect of 4 h of sodium butyrate treatment.
Understanding the number and abundance of proteoforms is important, especially in systems such as chromatin, which fundamentally operates at the proteoform level. This method is the first to present quantitative full proteoform information in a high-throughput manner for histone H4. To achieve this, we sought to develop a method that separated proteoforms that were not characterized by middle-down approaches. Specifically, we targeted K31 acetylation and M84 oxidation. We achieve near-baseline separation of M84 oxidation from other proteoforms. This is important even when M84ox is not of interest because these proteoforms are potentially confounding isomers or isobars of other proteoforms. Further, M84ox comprises about 47% of the total number of proteoforms. Thus, chromatographic separation of M84ox proteoforms allows for more accurate quantitation and identification of all proteoforms. More importantly to the value of a true top-down method, K31ac is found in 45% of proteoforms and likely has important biological functions.
The quality of identification and precision of quantitation of proteoforms is inherently linked to abundance; thus, the relative abundance of high confidence proteoforms is readily determined and low-abundance proteoforms are more prone to error. An abundant proteoform will typically be identified with spectra that unambiguously localize all PTMs present, but due to the extensive overlap of fragment ions between closely related proteoforms, one cannot conclude that all signal from an MS2 spectrum exclusively represents the identified proteoform. We address this issue with a mixture model that uses competing ions to distribute non-competing fragment ions signal proportionally. This more accurately determines the contributions of proteoforms to mixed spectra. While clearly more reproducible and accurate, it is not devoid of error in quantity or identity. For this study, as is the currently accepted standard, the metric of identity is manual validation. However, a key principle of our work is that absolute discrimination of proteoforms is not a hard prerequisite when studying biological processes given sufficient and reproducible sampling. We reach this consistency by having multiple MS2 for the same precursor, 3 technical replicates, and 4 biological replicates. Changes in abundance of proteoforms are the paramount metric we aim to measure. To this end, it does not matter if we perfectly identify and quantitate every low-abundance proteoform-containing spectrum. As long as our quantitation is reproducible, we can detect changes in proteoform abundance from a minor, difficult to accurately measure, proteoform to a moderately abundant more confidently identified proteoform, or vice versa. Our capacity to be quantitative hinges on our sampling rate where each precursor is fragmented and identified multiple times, as well as separating proteoforms such that we are not constrained by instrument duty cycle. This approach requires the method to have consistent cadence. Intact proteins, while being considerably more challenging to chromatographically separate and analyze, are relatively more robust against ionization efficiency differences induced by PTMs. After butyrate treatment, we manually validate the identity and precursor abundance for specific proteoforms that significantly changed due to the treatment. Manual interpretation of an annotated spectra remains the gold standard for top-down identification of proteoforms; however, this is fundamentally anecdotal and we find that it is also not without susceptibility to false identifications. Nonetheless, our manual interpretation validates the conclusions generated by our data analysis pipeline. Thus, the method reproducibly quantitates at the proteoform level.
The abundance of M84 oxidation and R39 sulfate adducts are precisely quantitated on the technical level but exhibit large variance on the biological level. All other modifications are highly reproducible on both technical and the biological level. It may be that the abundances we are detecting are accurate and M84 has high biological variance or is linked to a biological pathway that we are not able to control. We observe changes in M84ox with increased time and exposure to air, concluding that at least part of the signal is due to non-enzymatic reactions. Failure to include R39 sulfate adduct results in increased variance and altered proportions in K31ac, and K20 methylations. The sulfate adduct is largely due to the use of sulfuric acid in the acid extraction process. While this issue is easily addressed by replacing sulfuric acid with hydrochloric acid, it has been the standard histone precipitation method for decades. With our chromatography, multiple-MS2 scans and mixture model, we are able to account for this adduct and achieve accurate quantitation of PTMs.
With the addition of K31ac, M84ox, and the R39 sulfate adduct, our proteoform space is considerably larger than any previous study and we identify and quantitate around 350 proteoforms per run. Removing M84ox and R39 sulfate adduct, we regularly quantitate around 160 proteoforms. The previous consortium for top-down proteomics analysis of histone H4 resulted in 74 proteoforms identified by the seven laboratories, 21 of which are without ambiguity [3]. With the inclusion of D68 to succinimide, and allysine in the C-terminal tail, we estimate around ~ 600 proteoforms possible, with around 200 being real, biologically driven (enzymatically added). To our knowledge, there are no available quantitative estimates of histone H4 full proteoforms. Previous middle-down efforts have found 70 proteoforms from a 1–23 amino acid fragment, with 1 μg loading onto a column, a 2 h HILIC gradient [7]. The proteoforms observed are consistent with previous studies. We observe 81% (58/72) of the proteoforms observed by Phanstiel [44]. Compared with Young [7], we also observe 81% (42/52) proteoforms identified in each study. These concordant proteoforms are typically abundant in our analysis. It should be noted that both these studies used different cell lines and different butyrate treatments and that we exclude N-terminally unacetylated proteoforms when making this comparison. While quantitative abundances are presented, to our knowledge, there is no study that has multiple replicates and thus, no study provides any statistics for precision and reproducibility making these comparisons indirect. Despite this, we expect much greater sensitivity from a peptide, and our results have ~ 20× higher sensitivity with an intact protein (from 70 H4 proteoforms with 1 μg to 160 with 55 ng). By having reproducible chromatography, high sampling cadence, fewer ionization effects, and mixture model-based identification and quantitation, the method is competitive with bottom-up [45].
A significant portion of these proteoforms have low abundances and therefore high variance, yet are reproducibly detected across multiple replicates. Approximate abundance metrics for these proteoforms are still valuable. Of these proteoforms, many may increase drastically in certain conditions, such as after butyrate treatment. With CVs of around 100%, proteoforms must change drastically in abundance to be considered statistically meaningful; however, this change is approximately the standard 2-fold increase arbitrarily used in many studies. We have found that filtering proteoforms for those that appear in more than half of biological replicates greatly improves consistent quantitation. While undoubtedly our false-negative rate is higher with this approach, we are still able to observe subtle changes.
The method presented is capable of relative quantitation of full proteoforms and is robust and high throughput. The use of this method for probing chromatin dynamics and interactions will allow for an unprecedented understanding of molecular mechanisms.
Conclusion
Current prevalent quantitative proteomic approaches fail to capture proteoforms. There is growing evidence that proteoform-level information is vital in understanding molecular functions and mechanisms. In particular, chromatin biology is heavily dependent on proteoform biochemistry. Here we have presented the first quantitative high-throughput top-down histone H4 method that successfully addresses this issue. The method and results presented have multiple degrees of advancements on previous approaches in regard to sample requirements, processing time, reproducibility, and precision in quantitation. We show the capabilities of this method to analyze triple-negative breast cancer cells and the effect of HDAC inhibition on H4 proteoforms.
References
Toby, T.K., Fornelli, L., Kelleher, N.L.: Progress in top-down proteomics and the analysis of proteoforms. Annu Rev Anal Chem (Palo Alto, Calif). 9, 499–519 (2016)
Ntai, I., Toby, T.K., LeDuc, R.D., Kelleher, N.L.: A method for label-free, differential top-down proteomics. Methods Mol. Biol. 1410, 121–133 (2016)
Dang, X., Scotcher, J., Wu, S., Chu, R.K., Tolić, N., Ntai, I., Thomas, P.M., Fellers, R.T., Early, B.P., Zheng, Y., et al.: The first pilot project of the consortium for top-down proteomics: a status report. Proteomics. 14, 1130–1140 (2014)
Fraga, M.F., Ballestar, E., Villar-Garea, A., Boix-Chornet, M., Espada, J., Schotta, G., Bonaldi, T., Haydon, C., Ropero, S., Petrie, K., et al.: Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 37, 391–400 (2005)
Sidoli, S., Bhanu, N.V., Karch, K.R., Wang, X., Garcia, B.A.: Complete workflow for analysis of histone post-translational modifications using bottom-up mass spectrometry: from histone extraction to data analysis. JoVE J. Vis. Exp. 10, 3791–54112 (2016)
Gao, Y., Wang, Y.: A method to determine the ionization efficiency change of peptides caused by phosphorylation. J. Am. Soc. Mass Spectrom. 18, 1973–1976 (2007)
Young, N.L., et al.: High throughput characterization of combinatorial histone codes. Mol. Cell. Proteomics. 8, 2266–2284 (2009)
Jiang, T., Hoover, M.E., Holt, M.V., Freitas, M.A., Marshall, A.G., Young, N.L.: Middle-down characterization of the cell cycle dependence of histone H4 posttranslational modifications and proteoforms. PROTEOMICS. 18, 1700442 (2018)
Holt, M.V., Wang, T., Young, N.L.: One pot quantitative top-and middle-down analysis of GluC digested histone H4. J. Am. Soc. Mass Spectrom. (2019). https://doi.org/10.1007/s13361-019-02219-1
Luger, K., Mäder, A.W., Richmond, R.K., Sargent, D.F., Richmond, T.J.: Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 389, 251–260 (1997)
Kornberg, R.D.: Chromatin structure: a repeating unit of histones and DNA. Science. 184, 868–871 (1974)
Chakravarthy, S., Park, Y.-J., Chodaparambil, J., Edayathumangalam, R.S., Luger, K.: Structure and dynamic properties of nucleosome core particles. FEBS Lett. 579, 895–898 (2005)
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M.L., Rehman, M., Walther, T.C., Olsen, J.V., Mann, M.: Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 325, 834–840 (2009)
Kouzarides, T.: Chromatin modifications and their function. Cell. 128, 693–705 (2007)
Jenuwein, T., Allis, C.D.: Translating the histone code. Science. 293, 1074–1080 (2001)
Vardabasso, C., Hasson, D., Ratnakumar, K., Chung, C.-Y., Duarte, L.F., Bernstein, E.: Histone variants: emerging players in cancer biology. Cell. Mol. Life Sci. 71, 379–404 (2014)
Dang, X., Singh, A., Spetman, B.D., Nolan, K.D., Isaacs, J.S., Dennis, J.H., Dalton, S., Marshall, A.G., Young, N.L.: Label-free relative quantitation of isobaric and isomeric human histone H2A and H2B variants by Fourier transform ion cyclotron resonance top-down MS/MS. J. Proteome Res. 15(9), 3196–203 (2016)
Rogakou, E.P., Pilch, D.R., Orr, A.H., Ivanova, V.S., Bonner, W.M.: DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868 (1998)
Faast, R., Thonglairoam, V., Schulz, T.C., Beall, J., Wells, J.R., Taylor, H., Matthaei, K., Rathjen, P.D., Tremethick, D.J., Lyons, I.: Histone variant H2A.Z is required for early mammalian development. Curr. Biol. 11, 1183–1187 (2001)
Binda, O., Sevilla, A., LeRoy, G., Lemischka, I.R., Garcia, B.A., Richard, S.: SETD6 monomethylates H2AZ on lysine 7 and is required for the maintenance of embryonic stem cell self-renewal. Epigenetics. 8, 177–183 (2013)
Allahverdi, A., Yang, R., Korolev, N., Fan, Y., Davey, C.A., Liu, C.-F., Nordenskiöld, L.: The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Res. 39, 1680–1691 (2011)
Dion, M.F., Altschuler, S.J., Wu, L.F., Rando, O.J.: Genomic characterization reveals a simple histone H4 acetylation code. PNAS. 102, 5501–5506 (2005)
Marushige, K.: Activation of chromatin by acetylation of histone side chains. Proc. Natl. Acad. Sci. U. S. A. 73, 3937–3941 (1976)
O’Neill, L.P., Turner, B.M.: Histone H4 acetylation distinguishes coding regions of the human genome from heterochromatin in a differentiation-dependent but transcription-independent manner. EMBO J. 14, 3946–3957 (1995)
Shogren-Knaak, M., Ishii, H., Sun, J.-M., Pazin, M.J., Davie, J.R., Peterson, C.L.: Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 311, 844–847 (2006)
Strahl, B.D., Allis, C.D.: The language of covalent histone modifications. Nature. 403, 41–45 (2000)
Tan, M., Luo, H., Lee, S., Jin, F., Yang, J.S., Montellier, E., Buchou, T., Cheng, Z., Rousseaux, S., Rajagopal, N., et al.: Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 146, 1016–1028 (2011)
Zhang, Y., Reinberg, D.: Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 15, 2343–2360 (2001)
Zhang, L., Eugeni, E.E., Parthun, M.R., Freitas, M.A.: Identification of novel histone post-translational modifications by peptide mass fingerprinting. Chromosoma. 112, 77–86 (2003)
Zhao, S., Xu, W., Jiang, W., Yu, W., Lin, Y., Zhang, T., Yao, J., Zhou, L., Zeng, Y., Li, H., et al.: Regulation of cellular metabolism by protein lysine acetylation. Science. 327, 1000–1004 (2010)
Pepenella, S., Murphy, K.J., Hayes, J.J.: Intra- and Inter-nucleosome interactions of the core histone tail domains in higher-order chromatin structure. Chromosoma. 123, 3–13 (2014)
Wang, T., Holt, M.V., Young, N.L.: The histone H4 proteoform dynamics in response to SUV4-20 inhibition reveals single molecule mechanisms of inhibitor resistance. Epigenetics Chromatin. 11, 29 (2018)
Wang, T., Holt, M.V., Young, N.L.: Early butyrate induced acetylation of histone H4 is proteoform specific and linked to methylation state. Epigenetics. 13, 519–535 (2018)
Sterner, D.E., Berger, S.L.: Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64, 435–459 (2000)
Barnard, J.A., Warwick, G.: Butyrate rapidly induces growth inhibition and differentiation in HT-29 cells. Cell Growth Differ. 4, 495–501 (1993)
Candido, E.P., Reeves, R., Davie, J.R.: Sodium butyrate inhibits histone deacetylation in cultured cells. Cell. 14, 105–113 (1978)
Chopin, V., Toillon, R.-A., Jouy, N., Le Bourhis, X.: Sodium butyrate induces P53-independent, Fas-mediated apoptosis in MCF-7 human breast cancer cells. Br. J. Pharmacol. 135, 79–86 (2002)
Kruh, J.: Effects of sodium butyrate, a new pharmacological agent, on cells in culture. Mol. Cell. Biochem. 42, 65–82 (1982)
Kuefer, R., Hofer, M.D., Altug, V., Zorn, C., Genze, F., Kunzi-Rapp, K., Hautmann, R.E., Gschwend, J.E.: Sodium butyrate and tributyrin induce in vivo growth inhibition and apoptosis in human prostate cancer. Br. J. Cancer. 90, 535–541 (2004)
Louis, M., Rosato, R.R., Brault, L., Osbild, S., Battaglia, E., Yang, X.-H., Grant, S., Bagrel, D.: The histone deacetylase inhibitor sodium butyrate induces breast cancer cell apoptosis through diverse cytotoxic actions including glutathione depletion and oxidative stress. Int. J. Oncol. 25, 1701–1711 (2004)
Medina, V., Afonso, J.J., Alvarez-Arguelles, H., Hernández, C., González, F.: Sodium butyrate inhibits carcinoma development in a 1,2-dimethylhydrazine-induced rat colon cancer. JPEN J. Parenter. Enteral Nutr. 22, 14–17 (1998)
DiMaggio Jr., P.A., Young, N.L., Baliban, R.C., Garcia, B.A., Floudas, C.A.: A mixed-integer linear optimization framework for the identification of post-translational modifications in histone H3 using ETD and ECD tandem mass spectrometry. Mol. Cell. Proteomics. 8, 2527–2543 (2009)
Zhou, M., Wu, S., Stenoien, D.L., Zhang, Z., Connolly, L., Freitag, M., Paša-Tolić, L.: Profiling changes in histone post-translational modifications by top-down mass spectrometry. Methods Mol. Biol. 1507, 153–168 (2017)
Phanstiel, D., Brumbaugh, J., Berggren, W.T., Conard, K., Feng, X., Levenstein, M.E., McAlister, G.C., Thomson, J.A., Coon, J.J.: Mass spectrometry identifies and quantifies 74 unique histone H4 isoforms in differentiating human embryonic stem cells. Proc. Nat. Acad. Sci. Mar. 105(11), 4093–4098 (2008)
Janssen, K.A., Coradin, M., Lu, C., et al.: Quantitation of Single and Combinatorial Histone Modifications by Integrated Chromatography of Bottom-up Peptides and Middle-down Polypeptide Tails. J. Am. Soc. Mass Spectrom. (2019)
Perez-Riverol, Y., Csordas, A., Bai, J., Bernal-Llinares, M., Hewapathirana, S., Kundu, D.J., Inuganti, A., Griss, J., Mayer, G., Eisenacher, M., Pérez, E., Uszkoreit, J., Pfeuffer, J., Sachsenberg, T., Yilmaz, S., Tiwary, S., Cox, J., Audain, E., Walzer, M., Jarnuczak, A.F., Ternent, T., Brazma, A., Vizcaíno, J.A.: The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47(D1), D442–D450 (2019)
Acknowledgements
We would like to thanks Dr. A. Assié for critical reviews on the manuscript.
Raw data and full proteoform lists:
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [46] partner repository with the dataset identifier PXD013766
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Materials
Supplementary Figure 1.
Normalized extracted ion chromatograms of 755.83 m/z (Nα-acK20me2M84ox). Black: technical replicates of biological replicate 1. Red: technical replicates of biological replicate 2. Blue: technical replicates of biological replicate 3. Orange: technical replicates of biological replicate 4. (PPTX 117 kb)
Supplementary Figure 2.
Offline HPLC UV standard curve for the quantitation of histone H4 present in each sample based on recombinant Xenopus laevis H4. (PPTX 60 kb)
Supplementary Figure 3.
A) A scatter plot of CVs vs. log10 of % abundance. Black: biological replicates, Red: average of technical replicates. B) Boxplots of the top 100 proteoform CVs. Technical CVs are significantly lower than biological (p<0.01) (PPTX 114 kb)
Supplementary Table 1.
Retention time CVs for technical replicates and the overall biological CV for retention time. (PPTX 55 kb)
Supplementary Figure 4.
A) Histogram of top 20 proteoforms scaled abundances for all control samples (240 measurements) is approximately normally distributed. B) Q-Q Plot of all proteoforms C) Anderson-Darling test for Normality p-value summary. The vast majority of proteoforms are most likely normally distributed. (PPTX 110 kb)
Supplementary Figure 5.
Histogram of all p-values from filtered proteoforms between Control and Butyrate treated SUM159 cells. (PPTX 84 kb)
Supplementary Figure 6.
A) Elution profile of a control SUM159 run that contained early eluting peaks “B” and “C” B) MS1 of early eluting peak “B”. The m/z distributions are similar to those of the predominant H4 proteoforms, but shifted -2 Da C) MS1 of early eluting peak “C”. The m/z distributions are similar to those of the predominant H4 proteoforms, but shifted -18 Da. D) Prosight PC ion map from the most abundant precursor in B. Histone H4 with a -1.027 Da modification is the best fit (p-value and ions matched), indicative of allysine on a C-terminal lysine. Accurate localization was not achieved. E) Prosight PC ion map from the most abundant precursor in C. Histone H4 with a -18 Da modification is the best fit (p-value and ions matched) by Succinimide on D68. (PPTX 558 kb)
Supplementary Table 2
Oxidation and sulfate marginalized proteoforms that are significantly increased by butyrate and have the highest fold change. The fully tail acetylated combination “K5acK8acK12acK16ac” is predominantly observed as the most increased. (PPTX 55 kb)
Supplementary Table 3
(PPTX 55 kb)
Rights and permissions
About this article

Cite this article
Holt, M.V., Wang, T. & Young, N.L. High-Throughput Quantitative Top-Down Proteomics: Histone H4. J. Am. Soc. Mass Spectrom. 30, 2548–2560 (2019). https://doi.org/10.1007/s13361-019-02350-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-019-02350-z