
Abstract
The abundance of an ion in an electrospray ionization mass spectrum is dependent on many factors beyond just solution concentration. Even in cases where the analytes of interest are permanently charged (under study here are ammonium and phosphonium ions) and do not rely on protonation or other chemical processes to acquire the necessary charge, factors such as cation structure, molecular weight, solvent, and the identity of the anion can affect results. Screening of a variety of combinations of cations, anions, and solvents provided insight into some of the more important factors. Rigid cations and anions that conferred high conductivity tended to provide the highest responses. The solvent that most closely reflected actual solution composition was acetonitrile, while methanol, acetonitrile/water, and dichloromethane produced a higher degree of discrimination between different ions. Functional groups that had affinity for the solvent tended to depress response. These observations will provide predictive power when accounting for analytes that for reasons of high reactivity can not be isolated.

Similar content being viewed by others


Introduction
Electrospray ionization (ESI) is well-known to exhibit differing ion responses depending on the nature of the analyte under study [1,2,3]. This effect extends beyond the propensity of the analyte to acquire a charge in the solution phase (the more basic a compound is, the more readily it is protonated) [4], as permanently charged ions provide different intensities depending on their structure. Prior studies in this area have principally focused on the most intuitively important factor, the extent of ionization of a given analyte, which depends on factors such as solution pH and the basicity of the molecule in question [5,6,7]. Signal strength in a mass spectrum does not transparently reflect solution composition [8].
The ion response describes the surface activity of species in a mass spectrum, which implies that surface-active analytes have a stronger ESI response than less surface-active analytes [9,10,11,12]. Possible parameters that correlate with the ESI response of analytes include morphology of the analyte, cation–anion interaction, and solvation strength [13, 14]. The nature of the solvent is a contributory factor to the manner of ESI response of species, given that the response is contingent on the solvation strength [13,14,15,16]. However, the extent of solvation of ions may differ in polar and non-polar solvents eligible for ESI-MS [17,18,19]. Hydrophilic and hydrophobic species tend to be more solvated in polar and non-polar solvents, respectively, thereby exhibiting poor surface activity. In this regard, we previously reported that chloride aggregates of 1-butyl-3-methylimidazolium [(BMIM)2 + Cl]+ showed a better ESI-MS response than bistriflimide aggregates of the same cation [(BMIM)2 + NTf2]+ in dichloromethane, due to lower solvation of the chloride aggregates in the non-polar solvent [20]. In an experiment to investigate the ESI response of ionic liquids, Bortolini et al. found that a cobalt-based ionic liquid with bistriflimide counterion was more responsive in methanol than in dichloromethane [21]. These findings apparently indicate that intensity is related to the solvation strength. Strong solvation of analytes can occur because of hydrogen bonding and other intermolecular forces [22,23,24,25,26,27,28]. For example, solvation of chloride in methanol or water/acetonitrile systems comes about because of ion–dipole interactions. Spatial effects in terms of source geometry can also affect relative ESI-MS response [29].
Analytes with flexible alkyl groups are identified as hydrophobic and are likely to occupy surface sites in a droplet of polar solvent. Considering the morphology of such analytes, Song and co-workers reported an anomaly in a trend of surface activity for quaternary ammonium species with flexible alkyl chains in an aqueous sodium chloride system [30]. They suggested that, as the alkyl chain length increases (above 14 carbon atoms), aggregates are formed with less or no surface activity depending on the environment of the species [30]. However, their assessment was not an MS study and involved gemini surfactants (more than one headgroup) [30].
In this work, we investigate the parameters that could influence the ESI response of an equimolar mixture of six cations (cesium, tetraethylammonium, tetrabutylammonium, tridodecylmethylammonium, bis(triphenylphosphoranylidene)ammonium, and tetradodecylammonium, i.e., Cs+, NEt4+ (Et = ethyl), NBu4+ (Bu = butyl), NDo3Me+ (Do = dodecyl; Me = methyl), N(PPh3)2+ (Ph = phenyl) and NDo4+), paired with various anions (chloride, hexafluorophosphate, tetrafluoroborate, and bistriflimide, i.e., Cl−, [PF6]−, [BF4]− and [NTf2]−) in standard ESI solvents (Figure 1).

Representation of various sizes of cations and anions employed in this study
These salts were selected to provide a variety of molecular weights and structural variety; also ionic surfactants, such as tetraalkylammonium halides, have been reported by Bruins and co-workers as suitable model analytes for fundamental ESI-MS studies, given their surface activity and insusceptibility to solution pH [31]. Salts with counterions larger than chloride were synthesized by switching the chloride counterion of the six cations by employment of silver tetrafluoroborate (AgBF4), silver hexafluorophosphate (AgPF6), and silver bistriflimide (AgNTf2), exploiting the very low solubility of AgCl. We also investigated the correlation between rigidity and ESI response of structurally different species. A solid understanding of these factors is important when using ESI-MS to gather kinetic data [32,33,34,35,36,37,38] because while corrections can be applied when studying solution components such as reactants and products (which can be individually isolated and measured), such precautions are often impossible when dealing with reactive intermediates [39].
Experimental
All chemicals were purchased from Sigma-Aldrich and employed as received. Salts with hexafluorophosphate [PF6]−, tetrafluoroborate [BF4]−, and bistriflimide [NTf2]− counterions were prepared by reaction of an equimolar mixture of the salts with AgBF4, AgPF6, and AgNTf2 in methanol (MeOH), acetonitrile (MeCN), water/acetonitrile (H2O/MeCN 1:1), and dichloromethane (CH2Cl2) at room temperature. For example, the salt with hexafluorophosphate counterion in methanol was prepared by reaction of an equimolar mixture of the chloride salts (1 equivalent, 10 mmol in methanol) with silver hexafluorophosphate (1 equivalent, 10 mmol in methanol) at room temperature; a gray precipitate was formed and removed by gravity filtration. The filtrate was characterized by electrospray ionization mass spectrometry (ESI-MS) whereby [PF6]− counterion was identified as m/z 145 in the negative ion mode (see supporting information). This synthesis procedure was also carried out in acetonitrile, water/acetonitrile, and dichloromethane.
Phosphonium salts were also synthesized by a general procedure as follows: Triphenylphosphine (10 mmol) and a bromobenzyl compound (10 mmol) were dissolved in 20 mL of toluene and were stirred at room temperature under nitrogen for 48 h. The precipitate was filtered out of solution by vacuum filtration which produced a white solid. A saturated aqueous solution of the product obtained was added to a saturated aqueous sodium hexafluorophosphate solution to swap the bromide counterion for hexafluorophosphate counterion at room temperature; a white precipitate was recovered through vacuum filtration.
Prior to ESI-MS analysis, all analytes were further diluted to 10 μM. The ESI-MS spectra were obtained by means of a Waters Acquity Triple Quadrupole Detector (TQD). The instrument parameters employed were capillary voltage (2.5 kV), cone voltage (15 V), extraction voltage (1.0 V); source temperature was 80 °C for methanol; 110 °C for water/acetonitrile; 90 °C for acetonitrile; and 50 °C for dichloromethane; desolvation temperature was 180 °C for methanol; 200 °C for water/acetonitrile; 190 °C for acetonitrile; and 150 °C for dichloromethane. Cone gas flow rate, 100 L/h; desolvation gas flow rate, 100 L/h. Scan time was 3 s. All diluted analytes were fed into the mass spectrometer with a gastight analytical syringe connected to PEEK tubing and a syringe pump at a flow rate of 10 μL/min. A conductivity experiment was also carried out by measuring the solution resistances of analytes at room temperature (see Table S1, supporting information) using a potentiostat (Gamry Instruments, Reference 600, Warminster, PA, USA); the solution resistances measured were converted to molar conductivities (see Table 1 and Table S1, supporting information).
Results and Discussion
An equimolar mixture of cations (Cs+, [NEt4]+, [NBu4]+, [NDo3Me]+, [N(PPh3)2]+, [NDo4]+) was paired with one of four anions (Cl−, [BF4]−, [PF6]−, [NTf2]−) in methanol, acetonitrile, water/acetonitrile, and dichloromethane; however, CsCl in dichloromethane was excluded, given its insolubility (see supporting information). This exclusion also included pairing of the other anions with cesium in dichloromethane. The relative responses of these species were measured in positive mode ESI-MS.
Figure 2 shows the relative intensities of salts in acetonitrile. The differential response with chloride as a counterion (Figure 2a) showed high intensity for [NEt4]+, [NBu4]+, and [N(PPh3)2]+; moderate intensity for [NDo3Me]+ and [NDo4]+; and near-zero response for Cs+. The low response of Cs+ is possibly due to a high level of solvation and/or ion-pairing for this ion [40], and the lower response of the ammonium salts with dodecyl groups could probably be related to Song’s explanation for the formation of non-surface active aggregates in relation to surface activity of long-chain ammonium salts [30]. Swapping the chloride for increasingly hydrophilic counterions (Figure 2b–d) changes the distribution in favor of the lowest molecular weight species, and for [NTf2]−, the spectrum is dominated by [NEt4]+ with the next most intense ion, [NBu4]+; [N(PPh3)2]+ being less than 25% and all others at baseline levels. At the same time as ion differentiation increased, ion intensity dropped overall.

Positive ion ESI mass spectrum of an equimolar mixture of six cations [NEt4]+ (m/z 130), [Cs]+ (m/z 132), [NBu4]+ (m/z 242), [N(PPh3)2]+ (m/z 538), [NDo3Me]+ (m/z 536), and [NDo4]+ (m/z 690), paired with various counterions in acetonitrile: (a) [Cl]−; (b) [BF4]−; (c) [PF6]−; and (d) [NTf2]−
A representation of reproducible peak area distribution for species in acetonitrile also depicted a similar trend (see Figure S5, supporting information). Also, in a dichloromethane system (Figure 3), we observed that all cations paired with chloride were over-represented compared with cations paired with relatively hydrophobic anions ([BF4]−, [PF6]−, and [NTf2]−), considering that the cations paired with chloride were less solvated. Comparing the relative intensity (Figure 2) and peak area (Figure 3 and Figure S5, supporting information) of the hydrophobic rigid cation [N(PPh3)2]+ with that of the bulky hydrophobic flexible cations [NDo3Me+ and NDo4+] in both acetonitrile and dichloromethane, the hydrophobic flexible species were under-represented, though less so than the Cs+ (in acetonitrile), which was almost invisible, suppressed by the more surface-active ions.

Peak area distribution in positive ion ESI-MS of salts in dichloromethane. The cations are represented: 1 = [NEt4]+; 2 = [Cs]+; 3 = [NBu4]+; 4 = [N(PPh3)2]+; 5 = [NDo3Me]+; 6 = [NDo4]+. The anions are represented: a = [Cl]−; b = [BF4]−; c = [PF6]−; and d = [NTf2]−. There is no peak area representation for all cesium salts given that cesium chloride is insoluble in dichloromethane; hence, salt metathesis for the other counterions was not conducted. The standard deviation of the mean (n = 3) is represented by the vertical bars
In polar solvents, such as methanol and water/acetonitrile systems, polarity of the solvents could have influenced the solvation strength (between chloride and the solvents); as well, hydrogen bonding could also contribute to the solvation strength [20]. Figure 4 and Figure 5 show that cations paired with chloride anions as providing the highest ion counts. It is possible that this is the case due to chloride being more easily oxidized than any of the other counterions; thus, the ESI process generates a greater excess of charge for a given capillary voltage [41,42,43,44].

Peak area distribution in positive ion ESI-MS of salts in methanol. The cations are represented: 1 = [NEt4]+; 2 = [Cs]+; 3 = [NBu4]+; 4 = [N(PPh3)2]+; 5 = [NDo3Me]+; 6 = [NDo4]+. The anions are represented: a = [Cl]−; b = [BF4]−; c = [PF6]−; and d = [NTf2]−. The standard deviation of the mean (n = 3) is represented by the vertical bars

Peak area distribution in positive ion ESI-MS of salts in water/acetonitrile. The cations are represented: 1 = [NEt4]+; 2 = [Cs]+; 3 = [NBu4]+; 4 = [N(PPh3)2]+; 5 = [NDo3Me]+; 6 = [NDo4]+. The anions are represented: a = [cl]−; b = [BF4]−; c = [PF6]−; and d = [NTf2]−. The standard deviation of the mean (n = 3) is represented by the vertical bars
Also, the highest response exhibited by species paired with chloride anion could be attributed to their increase in conductivity as shown in Table 1. In this regard, the molar conductivity of all electrolytes examined in this study followed a trend whereby halide species (except CsCl in acetonitrile) had a relatively higher molar conductivity as compared to polyatomic species (see Table 1 and Table S1, supporting information). This result could be interpreted as halide species being more conductive in solution, possibly due to a relatively less ion–ion interaction [45, 46] in all solvent systems employed.
In Figure 4 and Figure 5, it can be observed that the bulky hydrophobic flexible cation species [NDo3Me]+ and [NDo4]+ had a low response again, probably due to the formation of less surface-active premicellar species [30]. Also, as shown in Figure 4, Figure S5, and Figure 5, tetraethylammonium and cesium ions (m/z 130 and m/z 132, respectively) have different peak area distributions in methanol, acetonitrile, and water/acetonitrile systems. Cs+ had a poor response in the polar solvents, probably due to a strong ion pairing that existed in the cesium species (e.g., CsCl) [40, 47], as well as the overall relatively small size of the cesium species; thereby, the solvation strength was larger as compared to the tetraethylammonium species.
Furthermore, [NBu4]+ species had a better response in acetonitrile and dichloromethane as compared with [N(PPh3)2]+ species, possibly due to a relatively higher molar conductivity in acetonitrile and dichloromethane (see Table S1, supporting information); however, this was less favored in methanol and water/acetonitrile systems. This study has clearly revealed that when an equimolar mixture of cations of different sizes was paired with weakly coordinating counterions ([BF4]−, [PF6]−, [NTf2]−) in polar and non-polar standard ESI solvents, there was no striking effect in their ESI response; and as well, discrimination between masses of all species employed can be observed regardless of their equal amounts in solution due to possible non-uniform ion transmission in the mass analyzer (triple quadrupole) employed [48, 49]. Another notable observation was that the highest relative intensity and peak area distribution for analytes in acetonitrile (Figure S5, supporting information and Figure 2) compared with analytes in the other solvent systems. This result agreed with our previous findings where we observed that the aprotic polar solvent (acetonitrile) could solvate ions to a somewhat similar extent regardless of their morphology [20].
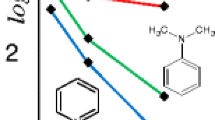
The correlation between rigidity and response of analytes was investigated by assessing an equimolar mixture of structurally different species of approximately the same molecular weights (m/z 353± 14) in both polar and non-polar ESI solvents as shown in Figure 6.

Positive ion ESI mass spectrum of an equimolar mixture of four cations paired with bromide counterion in (a) acetonitrile, (b) water/acetonitrile, (c) methanol, and (d) dichloromethane
In Figure 6, it can be observed that phosphonium species with rigid groups at m/z 339 and m/z 347 were more responsive than the ammonium species with flexible alkyl chains at m/z 354 in both polar and non-polar solvents; however, a difference in design of the phosphonium species at m/z 367 affected its response in all solvents employed. These observations were also apparent in Figure S9 (supporting information), where [PF6]− was the counterion, i.e., the counterion seems to play only a minor role in affecting the relative response of the various cations. We therefore considered probing the structural effect further by evaluating an equimolar mixture of phosphonium salts in the form of [Ph3PCH2C6H4R]+[X]ˉ, where R = H, CH3, CH(CH3)2, CO2CH3; X = [PF6]−, in both polar and non-polar solvents. In this case, all four ions share a common structural feature. We have previously shown that such ions tend to show similar responses in reactions tracked by ESI-MS, whether measured directly [50] or observable by a flat total ion current for the duration of the transformation of one ion into another [32, 51, 52]. Figure 7 captures the relative intensities of the phosphonium analytes examined.

Positive ion ESI mass spectrum of an equimolar mixture of four phosphonium species paired with hexafluorophosphate counterion in (a) acetonitrile; (b) water/acetonitrile; (c) methanol; and (d) dichloromethane
In relation to Figure 7, the relative intensities of the species decreased upon changes in the R group (H > CH3 > CH(CH3)2 > CO2CH3) in acetonitrile, water/acetonitrile, and dichloromethane solvents. In methanol, hydrophobic species at m/z 395, m/z 367, and m/z 353 (decreasing hydrophobicity from higher mass to lower mass) were all more responsive than the ester-functionalized phosphonium ion at m/z 411. The higher polarity of this species increases its affinity for the solvent, making it less likely to migrate to the surface of the droplet and in turn migrate to the gas phase through ion evaporation.
Conclusions
An examination of a variety of solvents, cation structures, cation functionality, and counterion identities in the context of electrospray ionization response has revealed that all of these factors play a part in dictating peak height. Polar protic solvents tend to discriminate between different ions most strongly; contrastingly, acetonitrile was the best in terms of most closely representing concentration with peak area. Smaller species tend to have higher responses overall, most likely due to an increase in conductivity and/or ease of oxidation. These effects were particularly marked for species paired with chloride over the other analytes investigated (all polyatomic). Any effect due to reduced ion pairing due to a larger counterion seems very minor. Cations that have long flexible groups are significantly under-represented in mass spectra compared with rigid ions of the same m/z ratio. Cations with functional groups capable of forming stronger intermolecular forces with the solvent are also likely to be under-represented. Collectively, these observations should provide experimentalists with some explanatory power in explaining discrepancies between different analytes and should help them design experiments to either mitigate or exacerbate these effects as desired.
References
Zhou, S., Cook, K.D.: A mechanistic study of electrospray mass spectrometry: charge gradients within electrospray droplets and their influence on ion response. J. Am. Soc. Mass Spectrom. 12, 206–214 (2001)
Cech, N.B., Enke, C.G.: Practical implications of some recent studies in electrospray ionization fundamentals. Mass Spectrom. Rev. 20, 362–387 (2001)
Amad, M.H., Cech, N.B., Jackson, G.S., Enke, C.G.: Importance of gas-phase proton affinities in determining the electrospray ionization response for analytes and solvents. J. Mass Spectrom. 35, 784–789 (2000)
Chalcraft, K.R., Lee, R., Mills, C., Britz-McKibbin, P.: Virtual quantification of metabolites by capillary electrophoresis-electrospray ionization-mass spectrometry: predicting ionization efficiency without chemical standards. Anal. Chem. 81, 2506–2515 (2009)
Kiontke, A., Oliveira-Birkmeier, A., Opitz, A., Birkemeyer, C.: Electrospray ionization efficiency is dependent on different molecular descriptors with respect to solvent pH and instrumental configuration. PLoS One. 11, e0167502 (2016)
Henriksen, T., Juhler, R.K., Svensmark, B., Cech, N.B.: The relative influences of acidity and polarity on responsiveness of small organic molecules to analysis with negative ion electrospray ionization mass spectrometry (ESI-MS). J. Am. Soc. Mass Spectrom. 16, 446–455 (2005)
Mandra, V.J., Kouskoura, M.G., Markopoulou, C.K.: Using the partial least squares method to model the electrospray ionization response produced by small pharmaceutical molecules in positive mode. Rapid Commun. Mass Spectrom. 29, 1661–1675 (2015)
Cheng, Z.L., Siu, K.W.M., Guevremont, R., Berman, S.S.: Electrospray mass spectrometry: a study on some aqueous solutions of metal salts☆. J. Am. Soc. Mass Spectrom. 3, 281–288 (1992)
Kebarle, P., Peschke, M.: On the mechanisms by which the charged droplets produced by electrospray lead to gas phase ions. Anal. Chim. Acta. 406, 11–35 (2000)
Fenn, J.B.: Ion formation from charged droplets: roles of geometry, energy, and time. J. Am. Soc. Mass Spectrom. 4, 524–535 (1993)
Wang, G., Cole, R.B.: Charged residue versus ion evaporation for formation of alkali metal halide cluster ions in ESI. Anal. Chim. Acta. 406, 53–65 (2000)
Tang, L., Kebarle, P.: Dependence of ion intensity in electrospray mass spectrometry on the concentration of the analytes in the electrosprayed solution. Anal. Chem. 65, 3654–3668 (1993)
Fei, Z., Zhu, D.-R., Yan, N., Scopelliti, R., Katsuba, S.A., Laurenczy, G., Chisholm, D.M., McIndoe, J.S., Seddon, K.R., Dyson, P.J.: Electrostatic and non-covalent interactions in dicationic imidazolium-sulfonium salts with mixed anions. Chem. - A Eur. J. 20, 4273–4283 (2014)
Bini, R., Bortolini, O., Chiappe, C., Pieraccini, D., Siciliano, T.: Development of cation/anion “interaction” scales for ionic liquids through ESI-MS measurements. J. Phys. Chem. B. 111, 598–604 (2007)
Bruins, A.P.: Mechanistic aspects of electrospray ionization. J. Chromatogr. A. 794, 345–357 (1998)
Kebarle, P., Tang, L.: From ions in solution to ions in the gas phase - the mechanism of electrospray mass spectrometry. Anal. Chem. 65, 972A–986A (1993)
Cole, R.B., Harrata, A.K.: Charge-state distributuion and electric-discharge suppression in negative-ion electrospray mass spectrometry using/chlorinated solvents. Rapid Commun. Mass Spectrom. 6, 536–539 (1992)
Cole, R.B., Harrata, A.K.: Solvent effect on analyte charge state, signal intensity, and stability in negative ion electrospray mass spectrometry; implications for the mechanism of negative ion formation. J. Am. Soc. Mass Spectrom. 4, 546–556 (1993)
Stassen, H.K., Ludwig, R., Wulf, A., Dupont, J.: Imidazolium salt ion pairs in solution. Chem. - A Eur. J. 21, 8324–8335 (2015)
Pape, J., Vikse, K.L., Janusson, E., Taylor, N., McIndoe, J.S.: Solvent effects on surface activity of aggregate ions in electrospray ionization. Int. J. Mass Spectrom. 373, 66–71 (2014)
Bortolini, O., Chiappe, C., Ghilardi, T., Massi, A., Pomelli, C.S.: Dissolution of metal salts in bis(trifluoromethylsulfonyl)imide-based ionic liquids: studying the affinity of metal cations toward a “weakly coordinating” anion. J. Phys. Chem. A. 119, 5078–5087 (2015)
Hunt, P.A.: Why does a reduction in hydrogen bonding lead to an increase in viscosity for the 1-butyl-2,3-dimethyl-imidazolium-based ionic liquids? †. J. Phys. Chem. B. 111, 4844–4853 (2007)
Kohagen, M., Brehm, M., Lingscheid, Y., Giernoth, R., Sangoro, J., Kremer, F., Naumov, S., Iacob, C., Kärger, J., Valiullin, R., Kirchner, B.: How hydrogen bonds influence the mobility of imidazolium-based ionic liquids. A combined theoretical and experimental study of 1-n-butyl-3-methylimidazolium bromide. J. Phys. Chem. B. 115, 15280–15288 (2011)
Izgorodina, E.I., MacFarlane, D.R.: Nature of hydrogen bonding in charged hydrogen-bonded complexes and imidazolium-based ionic liquids. J. Phys. Chem. B. 115, 14659–14667 (2011)
Katsyuba, S.A., Vener, M.V., Zvereva, E.E., Fei, Z., Scopelliti, R., Laurenczy, G., Yan, N., Paunescu, E., Dyson, P.J.: How strong is hydrogen bonding in ionic liquids? Combined X-ray crystallographic, infrared/Raman spectroscopic, and density functional theory study. J. Phys. Chem. B. 117, 9094–9105 (2013)
Fumino, K., Reimann, S., Ludwig, R.: Probing molecular interaction in ionic liquids by low frequency spectroscopy: coulomb energy, hydrogen bonding and dispersion forces. Phys. Chem. Chem. Phys. 16, 21903–21929 (2014)
Dhumal, N.R., Noack, K., Kiefer, J., Kim, H.J.: Molecular structure and interactions in the ionic liquid 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide. J. Phys. Chem. A. 118, 2547–2557 (2014)
Skarmoutsos, I., Welton, T., Chemical, P.H.-P.C.: U.: The importance of timescale for hydrogen bonding in imidazolium chloride ionic liquids. Phys. Chem. Chem. Phys. 16, 3675–3685 (2014)
Janusson, E., Hesketh, A.V., Bamford, K.L., Hatlelid, K., Higgins, R., McIndoe, J.S.: Spatial effects on electrospray ionization response. Int. J. Mass Spectrom. 388, 1–8 (2015)
Song, L.D., Rosen, M.J.: Surface properties, micellization, and premicellar aggregation of gemini surfactants with rigid and flexible spacers. Langmuir. 12, 1149–1153 (1996)
Zook, D.R., Bruins, A.P.: On cluster ions, ion transmission, and linear dynamic range limitations in electrospray (ionspray) mass spectrometry. Int. J. Mass Spectrom. Ion Process. 162, 129–147 (1997)
Yunker, L.P.E., Ahmadi, Z., Logan, J.R., Wu, W., Li, T., Martindale, A., Oliver, A.G., McIndoe, J.S.: Real-time mass spectrometric investigations into the mechanism of the Suzuki–Miyaura reaction. Organometallics. 37, 4297–4308 (2018)
Belli, R.G., Wu, Y., Ji, H., Joshi, A., Yunker, L.P.E., McIndoe, J.S., Rosenberg, L.: Competitive ligand exchange and dissociation in Ru indenyl complexes. Inorg. Chem. 58, 747–755 (2019)
Luo, J., Wu, Y., Zijlstra, H.S., Harrington, D.A., McIndoe, J.S.: Mass transfer and convection effects in small-scale catalytic hydrogenation. Catal. Sci. Technol. 7, 2609–2615 (2017)
Yan, X., Sokol, E., Li, X., Li, G., Xu, S., Cooks, R.G.: On-line reaction monitoring and mechanistic studies by mass spectrometry: Negishi cross-coupling, hydrogenolysis, and reductive amination. Angew. Chemie Int. Ed. 53, 5931–5935 (2014)
Medeiros, G.A., da Silva, W.A., Bataglion, G.A., Ferreira, D.A.C., de Oliveira, H.C.B., Eberlin, M.N., Neto, B.A.D.: Probing the mechanism of the Ugi four-component reaction with charge-tagged reagents by ESI-MS(/MS). Chem. Commun. 50, 338–340 (2014)
Rauf, W., Brown, J.M.: Reactive intermediates in catalytic alkenylation; pathways for Mizoroki–Heck, oxidative Heck and Fujiwara–Moritani reactions. Chem. Commun. 49, 8430 (2013)
Ingram, A.J., Boeser, C.L., Zare, R.N.: Going beyond electrospray: mass spectrometric studies of chemical reactions in and on liquids. Chem. Sci. 7, 39–55 (2016)
Theron, R., Wu, Y., Yunker, L.P.E., Hesketh, A.V., Pernik, I., Weller, A.S., McIndoe, J.S.: Simultaneous orthogonal methods for the real-time analysis of catalytic reactions. ACS Catal. 6, 6911–6917 (2016)
Chantooni, M.K., Kolthoff, I.M.: Hydration of ions in acetonitrile. J. Am. Chem. Soc. 89, 1582–1586 (1967)
De Vos, N., Maton, C., Stevens, C.V.: Electrochemical stability of ionic liquids: general influences and degradation mechanisms. ChemElectroChem. 1, 1258–1270 (2014)
Abbott, A.P., Mckenzie, K.J.: Application of ionic liquids to the electrodeposition of metals. Phys. Chem. Chem. Phys. 8, 4265–4279 (2006)
Rohner, T.C., Lion, N., Girault, H.H.: Electrochemical and theoretical aspects of electrospray ionisation. Phys. Chem. Chem. Phys. 6, 3056–3068 (2004)
Liu, P., Lu, M., Zheng, Q., Zhang, Y., Dewald, H.D., Chen, H.: Recent advances of electrochemical mass spectrometry. Analyst. 138, 5519 (2013)
Kay, R.L., Zawoyski, C., Evans, D.F.: The conductance of the symmetrical tetraalkylammonium halides and picrates in methanol at 25 and 10° 1. J. Phys. Chem. 69, 4208–4215 (1965)
Evans, D.F., Zawoyski, C., Kay, R.L.: The conductance of the symmetrical tetraalkylammonium halides and picrates in acetonitrile at 25° 1. J. Phys. Chem. 69, 3878–3885 (1965)
Kay, R.L., Hawes, J.L.: The association of cesium chloride in anhydrous methanol at 25°. J. Phys. Chem. 69, 2787–2788 (1965)
Miller, P.E., Denton, M.B.: The transmission properties of an RF-only quadrupole mass filter. Int. J. Mass Spectrom. Ion Process. 72, 223–238 (1986)
Turecek, F., Gu, M., Shaffer, S.A.: Novel tandem quadrupole-acceleration-deceleration mass spectrometer for neutralization-reionization studies. J. Am. Soc. Mass Spectrom. 3, 493–501 (1992)
Vikse, K.L., Ahmadi, Z., Manning, C.C., Harrington, D.A., McIndoe, J.S.: Powerful insight into catalytic mechanisms through simultaneous monitoring of reactants, products, and intermediates. Angew. Chemie Int. Ed. 50, 8304–8306 (2011)
Ahmadi, Z., Yunker, L.P.E., Oliver, A.G., McIndoe, J.S.: Mechanistic features of the copper-free Sonogashira reaction from ESI-MS. Dalt. Trans. 44, 20367–20375 (2015)
Ahmadi, Z., McIndoe, J.S.: A mechanistic investigation of hydrodehalogenation using ESI-MS. Chem. Commun. 49, 11488 (2013)
Acknowledgements
JSM thanks NSERC (Discovery and Discovery Accelerator Supplement) for operational funding and CFI, BCKDF, and the University of Victoria for infrastructural support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(DOCX 437 kb)
Rights and permissions
About this article

Cite this article
Omari, I., Randhawa, P., Randhawa, J. et al. Structure, Anion, and Solvent Effects on Cation Response in ESI-MS. J. Am. Soc. Mass Spectrom. 30, 1750–1757 (2019). https://doi.org/10.1007/s13361-019-02252-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-019-02252-0