
Abstract
G protein–coupled receptors (GPCRs) constitute the largest class of cell surface signaling receptors and regulate major neurobiological processes. Accordingly, GPCRs represent primary targets for the treatment of brain disorders. Several human genetic polymorphisms affecting GPCRs have been associated to different components of alcohol use disorder (AUD). Moreover, GPCRs have been reported to contribute to several features of alcohol-related behaviors in animal models. Besides traditional pharmacological tools, genetic-based approaches mostly aimed at deleting GPCR genes provided substantial information on how key GPCRs drive alcohol-related behaviors. In this review, we summarize the alcohol phenotypes that ensue from genetic manipulation, in particular gene deletion, of key GPCRs in rodents. We focused on GPCRs that belong to fundamental neuronal systems that have been shown as potential targets for the development of AUD treatment. Data are reviewed with particular emphasis on alcohol reward, seeking, and consumption which are behaviors that capture essential aspects of AUD. Literature survey indicates that in most cases, there is still a gap in defining the intracellular transducers and the functional crosstalk of GPCRs as well as the neuronal populations in which their signaling regulates alcohol actions. Further, the implication of only a few orphan GPCRs has been so far investigated in animal models. Combining advanced pharmacological technologies with more specific genetically modified animals and behavioral preclinical models is likely necessary to deepen our understanding in how GPCR signaling contributes to AUD and for drug discovery.
Similar content being viewed by others


Introduction
G protein–coupled receptors (GPCRs), which are also called seven-transmembrane (7TM) domain receptors, represent a large class of cell surface proteins encoded by more than 800 genes in human. These receptors are classically bound by a ligand that triggers changes of the highly dynamic GPCR conformation ultimately leading to the modulation of G protein–dependent and/or G protein–independent signaling cascades [1]. GPCR ligands include a broad panel of endogenous signaling molecules such as cytokines, metabolites, hormones, or neurotransmitters. The majority of GPCR endogenous ligands are agonists, and thus their binding to GPCRs induces G protein activation which may, in turn, modulate several second messengers and signaling cascades such as adenylyl cyclase, cyclic adenosine monophosphate (cAMP), mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), and enzyme phospholipase A2 (PLA2) pathways [2]. G proteins mediate a wide variety of cellular functions, including ion channel activity and modulation of neurotransmitter release, and thus regulate a remarkable variety of biological outputs. Alternatively, the multifunctional proteins β-arrestins 1 and 2 can also mediate GPCR signaling [3]. GPCRs are the main receptor targets for the development of pharmaceutical drugs (see guide to pharmacology at http://www.guidetopharmacology.org/about.jsp) [4]. GPCRs have also the capacity to form oligomer by covalent bonds or by intermolecular forces and may be homodimer (two identical GPCRs) or heterodimer (two different GPCRs). Oligomerization may change the ligand binding and signaling properties of the GPCRs and thus can result in unique functional entities [5, 6]. Interestingly, GPCR heterodimers have been described in different structures of the rodent brain and are believed to contribute to several features of substance use disorder [7].
More than 30% of the drugs approved by the Food and Drug Administration (FDA) of the USA target GPCRs with a total sales volume estimated to be 180 billion USD [8,9,10]. These pharmaceutical drugs bind to GPCRs expressed notably in the nervous system such as opioid, endocannabinoid, and aminergic (5-hydroxytryptamine (5-HT), serotonin, and dopamine (DA)) receptors [9]. Accordingly, many drugs used to treat psychiatric disorders target GPCRs or proteins that modulate the extracellular levels of neurotransmitters (e.g., selective serotonin reuptake inhibitors). In this context, innovative strategies including location biased [11], system biased [12, 13], allostery [14], and structure-based docking approaches [15] have being implemented to design safer and more efficient drugs targeting GPCRs. In addition, GPCRs with no endogenous ligand identified, termed orphan GPCRs, also represent promising targets for drug development of psychiatric diseases [16]. The majority of orphan GPCRs are also expressed in the brain [17,18,19] and are located at the cell surface allowing for ease of access. Today, GPCRs are still considered promising therapeutic targets for the treatment of psychiatric diseases. Through the advent of transgenic tools, the generation of genetically modified mice has constituted a fruitful approach to tackle GPCR function in vivo and to examine their potential as drug targets.
Targeted Gene Disruption to Study GPCR Function In Vivo
Cloning of the genes encoding GPCRs in the late 1980s and 1990s along with the development of transgenic technologies to engineer mice with targeted gene disruption propelled scientists to address GPCR function in health and disease through a brand new experimental framework [20, 21]. Until then, traditional pharmacological methods had provided many valuable insights but were limited in certain cases owing to the low selectivity of the available ligands. This is particularly true when it is necessary to untangle the functional characteristics of highly related GPCR subtypes in vivo where it is arduous to control tissue concentration of an active molecule and its selective binding to the targeted receptor following systemic or local administration. Similarly and regarding orphan GPCRs, the lack of potent synthetic brain-penetrant ligands constitutes additional obstacles to probe their function in vivo. In this context, in vivo manipulation of genes encoding GPCRs represented a powerful alternative and/or complementary approach to explore individually the contribution of each GPCR subtype and orphan GPCRs in physiological and pathological processes. Although the use of GPCR null mutant mice provided a way to circumvent the lack of ligand selectivity, these models do have their own drawbacks [21]. In particular, deletion of a specific gene can be lethal or give rise to unexpected genetic compensation that could distort or mask the actual function of the targeted GPCR. Moreover, specific neuronal or brain region localizations of receptors, as exemplified by DA receptors, can lead to various and complex actions of GPCRs that will not be fully captured by a null mutant approach. In such instances, tissue-specific, conditional, and inducible knockouts can substitute traditional null mutant animals [21]. In addition to the use of GPCR null mutant mice, targeted disruption of genes encoding GPCR ligands or encoding enzymes involved in their production or degradation has been also exploited to explore the contribution of GPCR signaling in vivo. Importantly, combining such genetic animal models with pharmacological compounds has made it possible to interrogate in vivo the functional relevance of GPCR constitutive signaling, GPCR oligomerization, and the crosstalk between GPCR systems [22,23,24,25]. Thus, genetic rodent models must not be regarded only as a new way to verify or clarify GPCR function but as complementary tools and new opportunities to deepen our understanding in GPCR biology.
GPCRs and Alcohol-Related Behaviors: Studies in Mutant Knockout Mice
Drugs of abuse affect neuromodulation mediated by GPCRs, with multiple consequences for acute intoxication, drug seeking, and consumption. Moreover, substantial literature indicates that several GPCRs are involved in prolonged drug exposure–induced neuroadaptations that play a critical role in withdrawal symptom and drug relapse [26, 27]. Alcohol use disorder (AUD) is today among the most pervasive psychiatric disorder that encompasses multiple and complex physiological and behavioral phenotypes. The familial incidence of alcoholism indicates that heredity significantly increases the likelihood of heavy drinking behavior and the development of the disorder [28]. Indeed, individuals who have a family history of alcoholism are of high risk to develop and express alcohol addiction, relative to those who are family history negative [29]. This genetic association has been confirmed by multiple gene studies including the Collaborative Study on the Genetics of Alcoholism (COGA) [30]. Thus, one major goal for neuroscientists has been to identify genes and proteins in the brain that influence the development and/or the expression of AUD. In this regard, the use of genetically engineered mice, particularly global homozygous knockouts, has provided a productive and straightforward way to individually evaluate the contribution of protein-coding genes in alcohol-related behaviors [31,32,33]. Thus, shortly after the cloning of GPCR genes, the use of null mutant mice models of alcohol abuse naturally emerged as an obvious strategy to dissect out the contribution of GPCR signaling in alcohol actions [21, 34]. Hence, mice with targeted GPCR gene disruption were tested under diverse typical procedures to evaluate their sensitivity to acute and chronic alcohol exposure and alcohol consumption. The applicability of this genetic-based approach was first illustrated with the use of null mutant mice lacking the 5-HT1B serotonin receptor gene which displayed enhanced voluntary alcohol intake and lower sensitivity to acute alcohol action [35]. In the present review, we will give updated progress that has been made in studying the role of GPCRs in alcohol actions using transgenic mouse models and in particular knockout animals. We decided to discuss below only 1) GPCRs that have recognized a role in drug use disorders and that represent key neurotransmitter systems (DA, serotonin, glutamate, and γ-aminobutyric acid (GABA) receptors) and 2), based on our current knowledge, GPCRs that constitute current therapeutic or emerging targets for the treatment of AUD (opioid [36], endocannabinoid [37], ghrelin receptor [215], corticotropin-releasing factor (CRF) [38], melanocortin receptors [39], and orphan GPCRs). However, we have also broadly catalogued reports that addressed GPCR function in alcohol drinking using knockout mice (see Table 1) which will enhance the system-wide view of GPCR genes role in alcohol actions.
Opioid Receptors
The opioid system is composed of three homologous GPCRs, the mu opioid receptors (MORs), delta opioid receptors (DORs), and kappa opioid receptors (KORs) [109] principally coupled to Gi/o. These three GPCRs are activated by endogenous opioid peptides such as β-endorphin, enkephalins, and dynorphins [110]. Activation of the opioid system by opioid peptides may diminish responses to stress and painful feelings and modulate both mood and reward processing [109, 111]. Specifically, activation of MORs produces analgesia and euphoria and MORs are critical for developmental, social, and drug rewards [112]. DORs are modulating anxiety and mood functions [113], and activation of DORs has been considered as a mood enhancer. Dysfunction of the DOR system has been associated with mood disorders, depression, and chronic pain [114, 115]. Finally, KORs are mediating highly aversive and psychotomimetic effects [116, 117]. The different components of the endogenous opioid system are highly expressed in brain areas involved in reward and motivation (for review, see [109]), and opioid system dysfunction may thus cause addiction-related mood and cognitive disorders [112]. Here, we will review the role of the three opioid receptors in alcoholism identified using genetically modified mice.
MORs
The MORs are the primary target of morphine, as the deletion of MORs (encoded by the Oprm1 gene) in mice abolished the analgesic (therapeutic effect), dependence, and rewarding properties of this drug [118]. Later, it was demonstrated that MORs are the main receptors to mediate effects of other opiates, both those used in clinic to treat pain or those abused in the street for their euphoric properties (such as oxycodone, fentanyl, methadone, buprenorphine, and heroin) [109, 120]. Misuse of opioids may cause addiction and is a leading cause of the rising opioid epidemic in North America [109, 111, 119]. Furthermore, studies using MOR knockout mice demonstrated that MORs are essential to drive reward properties of other drugs of abuse including cocaine, Δ9-tetrahydrocannabinol (THC), nicotine, and alcohol [121, 122]. Deletion of MORs in mice abolished self-administration and reduced voluntary drinking of alcohol and alcohol rewarding effect [41, 123]. Furthermore, deletion of MORs abolished the acute anxiolytic-like and stimulant effects of alcohol and accelerated the development of alcohol dependence [124], indicating that MORs are essential for anxiolytic, locomotor, rewarding, and addictive properties of alcohol. Recently, circuit mechanisms underlying the role of MORs in alcohol-related behaviors have been investigated by genetic approaches. Conditional MOR deletion in GABAergic forebrain neurons using a Dlx5/6-Cre driver line (Cre expressed in GABAergic neurons [125] crossed with floxed Oprm1 mice) diminished voluntary alcohol drinking and alcohol rewarding properties [126]. Endogenous peptides binding to MORs such as β-endorphin and his precursor proopiomelanocortin (POMC) have also been implicated in alcohol use and abuse disorders [121, 127, 128]. Specifically, β-endorphin heterozygote knockout mice showed an increased voluntary alcohol consumption in a two-bottle choice paradigm [46]. In another study, voluntary consumption was reduced in β-endorphin knockout mice with a stronger reduction in female mice [47]. Recently, a mouse line with a genome-wide deletion of neuronal Pomc enhancer 1 was generated [127]. This deletion reduces specifically β-endorphin levels in the hypothalamus and decreases significantly alcohol consumption in two alcohol drinking paradigms: drinking in the dark and intermittent access [127]. Together, these studies revealed a contribution of β-endorphin in alcohol consumption.
The most studied MOR single nucleotide polymorphism (SNP) is the A118G substitution of opioid receptor mu gene (OPRM1; rs1799971, exon 1), which replaces asparagine at position 40 by aspartate (Asn40Asp) [129]. Functionally, this substitution has been characterized as creating a new CpG site for methylation and may explain the reduced MOR messenger RNA (mRNA) levels identified in vivo and in vitro [129]. In parallel, it was also shown to remove a potential N-glycosylation site in the extracellular domain altering β-endorphin binding and signaling [130]. The human A118G variant has also been studied in animal genetic models of alcoholism. In this model, exon 1 of the human OPRM1 with or without the variant A118G was inserted into mice [131]. Striatal dopamine release induced by alcohol was increased in A118G carrier in both humanized mice and humans [131]. Moreover, 118GG mice drank significantly more alcohol using a continuous access paradigm compared to control animals [132, 42].
Altogether, studies using knockout, conditional knockout, and knock-in strategies demonstrate that MORs are crucial in the development and maintenance of alcohol drinking and support the fact that naltrexone (a MOR antagonist) is one of the few available authorized medications to treat alcohol abuse [133].
DORs
Considering that DORs play a role in mood disorders such as depression and anxiety [113, 134] and that depression-like and anxiety-like behaviors are comorbid with AUD [135], the role of DORs in alcohol addiction has been investigated as a potential target for treatment of AUD. It has been suggested that acute and chronic alcohol exposure can affect the expression, pharmacology, and function of DORs [136, 137]. For instance, prolonged alcohol exposure using two-bottle limited access [138] or alcohol-induced conditioned place preference (CPP) procedures increased expression of DORs [139]. Moreover, acute alcohol injection in rats alters the binding of selective ligand of the DORs ([3H][d-Pen2,d-Pen5]-enkephalin ([3H]-DPDPE)) in the prefrontal cortex (PFC), caudate putamen (CP), nucleus accumbens (NAc) core and shell, and substantia nigra [140]. At the behavioral level, mice with total deletion of DORs showed an increased preference for alcohol measured in the two-bottle choice procedure and have an increased alcohol intake in this same paradigm only after a long exposure to an operant alcohol administration procedure [44]. Importantly, mice with C320T polymorphism (Ala107Val) in exon 2 of the DOR gene (Oprd1) showed an enhanced alcohol preference in CT heterozygous mice compared to homozygous CC mice [141]. Regarding the endogenous agonists, mice lacking the precursor preproenkephalin (Penk) learned to self-administer alcohol similarly to wild-type mice [142] and showed an intact rewarding effect of alcohol and a normal pattern of alcohol consumption [48]. However, alcohol drinking was reduced in Penk knockout mice under stressful conditions [47]. Given the important role of DORs in anxiety-like responses [113], the latter observations support a role for DOR/pEnk signaling in regulating emotional responses that may impact alcohol-intake behavior. In alcoholics, however, no change in PENK mRNA was detected in any brain structures tested including the dorsolateral PFC, orbitofrontal cortex, and hippocampus [143]. Furthermore, a recent analysis of SNPs in PENK in multiplex alcohol-dependent Caucasian American families provided no support for the association of variations in PENK with alcohol dependence [144]. Further studies to investigate the role of pEnk and DORs in alcohol dependence in different populations will be for high interest. Altogether, these findings show that DORs may promote alcohol consumption via regulation of emotional processes.
KORs
KORs are implicated in many biological processes, such as pain, stress, anxiety, and depression [145, 146]. The role of KORs in alcohol-related behaviors has been reviewed in detail in [147,147,149]. While study in rats showed that blockade of KORs with n-BNI, a KOR antagonist, increased alcohol consumption in a two-bottle choice paradigm [150], total deletion of KORs in mice was associated with a reduced alcohol intake in the same paradigm [45, 151]. Interestingly, conditional knockout of KORs in the central nucleus of the amygdala (CeA) resulted in reductions of alcohol intake in male but not female mice [152]. The fact that KOR conditional knockout within the CeA did not affect alcohol drinking in female mice may suggest that the mechanism of protective action of KOR deletion is likely different between sexes. Moreover, KOR total knockout mice showed a potentiation of the alcohol-evoked dialysate DA levels in the NAc, suggesting that reduction of endogenous activity of KOR systems may lead to an enhancement of the reinforcing efficacy of alcohol [153]. The reasons for these incongruent findings are still not clear. It is, however, worth noting that the amount of alcohol consumed depends on the efficacy of alcohol’s reinforcing effects. Thus, increased reward value of alcohol could result in reduced drinking when high concentrations of alcohol are used, as lower amounts of alcohol would be sufficient to reach the same rewarding effects. Further studies using instrumental learning tasks may be critical for differentiating the role of KOR systems in modulating the reinforcing efficacy of alcohol.
At the peptide level, inhibition of dynorphin (an opioid peptide that acts primarily at KORs) through total deletion of the prodynorphin Pdyn gene increased voluntary consumption in two-bottle choice procedure in mice [49, 50]. In addition, rewarding effect of alcohol was unchanged [154,154,156] or increased [49] in mice lacking Pdyn as measured by the CPP procedure. The latter observation supports the drinking data and the reported aversive-like activity of dynorphin peptides. Pdyn knockout mice otherwise showed a normal increase in stress-induced alcohol preference [50, 156] but developed stronger withdrawal signs after chronic alcohol [49]. Conditional knockout of Pdyn in CeA neurons resulted in decreased alcohol consumption in males and females and reduced pDyn neuron excitability in female mice following alcohol drinking [152]. In summary, these studies synthesize what is known about the role of KOR/pDyn in alcohol phenotype and point out the sex differences in dynorphin and KOR systems in the regulation of alcohol drinking behaviors.
Nociceptin/Orphanin FQ Receptor
The closest homolog of the opioid receptors is the opioid-like receptor (ORL1 or nociceptin/orphanin FQ receptor (NOR)) [157]. This GPCR is targeted by the 17-amino acid neuropeptide nociceptin also termed orphanin FQ [158, 159]. Nociceptin and its receptor which couple to Gi/o proteins are widely distributed in brain areas that have been associated with reward-seeking behaviors. In human, in a Scandinavian population, SNP rs6010718 in the gene encoding NOR (OPRL1) showed significant association with alcohol dependence [160]. In rodents, several pharmacological studies have suggested that NOR signaling is involved in the regulation of addiction-related phenotypes, including drug wanting and taking and relapse to several drugs, including opioids, cocaine, and alcohol [157, 161,161,163]. When tested for alcohol intake, NOR knockout mice showed a lower preference for alcohol than wild-type mice [51]. However, this observation needs to be consolidated with further studies. Indeed, mutant mice also showed a disturbed water intake which may not conclusively prove the presence of any specific association between deletion of NOR and reduced alcohol intake. In addition, female mice lacking NOR and their control animals showed similar alcohol-induced CPP, suggesting a rewarding effect of alcohol in those not affected by NOR deletion [164]. Interestingly, rats lacking NOR showed reduced alcohol operant self-administration compared to outbred controls [52]. In summary, while pharmacological studies showed a promising finding that supports a role for NOR antagonism as a potential treatment option for AUD, data from engineered rodent models are still preliminary and need further investigations.
Altogether, preclinical research using genetically modified rodents has revealed the crucial roles of opioid receptors in the development and expression of alcohol-related behaviors that may be important in humans. However, given the impact of the opioid system in the acute and chronic effects of alcohol, further molecular studies identifying functional polymorphisms in opioid genes are needed to afford a more focused pharmacogenetic investigation.
Endocannabinoid Receptors
The endocannabinoid system consists of cannabinoid type 1 receptor (CB1R) and cannabinoid type 2 receptor (CB2R), their two most characterized endogenous agonists anandamide (AEA) and 2-arachidonoylglycerol (2-AG), as well as the proteins that control endocannabinoid production and degradation such as the catabolic enzyme fatty acid amide hydrolase (FAAH). The endocannabinoids are lipid-signaling molecules that regulate numerous peripheral and central biological processes through the activation of CB1R and CB2R [165,165,167].
Gi/o proteins mediate the canonical mode of CB1R and CB2R signaling. CB1R is the most densely expressed GPCR in the brain, whereas until the last decade, neuronal CB2R distribution in the brain remained unclear [167,167,169]. The endocannabinoid system influences dopamine neurotransmission and thus associated behavioral responses including reward [169,169,171]. CB1R is abundant in brain areas that control motivation, impulsivity, or reinforcement processes, in particular in the cortex, the hippocampus, and the basal ganglia [167,167,169, 172]. At the subcellular level, CB1R is predominantly located at GABAergic and glutamatergic presynaptic terminals. Postsynaptic release of endocannabinoids typically mediates retrograde inhibitory signaling to rapidly limit neurotransmitter release [169, 172, 173]. CB2R was originally considered absent from healthy brain tissue but was observed in activated microglia [169, 174]. More recently, CB2R was also located in the midbrain and striatal neurons along with other structures of the mesocorticolimbic system and its activation inhibits firing of dopaminergic neurons [169, 170]. Owing to its key role in neurotransmission, synaptic plasticity, and addiction-related behaviors, the endocannabinoid system has emerged as a reasonable target for the treatment of alcohol abuse [169, 171, 175]. This possibility has been further strengthened by human studies indicating that genetic polymorphisms including SNPs (e.g., rs1049353 and rs2023239) within the CNR1 gene encoding CB1R were associated with several features of AUD [37, 176, 177]. Thus, the development of endocannabinoid-based compounds and the generation of CB1R and CB2R mutant mice have understandably spurred scientists to preclinical and clinical initiatives [37, 175, 178,178,179,181].
Different CB1R lacking mice on distinct genetic backgrounds were generated by targeted disruption of the Cnr1 gene. Animals appeared healthy but exhibit some behavioral alterations with discrepancies between studies. For instance, Zimmer et al. [182] observed hypoactivity in mutant mice whereas other reports did not [56, 183]. Nonetheless, many studies converged to the finding that deletion of the Cnr1 gene altered physiological actions of alcohol along with alcohol-related behaviors. Mutant mice showed a strong inhibition of accumbal dopamine release following intraperitoneal administration of alcohol [55]. Alteration in dopaminergic signaling in response to lifelong suppression of CB1R is also substantiated by enhanced expression of the striatal DA receptor D2R [184]. In line with these findings, alcohol-induced hyperlocomotion [56] and CPP for alcohol were reduced in mice lacking CB1R [54, 184]. Importantly, genetic disruption of CB1R has been also reported to decrease voluntary alcohol intake in both male and female animals as assessed by different two-bottle choice paradigms. Specifically, CB1R knockout mice displayed lower alcohol consumption when they had a limited access to 10% alcohol for 6 h or 8 h [53, 54]. Similarly, alcohol drinking was also reduced under continuous access of solutions containing various concentrations of alcohol [23, 55, 56]. Except for one study [57], consistent results were also obtained regarding an attenuation of alcohol preference [23, 54, 56, 185, 186]. Of note, Racz et al. [57] also reported that unlike wild-type mice, CB1R mutant animals exposed to a stressor induced by a mild 5-min foot shock did not display any transient increase in alcohol intake and preference. In contrast to alcohol consumption, mutant mice expressed different drinking phenotypes with regard to sucrose intake. Poncelet et al. [53] reported a decrease of consumption using a 5% sucrose solution in a limited access procedure, whereas Naassila et al. [56] did not detect any difference in sucrose preference when mice were given a 48-h continuous access to 0.87% and 1.70% solutions. Besides voluntary alcohol consumption, CB1R deletion was shown to modulate signs that are indicators of alcohol withdrawal severity. However, once again depending of the genetic background or the procedure used, both enhancement [56] and reduction [57, 187] of withdrawal severity were described. Importantly, pharmacological blockade of CB1R signaling altered alcohol consumption and other alcohol-related behaviors in mice and rats under various procedures [169, 179], further highlighting that the endocannabinoid system is essential for the expression of behavioral effects of alcohol. Although detailed neurochemical and molecular mechanisms are largely lacking, pharmacological and genetic models provided together a global picture of CB1R implication in the effects of alcohol in which CB1R signaling, in brain structures that remain to be accurately defined, likely contributes to motivate and reinforce alcohol drinking [169, 171, 179]. Nonetheless, a prevailing role of peripheral CB1R in alcohol actions cannot be ruled out yet. Indeed, a recent study interrogated the contribution of CB1R signaling by taking advantage of peripherally restricted ligands in conjunction with null ghrelin and ghrelin receptor mice (see also the “Ghrelin Receptor” section below) and revealed a complex functional crosstalk between these two GPCR systems [23]. The data suggested that activation of peripheral CB1R promotes gastric production of the hormone ghrelin, thereby inducing vagus nerve stimulation which contributes to alcohol drinking. Consequently, preferential blockade of this periphery-to-brain signaling network with nonpenetrant brain CB1R or ghrelin receptor inverse agonists holds a promise for translation into clinical trials. This is particularly apropos for CB1R signaling because clinical trials conducted in alcohol abusers with the brain penetrant inverse agonist rimonabant (SR 141716) showed limited efficacy [175, 178, 181]. Most importantly, this compound was withdrawn from the market in 2008 due to serious psychiatric side effects. Consequently, more attention was naturally devoted to CB2R [37, 109].
Actually, an early genetic study in Japanese pointed out an association between a polymorphism in the CNR2 gene, which encodes CB2R, and alcoholism. This 2-bp replacement polymorphism (rs2501432) results in Q63R amino acid substitution and in a lower functional response of CB2R to its ligands [169, 188]. In addition, Cnr2 gene expression was found to be reduced in key brain structures of mice that exhibit higher alcohol preference [188, 189]. A significant progress in understanding how CB2R signaling could modulate alcohol-related behaviors has been made with the use of CB2R knockout mice that were tested under diverse procedures [58]. Mice with lifelong deletion of the Cnr2 gene appeared healthy but exhibited altered behavioral phenotypes such as decreased spontaneous locomotor activity and altered gene expression in the mesocorticolimbic system [58, 190]. Deletion of the Cnr2 gene resulted in enhanced severity of withdrawal signs revealed following administration of a single high dose of alcohol. With respect to wild-type mice, mutant mice also exhibit increased preference for the alcohol-conditioned place. Importantly, genetic blockade of CB2R enhanced voluntary alcohol intake and alcohol preference in male animals in a continuous access two-bottle choice drinking paradigm in which alcohol concentration was gradually increased from 2 to 8% within 16 days. In contrast, taste preference for saccharin and quinine was similar regardless of the genotype. Further, self-administration of alcohol using nose-spoke operant responding was enhanced in CB2R knockout mice originating from two different genetic backgrounds. Of note, when a progressive ratio schedule was conducted to quantify how much work can be provided by animals to earn a reinforcer, Ortega-Álvaro et al. [58] observed a higher breaking point (defined as the highest response rate accomplished to obtain a single reward [191]) for mutant mice, suggesting a greater sensitivity to the reinforcing properties of alcohol. Altogether, these behavioral tests indicated that CB2R signaling, in brain structures that remain to be precisely identified, modulates the motivation to consume and seek alcohol. Other tests using mutant mice also indicated that CB2R could be important in handling stress situations that are associated with alcohol drinking [192]. The neurochemical and molecular mechanisms by which CB2R signaling mediates alcohol effects remain unclear. However, by quantifying the mRNA encoding tyrosine hydroxylase (TH) and MOR in the ventral tegmental area (VTA) and the NAc, respectively, Ortega-Álvaro et al. [58] observed higher expression of MOR mRNA in naïve CB2R knockout mice and enhanced expression of both transcripts in response to a single oral administration of alcohol only in mutant animals. These data raise the possibility that a functional crosstalk between the dopaminergic, the opioidergic, and the endocannabinoid systems in mesolimbic structures could contribute to the neurobiological mechanisms underlying alcohol-reinforced behaviors. The therapeutic potential of targeting CB2R signaling was further strengthened by a complementary pharmacological study showing that the use of a selective agonist and inverse agonist of CB2R reduced and enhanced respectively alcohol self-administration in wild-type mice [193]. Finally, besides CB1R and CB2R, mutant mice lacking FAAH, which catabolizes AEA, showed increased alcohol drinking and preference in two-bottle choice paradigms [185, 194, 195].
The use of mutant mice clearly allowed light to be shed on the cannabinoid receptors as promising targets to modulate pathological drinking. Despite discouraging clinical data regarding the use of rimonabant, the endocannabinoid system, and in particular CB2R, is still in the limelight for the treatment of alcohol abuse.
Ghrelin Receptor
Ghrelin is an appetite-stimulating hormone which consists of a 28-amino acid octanoylated peptide mainly produced by endocrine cells located in the gastric fundus. Ghrelin is the endogenous agonist of the growth hormone secretagogue receptor (GHS-R1a) whose activation promotes growth hormone (GH) release from the pituitary. Currently, ghrelin is principally regarded as a multifaceted controller of energy homeostasis, metabolic functions, and neurobiological processes by acting on both peripheral and central circuitries [196,196,197,198,200]. Human and rodent ghrelin genes encode preproghrelin that is the precursor of obestatin, ghrelin, and unacylated ghrelin peptides. The biological activities of ghrelin require acylation of its serine 3 residue that is catalyzed by the ghrelin O-acyltransferase (GOAT) encoded by the Mboat4 gene in mice [23, 200,200,202]. Besides ghrelin, a new physiologically relevant endogenous ligand for GHS-R1a, namely liver-expressed antimicrobial peptide 2 (LEAP2), was recently uncovered [203], and unexpectedly, LEAP2 exhibits potent inverse agonism toward GHS-R1a [204].
GHS-R1a predominantly mediates Gq signaling. GHS-R1a exhibits strong constitutive activity in vitro [205,205,207] which is thought to also have physiological implications in vivo [22, 208, 209]. GHS-R1a is enriched in the anterior pituitary gland and in different nuclei of the hypothalamus. GHS-R1a is also expressed in brain circuits involved in motivation, reinforcement, impulsivity, and emotional processes [199, 200, 210]. Of note, brain and peripheral administration of ghrelin stimulates the mesolimbic dopamine system and brain GHS-R1a activation induces hyperlocomotion and enhances motivation-related behaviors [211].
While ghrelin regulates various neurobiological functions, its synthesis within the brain remains debated [212] and it is also unclear whether endogenous blood ghrelin can access deep brain structures [213]. Nonetheless, the ghrelin system is largely considered as a key element of the gut–brain axis and represents a potential druggable target for drug- and alcohol-related disorders [211, 214,214,215,216,217,219]. Interestingly, genetic human studies have detected associations between SNPs in both GHRL (e.g., rs696217) and GHSR (e.g., rs2948694 and rs2232165) genes and different features of AUD [211, 220,220,222]. In addition, preclinical and clinical studies substantiate a causal implication of the ghrelin system in key alcohol-related outcomes [211, 215, 223].
Mutant mice along with pharmacological agents were appropriately combined to explore the contribution of endogenous ghrelin and its receptor in the development and the expression of key alcohol-related behaviors. Both heterozygous and homozygous mutant mice with gene disruption of the Ghsr gene, encoding GHS-R1a, showed a reduction in locomotor activity and a strong inhibition of accumbal dopamine release following intraperitoneal administration of alcohol. In addition, alcohol CPP is reduced in Ghsr null mice. However, compared to wild-type mice, knockout mice consumed approximately the same amount of alcohol in a two-bottle choice limited access [59]. In contrast, Godlewski et al. [23] recently observed that Ghsr null mice had lower alcohol preference and consumption in a continuous access procedure with no difference in total fluid and food intake. Using three different paradigms, Zallar et al. [223, 224] also recently evaluated alcohol drinking in a newly developed GHS-R1a knockout rat model. These animals showed a reduction in sweetened alcohol self-administration and break point while water and non-alcoholic sweet solution responding were not affected. In the drinking in the dark limited access paradigm, only high drinker mutant rats consumed less unsweetened alcohol compared to the wild-type high drinkers. However, no difference in total alcohol intake was detected between the two rat genotypes in a two-bottle choice 24-h intermittent access paradigm. Fluid intake was not measured 30 min after the beginning of the drinking session which is an alternative way to evaluate excessive alcohol consumption [225,225,226,228]. Together, these results suggest that the GHS-R1a/alcohol consumption interplay is complex in rodents and could be revealed using only certain paradigms such as the ones that model the initial intake of a high amount of alcohol [224]. This is a possibility in line with the physiological role of neuronal GHS-R1a signaling which is important for inducing feeding and is sensitive to the palatable and energetic properties of food [200, 223, 229]. Besides the use of mutant mice, pharmacological disruption of GHS-R1a signaling with antagonists consistently led to the reduction of alcohol consumption as well as other alcohol-reinforced behaviors in rodent models [198, 215, 223].
Although the Ghrl gene encodes preproghrelin which gives rise to several biologically active peptides, its disruption was also implemented to evaluate the contribution of endogenous ghrelin in alcohol-related behaviors [23, 61, 62]. Heterozygous and homozygous Ghrl mutant mice exhibited a reduction in alcohol-induced locomotor activity and accumbal dopamine release. In addition, ghrelin suppression led to a decrease in alcohol-induced CPP. Regarding voluntary alcohol consumption, once again, varying results were obtained depending on the alcohol drinking paradigm. Jerlhag et al. [61] did not observe any genotype effect in a two-bottle choice limited access (90-min) procedure. In contrast, two other studies reported that alcohol consumption and preference were lower in mutant mice subjected to a two-bottle choice continuous access paradigm [23, 62]. Interestingly, this decrease appeared to be selective for alcohol as no genotype effect was statistically detected for total fluid, saccharin, and quinine consumption [23, 62]. Besides preproghrelin and GHS-R1a mutant rodents, voluntary alcohol drinking was also recently examined in mice lacking the GOAT enzyme, which is responsible for the acylation of ghrelin. Targeted disruption of Mboat4, using CRISPR/Cas9-mediated genome engineering, generated Mboat4−/− mice with reduced alcohol preference and consumption in a continuous access procedure but with similar fluid and food intakes with respect to their wild-type littermates [23]. These data largely mirrored the abovementioned studies undertaken with GHS-R1a and ghrelin mutant animals. Overall, both genetic and pharmacological inactivations of the ghrelin system obviously helped to apprehend its neurobiological contribution in the reinforcing effects of alcohol. These preclinical studies have been instrumental in the transition into clinical trials specifically for the GHS-R1a inverse agonist, PF-5190457 [214, 216, 230].
In light of recent studies identifying LEAP2 as an endogenous ligand of GHS-R1a that counter-regulates ghrelin action in vivo [203], the generation of mutant LEAP2 mice could be also exploited in an effort to further explore how the ghrelin system regulates physiological and behavioral responses for alcohol. Finally and as mentioned above, Godlewski et al. [23] recently reported that the endocannabinoid and the ghrelin systems act in concert peripherally to drive alcohol intake through activation of a gut–brain axis.
Serotonin Receptors
Serotonin (5-HT) is a monoamine neurotransmitter widely expressed throughout the central nervous system where it plays an important role in the regulation of mood. Serotonin acts via 7 classes of receptors (5-HT1–7). All 5-HT receptors are coupled to G proteins with the exception of the 5-HT3 receptor, which is an ion channel. The 5-HT system has been extensively implicated in the regulation of mood disorders and the reinforcing properties of drugs of abuse, including alcohol. Within the Gi-coupled 5-HT1 receptor class, the 5-HT1B receptor gene (HTR1B) SNPs, the G861C and the A-161T polymorphisms, were reported to play important roles in alcohol dependence in human [231, 232]. Moreover, SNPs within the 5-HT1A receptor gene (HTR1A) were also associated with alcohol dependence [233]. HTR2A and HTR2C genes were also reported as potential candidate genes for alcohol-related traits [234]. Due to the complexity of serotonergic neuromodulation at presynaptic and postsynaptic levels, as well as the plethora of serotoninergic receptor subtypes that leads to various and contrasting effects of serotoninergic ligands [33], transgenic methods appeared as very useful tools for the investigation of this system. However, and despite the fact that the first alcohol study using a genetic-based approach was performed with 5-HT1B receptor knockout mice [35], few studies have yet investigated the potential implication of specific serotoninergic receptors using transgenic animal models. Invalidation of the 5-HT1B receptor gene basically led to increased alcohol intake in two-bottle choice and operant procedures [35, 235]. However, this effect was controverted [74, 236], emphasizing the crucial importance of the mouse genetic background and the interaction with other alcohol-related genes (see [33] for more details). This discrepancy may also reflect the relatively modest implication of this receptor in the regulation of alcohol consumption. Finally, the potential implication of the Gs-coupled 5-HT6 receptor was also tested. Null mutant mice for this receptor did not differ from wild-type mice in terms of alcohol intake but were more sensitive to the stimulating effect as well as the sedative effect of alcohol [75]. Although these studies suggest the implication of 5-HT receptors in alcohol-related behaviors, this system remains under-investigated in the field. Undoubtedly, further in-depth investigations are necessary to dissect out the respective contribution of each receptor as well as the importance of their sublocalization, in order to fully capture the influence of this complex system on alcohol-related behaviors.
Dopamine Receptors
Although DA is clearly involved in the reinforcing effect of alcohol [237] as well as in the mechanisms underlying alcohol dependence and addiction [238], delineating the specific contribution of each DA receptor remains challenging. Indeed, it is difficult to find agonists or antagonists able to discriminate between DA receptor subtypes, as they are closely, structurally related within each subfamily (i.e., D1R and D5R for the D1R-like families which are coupled to Gs proteins, and D2R, D3R, and D4R for the D2R-like families which are coupled to Gi/o proteins). In addition, the complex presynaptic and postsynaptic sublocalization and neuroanatomical localization of DA receptors, as for 5-HT receptors, lead to strong paradoxical effects of DA ligands, as most of them can either increase or reduce alcohol-related behaviors depending on the dose and/or the model used (for review, see [239]). In this context, transgenic approaches, and more particularly conditional ones, appeared as a tool of choice to dissect the implication of a specific DA receptor subtype in alcohol drinking behaviors.
Dopamine D1R-Like Receptors
Although few studies have examined the consequence of deleting D1R (encoded by the Drd1 gene) on alcohol-related behaviors, evidence suggests its contribution to alcohol consumption. Indeed, alcohol intake and preference have been found to be reduced in D1R knockout mice in limited or continuous access two-bottle choice paradigms, while overall fluid intake is preserved [63]. Accordingly, selective inactivation of the metabotropic glutamate receptor 5 (mGluR5) in D1R-expressing neurons prevents escalation of alcohol drinking after a period of abstinence [240]. In addition, D1R knockout mice fail to develop locomotor sensitization to repeated administration of alcohol [241]. These results are also in line with the broad implication of D1R in reinforcement (e.g., [242]) and accumbal D1R-expressing neurons in the initiation of reinforcement learning associated with alcohol drinking [243]. As far as we know, transgenic approaches are yet to be used in mice to study D5R in relation to alcohol. However, the psychomotor effect of cocaine has been found to be reduced in D5R knockout mice [244], suggesting that this receptor might be implicated in some effects of drugs of abuse. DRD1 polymorphisms have been shown to play a role in alcohol dependence. Indeed, DRD1 polymorphism was associated with excessive alcohol drinking behaviors in nonhuman primates in the context of early environmental stress induced by maternal separation [245].
Dopamine D2R-Like Receptors
Several behaviors related to alcohol have been found to be reduced or abolished in D2R knockout mice, such as alcohol sensitization [241] (but see [65]) and place preference [246] and alcohol drinking in two-bottle choice paradigms [64,65,66,67] as well as in operant procedure [68]. These studies therefore point toward a pivotal role of D2R. However, due to the relative specific and complex localization of D2R (e.g., presynaptic autoreceptors on DA neurons or postsynaptic receptors of medium spiny neurons of the indirect pathway and cholinergic interneurons in the striatum), conditional approaches appear necessary to investigate the potential multifaceted implication of this receptor in alcohol consumption and addiction. For instance, a recent study has induced a specific overexpression of postsynaptic D2R in D2R-expressing neurons in the NAc, by using D2-Cre mice and a floxed virus coding for D2R, and subsequently found an increase in alcohol intake in a continuous but not intermittent access two-bottle choice paradigm [247].
Although pharmacological studies clearly indicate a strong implication of D3R in alcohol self-administration and relapse in rodents [239], the use of transgenic mice has yielded conflicting results. Whereas some studies have evidenced a near absence of alcohol sensitization [241] and reduced alcohol consumption in D3R knockout mice [69], others have found neither alteration of alcohol rewarding effects for alcohol nor alteration of alcohol drinking behaviors evaluated in two-bottle choice or operant self-administration procedures [70, 71]. Despite some methodological differences between these studies (see [69] for details), the cause of these discrepancies remains unclear. It should be noted, however, that the D3R may be presynaptic or colocalized postsynaptically with D1R and D2R. Consequently, a complex implication of this receptor in the modulation of DA in response to alcohol may have mitigated the effects of the genetic intervention. For the future, and as for D2R, conditional approaches should be favored to disentangle the potential implication of D3R depending on its localization.
Ablation of D4R also modifies alcohol-related behaviors, but in an opposite direction than the other DA receptors. Indeed, D4R knockout mice are more responsive to the psychomotor effect of alcohol [248] and show increased alcohol consumption [72].
In human, the D2R gene (DRD2) on chromosome 2 [249] polymorphism and the D4R (DRD4) gene on chromosome 11 [250] polymorphism have been shown to play a role in alcohol dependence. Two splice variants of the D2R, which differ by a 29-amino acid insert in the third intracellular loop in D2-long (D2L) that is absent in D2-short (D2S), have been associated with alcohol dependence in human [251]. Furthermore, the DRD2 gene single nucleotide polymorphism rs1076560 which is involved in regulating the splicing of the gene and modified the presynaptic and postsynaptic ratio of D2R isoforms as well as TaqIA polymorphism of the DRD2 gene were both associated with an increased risk of developing alcohol dependence [252, 253]. In a mouse model, a study found an increased alcohol intake among D2L receptor knockout mice [254]. Polymorphisms of the DRD4 gene, a 48-bp variable number tandem repeat in the third exon, have been also reported to be associated with heavy-drinking behaviors when certain social–environmental factors were present [255, 256].
To conclude, these studies clearly point toward a significant implication of the different DA receptors in alcohol-related behaviors and, notably, in its reinforcing effect. However, the use of state-of-the art conditional approaches will be a necessary step in order to further decipher the complex contribution of each receptor in alcohol-motivated behaviors and AUD.
Metabotropic Glutamate Receptors
Glutamate receptors are divided into two categories: the ionotropic receptors (iGluR: N-methyl-d-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate) and the metabotropic receptors, named mGlu receptors (mGluR) classified in subgroups (groups I, II, and III), depending on their sequence homology, pharmacology, and intracellular signaling pathway activated. Group I contains the mGluR1 and mGluR5 which are primarily coupled to Gq proteins and mainly localized at the postsynaptic level. Group II (mGluR2 and mGluR3) and group III (mGluR4, mGluR6, mGluR7, and mGluR8) are coupled to Gi proteins and mainly localized at the presynaptic level where they can regulate neurotransmitter release [257]. The implication of glutamate receptors, iGluR and mGluR, in addiction has long been demonstrated for a large variety of drugs of abuse including alcohol [258, 259] (for reviews, see [260, 261]). In human, an association between a polymorphism in the variable (GT)(n) repeat in the 5′-regulatory region of NMDA GRIN2A subtype and alcoholism was previously detected [262]. Moreover, higher risk for developing alcohol dependence was observed in individuals with specific GRM5 (mGluR5-encoding gene) polymorphisms [263, 264].
Studies on consequences of mGluR deletion in mice on alcohol-related behaviors are relatively sparse. Concerning group I and, specifically, mGluR5, two studies were published in 2008 using mGluR5 null mice. First, Bird et al. [76] generated mGluR5 null mice on a C57Bl/6J background and submitted them to various alcohol-related behaviors. In this study, the authors only used male mice [76]. They found a reduced alcohol consumption and preference in a two-bottle choice paradigm as compared to their wild-type littermates using 5% and 10% alcohol solutions. For both concentrations, the reduction in consumption and in preference reached about 50%. It is noteworthy that the authors did not find any effect of the mGluR5 deletion on saccharin consumption, suggesting a specific role of mGluR5 in alcohol consumption [76]. Using a classical protocol of CPP with a relatively low dose of alcohol (1 g/kg), they found that the mGluR5 null mice were able to develop place preference for the alcohol-paired compartment whereas the wild-type mice, with such a low dose, did not show any preference after the conditioning sessions. This observation suggests a higher sensitivity of the mGluR5 null mice to the rewarding properties of alcohol. The same year, Blednov and Harris [77] published a study using mGluR5 null mice on a mixed C57Bl/6J × 129/SvJ genetic background and they used male and female mice. The authors evaluated alcohol consumption using five different paradigms of consumption: progressive two-bottle choice (sequentially 3%, 6%, 9%, and 12% ethanol solutions), four-bottle choice (simultaneously 0%, 4%, 8%, and 12% ethanol solutions), drinking in the dark with one bottle, drinking in the dark with two bottles, and a scheduled fluid access (5% alcohol solution). Overall, they found reduced alcohol consumption only in the four-bottle choice and the drinking in the dark procedures in the female mice. Males were tested only in the progressive two-bottle choice paradigm with no difference between the genotypes. Beside the aspects directly correlated to the consumption and the sedation induced by the administration of alcohol, the authors have shown in the mGluR5 null mice a basal hyperlocomotion that is not followed by an alcohol-induced hyperlocomotion. Indeed, after moderate to high doses of alcohol (1.5 g/kg, 2 g/kg, 2.5 g/kg), the mGluR5 null mice exhibit a significantly lower locomotor activity than their wild-type littermates in both males and females. All together, these results are in line with the ones observed by Bird and colleagues [76], demonstrating the importance of the mGluR5 in the responses to alcohol in a large variety of alcohol-related behaviors. Recently, Campbell and colleagues [78] studied the role of the mGluR5 specifically within the bed nucleus of the stria terminalis (BNST), a brain structure part of the extended amygdala that plays a role in response to stress and to alcohol [265, 266]. For this study, the authors used a pharmacological approach in transgenic mice expressing a point mutation of mGluR5 (GRM5AA/AAvs. their wild-type littermates (GRM5TS/TS)) that cannot be phosphorylated by extracellular signal–regulated kinases (ERKs) and thus block the association with the scaffolding protein Homer. By doing so, no further activation of intracellular signaling pathway is possible. For the purpose of this review, we will just focus on the behavioral aspects observed with the GRM5AA/AA mice. Campbell et al. [78] found that the mutated mice consumed more alcohol in different drinking paradigms (continuous progressive two-bottle choice paradigm, single-bottle drinking in the dark paradigm, and operant self-administration paradigm). In addition, in a CPP test with three different alcohol doses tested (1 g/kg, 2 g/kg, and 3 g/kg), the authors found that the GRM5AA/AA are less sensitive to the rewarding properties of alcohol at the lowest dose (1 g/kg) whereas they are insensitive to the aversive properties of the highest dose (3 g/kg). These results are on the opposite direction than the ones observed with the mGlu5R null mice, suggesting differential mechanisms of action of the mGluR5 that do not necessarily imply the association with scaffolding protein such as Homer in the regulation of alcohol-related behaviors. The specific localization of the inactivation of the mGluR5 pathway, specifically within the BNST, also suggests a brain region–specific role for this mGluR.
The involvement of mGluR2 has also been studied thanks to knockout mice. Zhou and colleagues [79] demonstrated that the mGluR2 null mice show a greater escalation of alcohol consumption in a progressive two-bottle choice paradigm (3%, 6%, 9%, 11%, 13%, 15%, and 17% over 80 days). The preference for alcohol was also increased, but more slightly, with the mGluR2 null mice for the highest concentrations. It is noteworthy that saccharin consumption (0.066%) is moderately decreased in the knockout mice as compared to their wild-type littermates. Quinine test does not reveal a difference between the genotypes.
In another study, Blednov et al. [80] evaluated the role of mGluR4 in a variety of alcohol-related behaviors. In regard to alcohol consumption, they did not find any difference between the mGluR4 null mice and their wild-type littermates (progressive two-bottle choice paradigm with 3%, 6%, 9%, and 12% alcohol solutions). Another receptor member of group III has been studied with mutated mice, namely the mGluR7 encoded by the Grm7 gene. In this study, the authors found that their B6.129/Ola-Grm7tmNovartis knockout male mice drank significantly more alcohol in a progressive two-bottle choice paradigm (3%, 6%, 12%, and 18% alcohol solutions) and found a trend in female mice [81]. These results should be interpreted with caution due to the small number of animals used in this study.
All these studies concerning members of different groups of mGluR confirmed the relevance of studying these receptors in alcohol-related behaviors. It is somehow difficult to generalize with this low number of studies, but knockout of mGluR from group I led to a decrease in alcohol consumption whereas the knockout of the mGluR from groups II and III led to an increase or no change in alcohol consumption. This difference might be related to the synaptic localization with specific segregation of group I receptors on the one side and groups II and III on the other side of the synaptic cleft. Together, these studies highlight the crucial role of metabotropic glutamate receptors in alcohol phenotypes. The use of conditional knockout will help decipher the role of each mGluR in the development of AUD.
Metabotropic GABA Receptors
GABA binds to an ionotropic receptor (GABAA) and to a metabotropic receptor (GABAB). The GABAergic system is the largest inhibitory neurotransmitter system in the brain. Accordingly, the GABAB receptors are involved in numerous behaviors, such as learning [267], and also in pathologies, such as depression [268, 269], anxiety [268], and drug disorders [270] (for review, see [271]). The GABAB receptors which coupled to Gi/o proteins are heterodimers that include a GABAB1 subunit and a GABAB2 subunit [272]. The GABAB2 subunit is very stable across species whereas the variability of the heterodimer comes from the different isoforms of the GABAB1 subunit. The two main evolutionarily conserved isoforms are the GABAB1a and GABAB1b subunits [273, 274]. This variability is accompanied by a difference in location at the synapse level. Indeed, the heterodimer B1a/B2 seems to be located at the presynaptic level while the B1b/B2 receptor is mostly postsynaptic [275].
Several pharmacological studies have underscored the therapeutic potential of manipulating the activity of GABAB receptors using, for instance, positive allosteric modulators to mitigate several alcohol-associated behaviors [276,276,278]. However, even though studies have implicated post-transcriptional modification of the GABAB1 messenger [279] and mice selectively expressing GABAB1a or GABAB1b subunits were generated [275], no studies were conducted to evaluate the role of each GABAB subunit in alcohol-related behaviors. The relevance to bridge this gap with further studies in the alcohol field is pinpointed by the results obtained by Ribeiro et al. [280]. Indeed, in this study, the authors revealed a differential regulation of the two subunits of the GABAB receptors in mice submitted to a three-bottle choice paradigm and developing a resistance to the alcohol adulteration. The authors found a greater upregulation of the transcript for GABAB1 in the PFC, hippocampus, hypothalamus, and striatum [280]. In addition, Jacobson et al. [281] demonstrated a differential response to cocaine in GABAB1a and GABAB1b knockout mice in an acute locomotor activity test and a place preference paradigm [281], suggesting specific roles for the different subunits in the response to drugs of abuse. Finally, the GABAB2 receptor gene (GABBR2) SNP rs2900512 was recently associated with an increased risk of developing alcohol dependence [282]. This association was, however, not confirmed in another study [283].
Despite the lack of studies using knockout mice, GABAB receptors seem to be very promising targets for AUD treatment as suggested by several pharmacological studies. Indeed, beside the controversial use of baclofen at high doses showing no or poor efficacy, a specific interest in the enantiomer use can be of particular interest. Few studies have demonstrated that the racemic form of baclofen (prescribed form) is always less efficient than the R(+)-baclofen form [284,284,286]. In addition, the use of positive allosteric modulators of GABAB receptors has demonstrated their efficiency in the reduction of alcohol consumption in preclinical models [277, 287,287,289]. Thus, overall, further studies investigating the GABAB receptors as therapeutic targets for AUD should show great potentials.
CRF Receptors
Corticotropin-releasing factor is a 41-amino acid neuropeptide critically involved in the regulation of neuroendocrine and behavioral responses to stress. CRF actions are exerted through two GPCRs: CRF type 1 (CRF1 encoded by the Crhr1 gene) and type 2 (CRF2 encoded by the Crhr2 gene), which share about 70% amino acid sequence identity [290, 291]. CRF1 expression is found in many brain regions and neuronal subtypes [292,292,293,295]. CRF2 is also widely expressed in the central nervous system and found peripherally [296]. The complexity of the CRF system is further increased by the existence of additional endogenous agonists. CRF receptors can be activated by the urocortin (Ucn) family of neuropeptides: Ucn1, Ucn2, and Ucn3. Ucn1 binds with similar affinity to CRF1 and CRF2, whereas Ucn2 and Ucn3 bind primarily to CRF2 [297, 298]. The role of CRF and their receptors in the context of addiction, and specifically its importance in alcohol intake and behavioral traits including locomotor sensitization that reflect neuroplasticity induced by chronic alcohol exposure, has been broadly studied [299]. Numerous pharmacological studies have identified CRF1 as a major driver of alcohol consumption in rodents. We do not review this extensive literature here but instead call attention to several reviews [300,300,301,302,304] and also to human genetic studies that have identified association between CRF1 (CRHR1 gene) polymorphisms and the risk of AUD in certain populations [305,305,306,308]. Studies using CRF knockout mice showed reduced rewarding effects of alcohol resulting in increased alcohol consumption compared to control mice in both 24-h continuous access and limited access paradigms [309]. On the other hand, reduced alcohol intake phenotype was observed in CRF-overexpressing transgenic mice using a 24-h continuous access procedure [310]. At the receptor level, at the initial phase, both CRF1 knockout mice and conditional neuronal knockout (CRF1NestinCre) mice consumed the similar amount of alcohol as their corresponding controls [82]. Notably, increased alcohol intake using the continuous access procedure was observed in CRF1 knockout mice in response to stress exposure, such as chronic social defeat and forced swim stress [82, 83]. In contrast, CRF1NestinCre mice showed a reduced stress-induced drinking behavior compared to their control littermates [82]. Moreover, following alcohol vapor exposure, a pronounced escalation of intake in the post-dependent state was detected in knockout animals compared to their control littermates [83]. In contrast to these findings, other studies have come to opposite conclusions and showed reduced alcohol intake in CRF1 knockout mice compared to control animals using binge-like drinking in the dark [84, 85] or 24-h 20% alcohol intake procedures [86]. Moreover, repeated swim stress-induced increase alcohol intake and preference were reduced in CRF1 mutant mice compared to control littermates [86]. These contradictory results are likely to be related to the use of distinct animal backgrounds and alcohol drinking paradigms in each study (see [299] for review). Importantly, CRF1 signaling seems to play a role in other alcohol-related behaviors. For instance, while alcohol-seeking behavior in operant alcohol self-administration procedure was similar between CRF1 knockout animals and their controls, withdrawal-induced increase in alcohol-seeking behavior was reduced in CRF1 knockout mice [87]. In addition, CRF1-deficient mice showed reduced development of locomotor sensitization to alcohol [309, 311]. Altogether, the majority of the data suggest that CRF1 function is important for the development of excessive alcohol consumption and critical for stress-induced changes in alcohol-related phenotypes. These effects on alcohol intake cannot be explained by differential taste palatability or differential sensitivity to the sedative–hypnotic effects of alcohol between CRF1-deficient mice and their control animals [86].
Few studies have been conducted to test the role of CRF2 on alcohol-related behaviors using mutant mice. A small and transient reduction in alcohol consumption was observed in CRF2 knockout mice that appeared to be mainly in males [85]. In addition, CRF2 mutant mice showed reduced expression of alcohol rewarding effect as measured by the CPP procedure [312]. However, CRF2-deficient mice develop normal levels of sensitization, which is in contrast with null CRF1 animals [311]. To summarize, the CRF1 receptor seems to be an interesting target in a specific context of stress that is a common feature of abstinence in patients. It is well known that this stress associated or not to anxiety can be the trigger by relapse and, thus, understanding the specific role of CRF1 could lead to better pharmacological tools to block stress-induced relapse.
Melanocortin Receptors
The melanocortin peptides are produced centrally by the cleavage of the polypeptide precursor POMC within the arcuate nucleus of the hypothalamus and the nucleus of the solitary tract [313, 314]. Among these peptides are α-, β-, and γ-melanocyte-stimulating hormones (MSHs), as well as adrenocorticotrophic hormone (ACTH) [315]. In rodents, melanocortin peptides act through five GPCR subtypes, i.e., MC1–5R, all are known to be coupled to Gs proteins (also to Gq or Gi in some instances (see [316] for review) that stimulate the adenylyl cyclase pathway [317]. MC3R and MC4R are the most predominant MCR subtypes expressed in the brain [318], and modulation of the activity of these receptors impacts hypothalamic-dependent physiological functions [319].
In rats, acute and chronic exposures to alcohol have been shown to alter the immunoreactivity of melanocortin peptides in several brain regions [320,320,322]. Moreover, AA (alko, alcohol) rats, which are selectively bred to prefer alcohol, exhibit abnormal expression patterns of MC3R in several brain regions compared to ANA (alko, non-alcohol) rats [323]. Multiple pharmacological studies have also revealed the involvement of the melanocortin system in the neurobiological response to alcohol intake (for review, see [324]). However, only few studies using genetic mouse models were reported. In fact, due to developmental compensation in constitutive knockout mice [325], the contribution of the deleted single MCR gene in modulating alcohol consumption could be masked. Thus, the possible role of these receptors in alcohol phenotypes cannot be totally ruled out by null data. However, in an effort to examine the role of MC3R and MC4R in alcohol intake behaviors, several studies have thus combined pharmacological and genetic approaches. For instance, Olney and colleagues [88] found that binge drinking using the drinking in the dark procedure was similar between Mc3r knockout mice and their corresponding controls. However, they also showed that reduced binge-like alcohol drinking induced by melanotan II (MTII, intracerebroventricular infusion), a nonselective MCR agonist, is more pronounced in Mc3r mutant mice compared to control animals [88]. This observation indicates that MC3R counteracts the protective action of MTII against alcohol intake [324]. On the other hand, intracerebroventricular infusion of MTII failed to reduce alcohol intake in mutant mice lacking MC4R [89], suggesting that this MCR subtype is necessary for the central actions of MCR agonists on alcohol drinking behaviors [89]. Interestingly, several studies indicated a possible interaction between MC4R signaling and opioid system [326, 327]. For instance, MTII increases the effectiveness of naltrexone, an FDA-approved drug used to treat alcohol dependence, in reducing excessive alcohol intake in C57BL/6J mice [326]. Thus, it is still unclear where and how the melanocortin system contributes to alcohol action and whether combined therapy with naltrexone holds promise for AUD treatment [324].
Orphan GPCRs
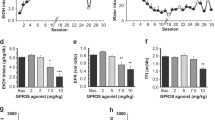
Orphan GPCRs are receptors for which endogenous ligands have not yet been fully identified. Of the 350 nonsensory GPCRs, nearly 120 remain orphan GPCRs, whose endogenous ligands and G protein signaling are unknown. Thus, discoveries of new ligands for these GPCRs profoundly impact our understanding of brain function [328]. Only few of these orphan receptors have been linked to drug addiction. Specifically, it was demonstrated that GPR139 receptor agonism in the habenula, a central structure connecting the forebrain to midbrain regions with a key role to integrate cognitive with emotional and sensory processing, decreases alcohol intake selectively in alcohol-dependent rats [329]. Regarding GPR26, another orphan GPCR, knockout mice drank more alcohol only at low concentration of alcohol (7%) compared to wild-type mice in a two-bottle choice paradigm. However, no difference between both genotypes was found for higher alcohol concentrations (i.e., 9% and 12%) [330]. Recently, the orphan GPCR GPR88 has been proposed as a new potential target for AUD. GPR88 is robustly expressed in the striatum throughout the dorsal and ventral areas [331, 332] as well as in the cerebral cortex, the amygdala, and the hypothalamus [333,333,334,336]. GPR88 expression was shown to be regulated in the CeA following treatment of several drugs of abuse, including morphine and alcohol [337]. More direct evidence of GPR88 neurobiological significance was obtained with knockout mice studies, suggesting that GPR88 plays an important role in regulating striatal-related processes. Gpr88 knockout mice demonstrated a disrupted prepulse inhibition of startle response, a phenotype mimicking symptoms of schizophrenia, and exhibited DA receptor hypersensitivity (as evidenced by increased sensitivity to apomorphine-induced climbing and stereotypy). Gpr88-deficient mice displayed increased locomotion, poor motor coordination and balance, impaired cue-based learning [338, 339], and decreased anxiety-related behaviors together with increased approach behaviors [340]. Gpr88 null mice drink more alcohol than control animals in continuous and intermittent two-bottle choice procedures [90]. The mutant mice also showed an increase alcohol self-administration and motivation to drink alcohol as shown by increased performance in progressive ratio test. Interestingly, lack of GPR88 decreased the rewarding effects of alcohol as measured by CPP and reduced alcohol-induced elevations in extracellular DA in the NAc in response to systemic alcohol injection. This suggests a reduction of alcohol reward in mutant mice that may explain, at least in part, enhanced alcohol drinking [90]. At present, our knowledge regarding the implication of orphan GPCRs in alcohol addiction is still limited but highly essential and a great deal of effort is needed to clarify the role of these receptors in the pathophysiology of AUD.
Conclusion and Future Directions
AUD involves long-lasting neurochemical, molecular, and structural adaptations in several neuronal networks that drive aberrant motivation to seek and consume alcohol [27]. As GPCRs constitute key receptors for nearly all major neurotransmitters, their crucial role in modulating alcohol actions was predictable. In this context, pharmacological approaches were crucial to identify potential GPCR targets and their contribution to diverse alcohol-related behaviors and to understand how modulation of GPCR signaling could be used for treating alcohol addiction. Then, genetic-based approaches mostly aimed at deleting individual GPCR genes allowed a more detailed exploration of the contribution of GPCR subtypes in alcohol phenotypes. Although such experimental frameworks are not devoid of weakness, they provided successful advances in our basic understanding of the neurobiology of alcohol addiction. In addition, these knockout approaches have been essential to characterize the function of inherited gene determinants of AUD and to identify candidate targets for therapeutic intervention. Importantly, data ensuing from GPCR null mice contributed, together with pharmacological studies, to the initiation of clinical trials. However, traditional knockout animals remain insufficient to delineate at the molecular level how GPCRs in specific circuits drive alcohol-related outcomes. For instance, these genetically modified animals are, by themselves, not sufficient to determine whether the constitutive activity of GPCRs, their oligomerization, or their coupling through G protein and/or β-arrestins mediates alcohol actions. Combining complex pharmacological, biophysical tools together with knockout and conditional mutant mice [7, 22, 24, 25] could help to cast light on the molecular mechanisms underlying GPCR signaling in alcohol-related behaviors. As mentioned above, the powerful potential of such combined strategies was recently illustrated by Godlewski et al. [23] reporting a functional crosstalk between the endocannabinoid and the ghrelin systems to promote alcohol intake through activation of the gastric vagal–brain axis. Newly genetically modified mice such as Cre lines [341] in combination of emerging technologies including opto/chemogenetic manipulations and neuroimaging will help to improve the knowledge on circuitry adaptation and response to alcohol in specific neuronal population and brain structure.
There is obviously still a substantial gap to unravel how other intertwined GPCR systems act in concert to induce alcohol-related neuroadaptations. In addition, in spite of significant progresses made to identify key GPCRs underlying pathological alcohol phenotypes, only a few GPCR ligands have been/are under clinical development. More translational studies using knock-in mice of SNP will be necessary to increase the accuracy to target potential GPCR to treat AUD. Nonetheless, GPCRs are still considered excellent potential targets for more selective, efficient, and safer drugs. Indeed, there is a growing interest in designing biased or allosteric signaling-selective molecules by taking advantage of the complex functional behavior of GPCRs. For example, GPCR signaling can involve distinct signaling transducers. This versatile signaling is thought to rely, at least in part, on the multitude of conformational intermediates that can be adopted by GPCRs. This conformational landscape could be modulated by biased ligands or allosteric modulators in order to preferentially engage distinct subsets of signaling pathways with more selective biological actions [342]. The therapeutic potential of biased ligands and allosteric modulators of GPCRs has to be thoroughly validated in animal models of alcohol disorder. In addition, deorphanization of the unexploited brain orphan GPCRs may provide valuable new therapeutic targets to treat AUD.
References
Trzaskowski B, Latek D, Yuan S, Ghoshdastider U, Debinski A, Filipek S. Action of molecular switches in GPCRs—theoretical and experimental studies. Curr Med Chem 2012;19(8):1090–109.
Wootten D, Christopoulos A, Marti-Solano M, Babu MM, Sexton PM. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat Rev Mol Cell Biol. 2018;19(10):638–53.
Smith JS, Rajagopal S. The beta-Arrestins: Multifunctional Regulators of G Protein-coupled Receptors. J Biol Chem 2016;291(17):8969–77.
Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res 2018;46(D1):D1091-D106.
Albizu L, Moreno JL, Gonzalez-Maeso J, Sealfon SC. Heteromerization of G protein-coupled receptors: relevance to neurological disorders and neurotherapeutics. CNS Neurol Disord Drug Targets 2010;9(5):636–50.
Damian M, Pons V, Renault P, M’Kadmi C, Delort B, Hartmann L, et al. GHSR-D2R heteromerization modulates dopamine signaling through an effect on G protein conformation. Proc Natl Acad Sci U S A 2018;115(17):4501–6.
Derouiche L, Massotte D. G protein-coupled receptor heteromers are key players in substance use disorder. Neurosci Biobehav Rev. 2018.
Ellis C, Smith A. Highlighting the pitfalls and possibilities of drug research. Nat Rev Drug Discov 2004;3(3):238–78.
Hauser AS, Attwood MM, Rask-Andersen M, Schioth HB, Gloriam DE. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov 2017;16(12):829–42.
Wacker D, Stevens RC, Roth BL. How Ligands Illuminate GPCR Molecular Pharmacology. Cell. 2017;170(3):414–27.
Stoeber M, Jullie D, Lobingier BT, Laeremans T, Steyaert J, Schiller PW, et al. A Genetically Encoded Biosensor Reveals Location Bias of Opioid Drug Action. Neuron. 2018;98(5):963–76 e5.
Urs NM, Gee SM, Pack TF, McCorvy JD, Evron T, Snyder JC, et al. Distinct cortical and striatal actions of a beta-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc Natl Acad Sci U S A 2016;113(50):E8178-E86.
Marcott PF, Gong S, Donthamsetti P, Grinnell SG, Nelson MN, Newman AH, et al. Regional Heterogeneity of D2-Receptor Signaling in the Dorsal Striatum and Nucleus Accumbens. Neuron. 2018;98(3):575–87 e4.
Lane JR, May LT, Parton RG, Sexton PM, Christopoulos A. A kinetic view of GPCR allostery and biased agonism. Nat Chem Biol 2017;13(9):929–37.
Shoichet BK, Kobilka BK. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol Sci 2012;33(5):268–72.
Fricker LD, Devi LA. Orphan neuropeptides and receptors: Novel therapeutic targets. Pharmacol Ther 2018;185:26–33.
Regard JB, Sato IT, Coughlin SR. Anatomical profiling of G protein-coupled receptor expression. Cell. 2008;135(3):561–71.
Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419.
Ehrlich AT, Maroteaux G, Robe A, Venteo L, Nasseef MT, van Kempen LC, et al. Expression map of 78 brain-expressed mouse orphan GPCRs provides a translational resource for neuropsychiatric research. Commun Biol 2018;1:102.
Rohrer DK, Kobilka BK. G Protein-Coupled Receptors: Functional and Mechanistic Insights Through Altered Gene Expression. Physiol Rev 1998;78(1):35–52.
Wehner JM, Bowers BJ. Use of Transgenics, Null Mutants, and Antisense Approaches to Study Ethanol’s Actions. Alcohol Clin Exp Res 1995;19(4):811–20.
Fernandez G, Cabral A, Andreoli MF, Labarthe A, M’Kadmi C, Ramos JG, et al. Evidence Supporting a Role for Constitutive Ghrelin Receptor Signaling in Fasting-Induced Hyperphagia in Male Mice. Endocrinology. 2018;159(2):1021–34.
Godlewski G, Cinar R, Coffey NJ, Liu J, Jourdan T, Mukhopadhyay B, et al. Targeting Peripheral CB1 Receptors Reduces Ethanol Intake via a Gut-Brain Axis. Cell Metab. 2019;29(6):1320–33.e8.
Kern A, Albarran-Zeckler R, Walsh Heidi E, Smith Roy G. Apo-Ghrelin Receptor Forms Heteromers with DRD2 in Hypothalamic Neurons and Is Essential for Anorexigenic Effects of DRD2 Agonism. Neuron. 2012;73(2):317–32.
Kern A, Mavrikaki M, Ullrich C, Albarran-Zeckler R, Brantley AF, Smith RG. Hippocampal Dopamine/DRD1 Signaling Dependent on the Ghrelin Receptor. Cell. 2015;163(5):1176–90.
Johnson KA, Lovinger DM. Presynaptic G Protein-Coupled Receptors: Gatekeepers of Addiction? Front Cell Neurosci 2016;10:264.
Ron D, Barak S. Molecular mechanisms underlying alcohol-drinking behaviours. Nat Rev Neurosci 2016;17(9):576–91.
Schuckit MA. Genetic aspects of alcoholism. Ann Emerg Med 1986;15(9):991–6.
Cservenka A. Neurobiological phenotypes associated with a family history of alcoholism. Drug Alcohol Depend 2016;158:8–21.
Agrawal A, Hinrichs AL, Dunn G, Bertelsen S, Dick DM, Saccone SF, et al. Linkage scan for quantitative traits identifies new regions of interest for substance dependence in the Collaborative Study on the Genetics of Alcoholism (COGA) sample. Drug Alcohol Depend 2008;93(1–2):12–20.
Mayfield J, Arends MA, Harris RA, Blednov YA. Genes and Alcohol Consumption: Studies with Mutant Mice. Int Rev Neurobiol 2016;126:293–355.
Bowers BJ. Applications of transgenic and knockout mice in alcohol research. Alcohol Res Health 2000;24(3):175–84.
Crabbe JC, Phillips TJ, Harris RA, Arends MA, Koob GF. Alcohol-related genes: contributions from studies with genetically engineered mice. Addict Biol 2006;11(3–4):195–269.
Crabbe J, Belknap J, Buck K. Genetic animal models of alcohol and drug abuse. Science. 1994;264(5166):1715–23.
Crabbe JC, Phillips TJ, Feller DJ, Hen R, Wenger CD, Lessov CN, et al. Elevated alcohol consumption in null mutant mice lacking 5-HT1B serotonin receptors. Nat Genet 1996;14(1):98–101.
Noble F, Lenoir M, Marie N. The opioid receptors as targets for drug abuse medication. Br J Pharmacol 2015;172(16):3964–79.
Basavarajappa BS, Joshi V, Shivakumar M, Subbanna S. Distinct functions of endogenous cannabinoid system in alcohol abuse disorders. Br J Pharmacol 2019;176(17):3085–109.
Haass-Koffler CL, Bartlett SE. Stress and addiction: contribution of the corticotropin releasing factor (CRF) system in neuroplasticity. Front Mol Neurosci 2012;5:91.
Olney JJ, Navarro M, Thiele TE. Targeting central melanocortin receptors: a promising novel approach for treating alcohol abuse disorders. Front Neurosci 2014;8:128.
Becker A, Grecksch G, Kraus J, Loh HH, Schroeder H, Hollt V. Rewarding effects of ethanol and cocaine in mu opioid receptor-deficient mice. Naunyn Schmiedeberg's Arch Pharmacol 2002;365(4):296–302.
Roberts AJ, McDonald JS, Heyser CJ, Kieffer BL, Matthes HW, Koob GF, et al. mu-Opioid receptor knockout mice do not self-administer alcohol. J Pharmacol Exp Ther 2000;293(3):1002–8.
Bilbao A, Robinson JE, Heilig M, Malanga CJ, Spanagel R, Sommer WH, et al. A pharmacogenetic determinant of mu-opioid receptor antagonist effects on alcohol reward and consumption: evidence from humanized mice. Biol Psychiatry 2015;77(10):850–8.
van Rijn RM, Whistler JL. The delta(1) opioid receptor is a heterodimer that opposes the actions of the delta(2) receptor on alcohol intake. Biol Psychiatry 2009;66(8):777–84.
Roberts AJ, Gold LH, Polis I, McDonald JS, Filliol D, Kieffer BL, et al. Increased ethanol self-administration in delta-opioid receptor knockout mice. Alcohol Clin Exp Res 2001;25(9):1249–56.
Kovacs KM, Szakall I, O’Brien D, Wang R, Vinod KY, Saito M, et al. Decreased oral self-administration of alcohol in kappa-opioid receptor knock-out mice. Alcohol Clin Exp Res 2005;29(5):730–8.
Grisel JE, Mogil JS, Grahame NJ, Rubinstein M, Belknap JK, Crabbe JC, et al. Ethanol oral self-administration is increased in mutant mice with decreased beta-endorphin expression. Brain Res 1999;835(1):62–7.
Racz I, Schurmann B, Karpushova A, Reuter M, Cichon S, Montag C, et al. The opioid peptides enkephalin and beta-endorphin in alcohol dependence. Biol Psychiatry 2008;64(11):989–97.
Koenig HN, Olive MF. Ethanol consumption patterns and conditioned place preference in mice lacking preproenkephalin. Neurosci Lett 2002;325(2):75–8.
Femenia T, Manzanares J. Increased ethanol intake in prodynorphin knockout mice is associated to changes in opioid receptor function and dopamine transmission. Addict Biol 2012;17(2):322–37.
Racz I, Markert A, Mauer D, Stoffel-Wagner B, Zimmer A. Long-term ethanol effects on acute stress responses: modulation by dynorphin. Addict Biol 2013;18(4):678–88.
Sakoori K, Murphy NP. Endogenous nociceptin (orphanin FQ) suppresses basal hedonic state and acute reward responses to methamphetamine and ethanol, but facilitates chronic responses. Neuropsychopharmacology. 2008;33(4):877–91.
Kallupi M, Scuppa G, de Guglielmo G, Calo G, Weiss F, Statnick MA, et al. Genetic Deletion of the Nociceptin/Orphanin FQ Receptor in the Rat Confers Resilience to the Development of Drug Addiction. Neuropsychopharmacology. 2017;42(3):695–706.
Poncelet M, Maruani J, Calassi R, Soubrié P. Overeating, alcohol and sucrose consumption decrease in CB1 receptor deleted mice. Neurosci Lett 2003;343(3):216–8.
Thanos PK, Dimitrakakis ES, Rice O, Gifford A, Volkow ND. Ethanol self-administration and ethanol conditioned place preference are reduced in mice lacking cannabinoid CB1 receptors. Behav Brain Res 2005;164(2):206–13.
Hungund BL, Szakall I, Adam A, Basavarajappa BS, Vadasz C. Cannabinoid CB1 receptor knockout mice exhibit markedly reduced voluntary alcohol consumption and lack alcohol-induced dopamine release in the nucleus accumbens. J Neurochem 2003;84(4):698–704.
Naassila M, Pierrefiche O, Ledent C, Daoust M. Decreased alcohol self-administration and increased alcohol sensitivity and withdrawal in CB1 receptor knockout mice. Neuropharmacology. 2004;46(2):243–53.
Racz I, Bilkei-Gorzo A, Toth ZE, Michel K, Palkovits M, Zimmer A. A Critical Role for the Cannabinoid CB1 Receptors in Alcohol Dependence and Stress-Stimulated Ethanol Drinking. J Neurosci 2003;23(6):2453–8.
Ortega-Álvaro A, Ternianov A, Aracil-Fernández A, Navarrete F, García-Gutiérrez MS, Manzanares J. Role of cannabinoid CB2 receptor in the reinforcing actions of ethanol. Addict Biol 2015;20(1):43–55.
Jerlhag E, Egecioglu E, Landgren S, Salomé N, Heilig M, Moechars D, et al. Requirement of central ghrelin signaling for alcohol reward. Proc Natl Acad Sci 2009;106(27):11318–23.
Zallar LJ, Tunstall BJ, Richie CT, Zhang YJ, You ZB, Gardner EL, et al. Development and initial characterization of a novel ghrelin receptor CRISPR/Cas9 knockout wistar rat model. Int J Obes (2005). 2018.
Jerlhag E, Landgren S, Egecioglu E, Dickson SL, Engel JA. The alcohol-induced locomotor stimulation and accumbal dopamine release is suppressed in ghrelin knockout mice. Alcohol. 2011;45(4):341–7.
Bahi A, Tolle V, Fehrentz J-A, Brunel L, Martinez J, Tomasetto C-L, et al. Ghrelin knockout mice show decreased voluntary alcohol consumption and reduced ethanol-induced conditioned place preference. Peptides. 2013;43:48–55.
El-Ghundi M, George SR, Drago J, Fletcher PJ, Fan T, Nguyen T, et al. Disruption of dopamine D1 receptor gene expression attenuates alcohol-seeking behavior. Eur J Pharmacol 1998;353(2–3):149–58.
Phillips TJ, Brown KJ, Burkhart-Kasch S, Wenger CD, Kelly MA, Rubinstein M, et al. Alcohol preference and sensitivity are markedly reduced in mice lacking dopamine D2 receptors. Nat Neurosci 1998;1(7):610–5.
Palmer AA, Low MJ, Grandy DK, Phillips TJ. Effects of a Drd2 deletion mutation on ethanol-induced locomotor stimulation and sensitization suggest a role for epistasis. Behav Genet 2003;33(3):311–24.
Delis F, Thanos PK, Rombola C, Rosko L, Grandy D, Wang GJ, et al. Chronic mild stress increases alcohol intake in mice with low dopamine D2 receptor levels. Behav Neurosci 2013;127(1):95–105.
Thanos PK, Rivera SN, Weaver K, Grandy DK, Rubinstein M, Umegaki H, et al. Dopamine D2R DNA transfer in dopamine D2 receptor-deficient mice: effects on ethanol drinking. Life Sci 2005;77(2):130–9.
Risinger FO, Freeman PA, Rubinstein M, Low MJ, Grandy DK. Lack of operant ethanol self-administration in dopamine D2 receptor knockout mice. Psychopharmacology 2000;152(3):343–50.
Leggio GM, Camillieri G, Platania CB, Castorina A, Marrazzo G, Torrisi SA, et al. Dopamine D3 receptor is necessary for ethanol consumption: an approach with buspirone. Neuropsychopharmacology. 2014;39(8):2017–28.
Boyce-Rustay JM, Risinger FO. Dopamine D3 receptor knockout mice and the motivational effects of ethanol. Pharmacol Biochem Behav 2003;75(2):373–9.
McQuade JA, Xu M, Woods SC, Seeley RJ, Benoit SC. Ethanol consumption in mice with a targeted disruption of the dopamine-3 receptor gene. Addict Biol 2003;8(3):295–303.
Thanos PK, Roushdy K, Sarwar Z, Rice O, Ashby CR, Jr., Grandy DK. The effect of dopamine D4 receptor density on novelty seeking, activity, social interaction, and alcohol binge drinking in adult mice. Synapse. 2015;69(7):356–64.
Risinger FO, Bormann NM, Oakes RA. Reduced Sensitivity to Ethanol Reward, But Not Ethanol Aversion, in Mice Lacking 5=HT1B Receptors. Alcohol Clin Exp Res 1996;20(8):1401–5.
Bouwknecht JA, Hijzen TH, van der Gugten J, Maes RA, Hen R, Olivier B. Ethanol intake is not elevated in male 5-HT(1B) receptor knockout mice. Eur J Pharmacol 2000;403(1–2):95–8.
Bonasera SJ, Chu HM, Brennan TJ, Tecott LH. A null mutation of the serotonin 6 receptor alters acute responses to ethanol. Neuropsychopharmacology. 2006;31(8):1801–13.
Bird MK, Kirchhoff J, Djouma E, Lawrence AJ. Metabotropic glutamate 5 receptors regulate sensitivity to ethanol in mice. Int J Neuropsychopharmacol 2008;11(6):765–74.
Blednov YA, Harris RA. Metabotropic glutamate receptor 5 (mGluR5) regulation of ethanol sedation, dependence and consumption: relationship to acamprosate actions. Int J Neuropsychopharmacol 2008;11(6):775–93.
Campbell RR, Domingo RD, Williams AR, Wroten MG, McGregor HA, Waltermire RS, et al. Increased Alcohol-Drinking Induced by Manipulations of mGlu5 Phosphorylation within the Bed Nucleus of the Stria Terminalis. J Neurosci 2019;39(14):2745–61.
Zhou Z, Karlsson C, Liang T, Xiong W, Kimura M, Tapocik JD, et al. Loss of metabotropic glutamate receptor 2 escalates alcohol consumption. Proc Natl Acad Sci U S A 2013;110(42):16963–8.
Blednov YA, Walker D, Osterndorf-Kahanek E, Harris RA. Mice lacking metabotropic glutamate receptor 4 do not show the motor stimulatory effect of ethanol. Alcohol. 2004;34(2–3):251–9.
Gyetvai B, Simonyi A, Oros M, Saito M, Smiley J, Vadasz C. mGluR7 genetics and alcohol: intersection yields clues for addiction. Neurochem Res 2011;36(6):1087–100.
Sillaber I, Rammes G, Zimmermann S, Mahal B, Zieglgansberger W, Wurst W, et al. Enhanced and delayed stress-induced alcohol drinking in mice lacking functional CRH1 receptors. Science. 2002;296(5569):931–3.
Molander A, Vengeliene V, Heilig M, Wurst W, Deussing JM, Spanagel R. Brain-specific inactivation of the Crhr1 gene inhibits post-dependent and stress-induced alcohol intake, but does not affect relapse-like drinking. Neuropsychopharmacology. 2012;37(4):1047–56.
Giardino WJ, Ryabinin AE. CRF1 receptor signaling regulates food and fluid intake in the drinking-in-the-dark model of binge alcohol consumption. Alcohol Clin Exp Res 2013;37(7):1161–70.
Kaur S, Li J, Stenzel-Poore MP, Ryabinin AE. Corticotropin-releasing factor acting on corticotropin-releasing factor receptor type 1 is critical for binge alcohol drinking in mice. Alcohol Clin Exp Res 2012;36(2):369–76.
Pastor R, Reed C, Burkhart-Kasch S, Li N, Sharpe AL, Coste SC, et al. Ethanol concentration-dependent effects and the role of stress on ethanol drinking in corticotropin-releasing factor type 1 and double type 1 and 2 receptor knockout mice. Psychopharmacology 2011;218(1):169–77.
Chu K, Koob GF, Cole M, Zorrilla EP, Roberts AJ. Dependence-induced increases in ethanol self-administration in mice are blocked by the CRF1 receptor antagonist antalarmin and by CRF1 receptor knockout. Pharmacol Biochem Behav 2007;86(4):813–21.
Olney JJ, Sprow GM, Navarro M, Thiele TE. The protective effects of the melanocortin receptor (MCR) agonist, melanotan-II (MTII), against binge-like ethanol drinking are facilitated by deletion of the MC3 receptor in mice. Neuropeptides. 2014;48(1):47–51.
Navarro M, Lerma-Cabrera JM, Carvajal F, Lowery EG, Cubero I, Thiele TE. Assessment of voluntary ethanol consumption and the effects of a melanocortin (MC) receptor agonist on ethanol intake in mutant C57BL/6J mice lacking the MC-4 receptor. Alcohol Clin Exp Res 2011;35(6):1058–66.
Ben Hamida S, Mendonca-Netto S, Arefin TM, Nasseef MT, Boulos LJ, McNicholas M, et al. Increased Alcohol Seeking in Mice Lacking Gpr88 Involves Dysfunctional Mesocorticolimbic Networks. Biol Psychiatry 2018;84(3):202–12.
de la Cour C, Sorensen G, Wortwein G, Weikop P, Dencker D, Fink-Jensen A, et al. Enhanced self-administration of alcohol in muscarinic acetylcholine M4 receptor knockout mice. Eur J Pharmacol 2015;746:1–5.
Nuutinen S, Lintunen M, Vanhanen J, Ojala T, Rozov S, Panula P. Evidence for the role of histamine H3 receptor in alcohol consumption and alcohol reward in mice. Neuropsychopharmacology. 2011;36(10):2030–40.
Duncan EA, Sorrell JE, Adamantidis A, Rider T, Jandacek RJ, Seeley RJ, et al. Alcohol drinking in MCH receptor-1-deficient mice. Alcohol Clin Exp Res 2007;31(8):1325–37.
Maul B, Krause W, Pankow K, Becker M, Gembardt F, Alenina N, et al. Central angiotensin II controls alcohol consumption via its AT1 receptor. FASEB J 2005;19(11):1474–81.
Sanbe A, Takagi N, Fujiwara Y, Yamauchi J, Endo T, Mizutani R, et al. Alcohol preference in mice lacking the Avpr1a vasopressin receptor. Am J Phys Regul Integr Comp Phys 2008;294(5):R1482–90.
Houchi H, Persyn W, Legastelois R, Naassila M. The adenosine A2A receptor agonist CGS 21680 decreases ethanol self-administration in both non-dependent and dependent animals. Addict Biol 2013;18(5):812–25.
Naassila M, Ledent C, Daoust M. Low ethanol sensitivity and increased ethanol consumption in mice lacking adenosine A2A receptors. J Neurosci 2002;22(23):10487–93.
Houchi H, Warnault V, Barbier E, Dubois C, Pierrefiche O, Ledent C, et al. Involvement of A2A receptors in anxiolytic, locomotor and motivational properties of ethanol in mice. Genes Brain Behav 2008;7(8):887–98.
Thiele TE, Koh MT, Pedrazzini T. Voluntary alcohol consumption is controlled via the neuropeptide Y Y1 receptor. J Neurosci 2002;22(3):RC208.
Thiele TE, Naveilhan P, Ernfors P. Assessment of ethanol consumption and water drinking by NPY Y(2) receptor knockout mice. Peptides. 2004;25(6):975–83.
Thiele TE, Miura GI, Marsh DJ, Bernstein IL, Palmiter RD. Neurobiological responses to ethanol in mutant mice lacking neuropeptide Y or the Y5 receptor. Pharmacol Biochem Behav 2000;67(4):683–91.
Blednov YA, Bergeson SE, Walker D, Ferreira VM, Kuziel WA, Harris RA. Perturbation of chemokine networks by gene deletion alters the reinforcing actions of ethanol. Behav Brain Res 2005;165(1):110–25.
Miyasaka K, Hosoya H, Takano S, Ohta M, Sekime A, Kanai S, et al. Differences in ethanol ingestion between cholecystokinin-A receptor deficient and -B receptor deficient mice. Alcohol Alcohol 2005;40(3):176–80.
Abramov U, Raud S, Innos J, Koks S, Matsui T, Vasar E. Gender specific effects of ethanol in mice, lacking CCK2 receptors. Behav Brain Res 2006;175(1):149–56.
George DT, Gilman J, Hersh J, Thorsell A, Herion D, Geyer C, et al. Neurokinin 1 receptor antagonism as a possible therapy for alcoholism. Science. 2008;319(5869):1536–9.
Thorsell A, Schank JR, Singley E, Hunt SP, Heilig M. Neurokinin-1 receptors (NK1R:s), alcohol consumption, and alcohol reward in mice. Psychopharmacology 2010;209(1):103–11.
Lee MR, Hinton DJ, Song JY, Lee KW, Choo C, Johng H, et al. Neurotensin receptor type 1 regulates ethanol intoxication and consumption in mice. Pharmacol Biochem Behav 2010;95(2):235–41.
Lee MR, Hinton DJ, Unal SS, Richelson E, Choi DS. Increased ethanol consumption and preference in mice lacking neurotensin receptor type 2. Alcohol Clin Exp Res 2011;35(1):99–107.
Darcq E, Kieffer BL. Opioid receptors: drivers to addiction? Nat Rev Neurosci 2018;19(8):499–514.
Bodnar RJ. Endogenous Opiates and Behavior: 2015. Peptides. 2017;88:126–88.
Ehrlich AT, Kieffer BL, Darcq E. Current strategies toward safer mu opioid receptor drugs for pain management. Expert Opin Ther Targets. 2019;23(4):315-26.
Lutz PE, Kieffer BL. The multiple facets of opioid receptor function: implications for addiction. Curr Opin Neurobiol 2013;23(4):473–9.
Filliol D, Ghozland S, Chluba J, Martin M, Matthes HW, Simonin F, et al. Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat Genet 2000;25(2):195–200.
Pradhan AA, Befort K, Nozaki C, Gaveriaux-Ruff C, Kieffer BL. The delta opioid receptor: an evolving target for the treatment of brain disorders. Trends Pharmacol Sci 2011;32(10):581–90.
Chu Sin Chung P, Kieffer BL. Delta opioid receptors in brain function and diseases. Pharmacol Ther 2013;140(1):112–20.
Tejeda HA, Shippenberg TS, Henriksson R. The dynorphin/kappa-opioid receptor system and its role in psychiatric disorders. Cell Mol Life Sci 2012;69(6):857–96.
Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by kappa opiate receptors. Science. 1986;233(4765):774–6.
Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383(6603):819–23.
Jones MR, Viswanath O, Peck J, Kaye AD, Gill JS, Simopoulos TT. A Brief History of the Opioid Epidemic and Strategies for Pain Medicine. Pain Ther 2018;7(1):13–21.
Ehrlich AT, Darcq E. Recommending buprenorphine for pain management. Pain Management 2019;9(1):13–16.
Charbogne P, Kieffer BL, Befort K. 15 years of genetic approaches in vivo for addiction research: Opioid receptor and peptide gene knockout in mouse models of drug abuse. Neuropharmacology. 2014;76 Pt B:204–17.
Contet C, Kieffer BL, Befort K. Mu opioid receptor: a gateway to drug addiction. Curr Opin Neurobiol 2004;14(3):370–8.
Hall FS, Sora I, Uhl GR. Ethanol consumption and reward are decreased in mu-opiate receptor knockout mice. Psychopharmacology 2001;154(1):43–9.
Ghozland S, Chu K, Kieffer BL, Roberts AJ. Lack of stimulant and anxiolytic-like effects of ethanol and accelerated development of ethanol dependence in mu-opioid receptor knockout mice. Neuropharmacology. 2005;49(4):493–501.
Monory K, Massa F, Egertova M, Eder M, Blaudzun H, Westenbroek R, et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51(4):455–66.
Ben Hamida S, Boulos LJ, McNicholas M, Charbogne P, Kieffer BL. Mu opioid receptors in GABAergic neurons of the forebrain promote alcohol reward and drinking. Addict Biol 2019;24(1):28–39.
Zhou Y, Liang Y, Low MJ, Kreek MJ. Nuclear transcriptional changes in hypothalamus of Pomc enhancer knockout mice after excessive alcohol drinking. Genes Brain Behav. 2019:e12600.
Angelogianni P, Gianoulakis C. Chronic ethanol increases proopiomelanocortin gene expression in the rat hypothalamus. Neuroendocrinology. 1993;57(1):106–14.
Crist RC, Berrettini WH. Pharmacogenetics of OPRM1. Pharmacol Biochem Behav 2014;123:25–33.
Reed B, Butelman ER, Yuferov V, Randesi M, Kreek MJ. Genetics of opiate addiction. Curr Psychiatry Rep 2014;16(11):504.
Ramchandani VA, Umhau J, Pavon FJ, Ruiz-Velasco V, Margas W, Sun H, et al. A genetic determinant of the striatal dopamine response to alcohol in men. Mol Psychiatry 2011;16(8):809–17.
Henderson-Redmond AN, Lowe TE, Tian XB, Morgan DJ. Increased ethanol drinking in “humanized” mice expressing the mu opioid receptor A118G polymorphism are mediated through sex-specific mechanisms. Brain Res Bull 2018;138:12–9.
Koob GF, Mason BJ. Existing and Future Drugs for the Treatment of the Dark Side of Addiction. Annu Rev Pharmacol Toxicol 2016;56:299–322.
Lutz PE, Kieffer BL. Opioid receptors: distinct roles in mood disorders. Trends Neurosci 2013;36(3):195–206.
Buckner JD, Schmidt NB, Lang AR, Small JW, Schlauch RC, Lewinsohn PM. Specificity of social anxiety disorder as a risk factor for alcohol and cannabis dependence. J Psychiatr Res 2008;42(3):230–9.
Saland LC, Abeyta A, Frausto S, Raymond-Stintz M, Hastings CM, Carta M, et al. Chronic ethanol consumption reduces delta-and mu-opioid receptor-stimulated G-protein coupling in rat brain. Alcohol Clin Exp Res 2004;28(1):98–104.
Alongkronrusmee D, Chiang T, van Rijn RM. Delta Opioid Pharmacology in Relation to Alcohol Behaviors. Handb Exp Pharmacol 2018;247:199–225.
van Rijn RM, Brissett DI, Whistler JL. Emergence of functional spinal delta opioid receptors after chronic ethanol exposure. Biol Psychiatry 2012;71(3):232–8.
Bie B, Zhu W, Pan ZZ. Ethanol-induced delta-opioid receptor modulation of glutamate synaptic transmission and conditioned place preference in central amygdala. Neuroscience. 2009;160(2):348–58.
Mendez M, Morales-Mulia M, Leriche M. [3H]DPDPE binding to delta opioid receptors in the rat mesocorticolimbic and nigrostriatal pathways is transiently increased by acute ethanol administration. Brain Res 2004;1028(2):180–90.
Sacharczuk M, Lesniak A, Lipkowski AW, Korostynski M, Przewlocki R, Sadowski B. Association between the A107V substitution in the delta-opioid receptors and ethanol drinking in mice selected for high and low analgesia. Addict Biol 2014;19(4):643–51.
Hayward MD, Hansen ST, Pintar JE, Low MJ. Operant self-administration of ethanol in C57BL/6 mice lacking beta-endorphin and enkephalin. Pharmacol Biochem Behav 2004;79(1):171–81.
Bazov I, Kononenko O, Watanabe H, Kuntic V, Sarkisyan D, Taqi MM, et al. The endogenous opioid system in human alcoholics: molecular adaptations in brain areas involved in cognitive control of addiction. Addict Biol 2013;18(1):161–9.
Xuei X, Flury-Wetherill L, Bierut L, Dick D, Nurnberger J, Jr., Foroud T, et al. The opioid system in alcohol and drug dependence: family-based association study. Am J Med Genet B Neuropsychiatr Genet 2007;144B(7):877–84.
Bruchas MR, Roth BL. New Technologies for Elucidating Opioid Receptor Function. Trends Pharmacol Sci 2016;37(4):279–89.
Al-Hasani R, Bruchas MR. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology. 2011;115(6):1363–81.
Anderson RI, Becker HC. Role of the Dynorphin/Kappa Opioid Receptor System in the Motivational Effects of Ethanol. Alcohol Clin Exp Res 2017;41(8):1402–18.
Walker BM, Koob GF. Pharmacological evidence for a motivational role of kappa-opioid systems in ethanol dependence. Neuropsychopharmacology. 2008;33(3):643–52.
Karkhanis A, Holleran KM, Jones SR. Dynorphin/Kappa Opioid Receptor Signaling in Preclinical Models of Alcohol, Drug, and Food Addiction. Int Rev Neurobiol 2017;136:53–88.
Mitchell JM, Liang MT, Fields HL. A single injection of the kappa opioid antagonist norbinaltorphimine increases ethanol consumption in rats. Psychopharmacology 2005;182(3):384–92.
Van’t Veer A, Smith KL, Cohen BM, Carlezon WA, Jr., Bechtholt AJ. Kappa-opioid receptors differentially regulate low and high levels of ethanol intake in female mice. Brain Behav 2016;6(9):e00523.
Bloodgood DW, Pati D, Pina MM, Neira S, Hardaway JA, Desai S, et al. Kappa Opioid Receptor and Dynorphin Signaling in the Central Amygdala Regulates Alcohol Intake. BioRxiv. 2019.
Zapata A, Shippenberg TS. Endogenous kappa opioid receptor systems modulate the responsiveness of mesoaccumbal dopamine neurons to ethanol. Alcohol Clin Exp Res 2006;30(4):592–7.
Blednov YA, Walker D, Martinez M, Harris RA. Reduced alcohol consumption in mice lacking preprodynorphin. Alcohol. 2006;40(2):73–86.
Nguyen K, Tseng A, Marquez P, Hamid A, Lutfy K. The role of endogenous dynorphin in ethanol-induced state-dependent CPP. Behav Brain Res 2012;227(1):58–63.
Sperling RE, Gomes SM, Sypek EI, Carey AN, McLaughlin JP. Endogenous kappa-opioid mediation of stress-induced potentiation of ethanol-conditioned place preference and self-administration. Psychopharmacology 2010;210(2):199–209.
Ciccocioppo R, Borruto AM, Domi A, Teshima K, Cannella N, Weiss F. NOP-Related Mechanisms in Substance Use Disorders. Handb Exp Pharmacol 2019;254:187–212.
Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, et al. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270(5237):792–4.
Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, et al. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377(6549):532–5.
Huang J, Young B, Pletcher MT, Heilig M, Wahlestedt C. Association between the nociceptin receptor gene (OPRL1) single nucleotide polymorphisms and alcohol dependence. Addict Biol 2008;13(1):88–94.
Zaveri NT. The nociceptin/orphanin FQ receptor (NOP) as a target for drug abuse medications. Curr Top Med Chem 2011;11(9):1151–6.
Witkin JM, Statnick MA, Rorick-Kehn LM, Pintar JE, Ansonoff M, Chen Y, et al. The biology of Nociceptin/Orphanin FQ (N/OFQ) related to obesity, stress, anxiety, mood, and drug dependence. Pharmacol Ther 2014;141(3):283–99.
Kallupi M, Varodayan FP, Oleata CS, Correia D, Luu G, Roberto M. Nociceptin/orphanin FQ decreases glutamate transmission and blocks ethanol-induced effects in the central amygdala of naive and ethanol-dependent rats. Neuropsychopharmacology. 2014;39(5):1081–92.
Zaveri NT, Marquez PV, Meyer ME, Polgar WE, Hamid A, Lutfy K. A Novel and Selective Nociceptin Receptor (NOP) Agonist (1-(1-((cis)-4-isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)methanol (AT-312) Decreases Acquisition of Ethanol-Induced Conditioned Place Preference in Mice. Alcohol Clin Exp Res 2018;42(2):461–71.
Garcia-Arencibia M, Molina-Holgado E, Molina-Holgado F. Effect of endocannabinoid signalling on cell fate: life, death, differentiation and proliferation of brain cells. Br J Pharmacol 2019;176(10):1361–9.
Montecucco F, Di Marzo V. At the heart of the matter: the endocannabinoid system in cardiovascular function and dysfunction. Trends Pharmacol Sci 2012;33(6):331–40.
Piazza PV, Cota D, Marsicano G. The CB1 Receptor as the Cornerstone of Exostasis. Neuron. 2017;93(6):1252–74.
Busquets-Garcia A, Bains J, Marsicano G. CB1 Receptor Signaling in the Brain: Extracting Specificity from Ubiquity. Neuropsychopharmacology. 2018;43(1):4–20.
Manzanares J, Cabañero D, Puente N, García-Gutiérrez MS, Grandes P, Maldonado R. Role of the endocannabinoid system in drug addiction. Biochem Pharmacol 2018;157:108–21.
Covey DP, Mateo Y, Sulzer D, Cheer JF, Lovinger DM. Endocannabinoid modulation of dopamine neurotransmission. Neuropharmacology. 2017;124:52–61.
Parsons LH, Hurd YL. Endocannabinoid signalling in reward and addiction. Nat Rev Neurosci 2015;16(10):579–94.
Moreira FA, Jupp B, Belin D, Dalley JW. Endocannabinoids and striatal function: implications for addiction-related behaviours. Behav Pharmacol 2015;26:59–72.
Lutz B, Marsicano G, Maldonado R, Hillard CJ. The endocannabinoid system in guarding against fear, anxiety and stress. Nat Rev Neurosci 2015;16(12):705–18.
Stella N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia. 2010;58(9):1017–30.
Sloan ME, Gowin JL, Ramchandani VA, Hurd YL, Le Foll B. The endocannabinoid system as a target for addiction treatment: Trials and tribulations. Neuropharmacology. 2017;124:73–83.
Schmidt LG, Samochowiec J, Finckh U, Fiszer-Piosik E, Horodnicki J, Wendel B, et al. Association of a CB1 cannabinoid receptor gene (CNR1) polymorphism with severe alcohol dependence. Drug Alcohol Depend 2002;65(3):221–4.
Hutchison KE, Haughey H, Niculescu M, Schacht J, Kaiser A, Stitzel J, et al. The incentive salience of alcohol: translating the effects of genetic variant in CNR1. Arch Gen Psychiatry 2008;65(7):841–50.
George DT, Herion DW, Jones CL, Phillips MJ, Hersh J, Hill D, et al. Rimonabant (SR141716) has no effect on alcohol self-administration or endocrine measures in nontreatment-seeking heavy alcohol drinkers. Psychopharmacology. 2010;208(1):37–44.
Henderson-Redmond AN, Guindon J, Morgan DJ. Roles for the endocannabinoid system in ethanol-motivated behavior. Prog Neuro-Psychopharmacol Biol Psychiatry 2016;65:330–9.
Kleczkowska P, Smaga I, Filip M, Bujalska-Zadrozny M. Cannabinoid Ligands and Alcohol Addiction: A Promising Therapeutic Tool or a Humbug? Neurotox Res 2016;29(1):173–96.
Soyka M, Koller G, Schmidt P, Lesch O-M, Leweke M, Fehr C, et al. Cannabinoid receptor 1 blocker rimonabant (SR 141716) for treatment of alcohol dependence: results from a placebo-controlled, double-blind trial. J Clin Psychopharmacol 2008;28(3):317–24.
Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci 1999;96(10):5780–5.
Ledent C. Unresponsiveness to Cannabinoids and Reduced Addictive Effects of Opiates in CB1 Receptor Knockout Mice. Science. 1999;283(5400):401–4.
Houchi H, Babovic D, Pierrefiche O, Ledent C, Daoust M, Naassila M. CB1 receptor knockout mice display reduced ethanol-induced conditioned place preference and increased striatal dopamine D2 receptors. Neuropsychopharmacology. 2005;30(2):339–49.
Vinod KY, Yalamanchili R, Thanos PK, Vadasz C, Cooper TB, Volkow ND, et al. Genetic and pharmacological manipulations of the CB1 receptor alter ethanol preference and dependence in ethanol preferring and nonpreferring mice. Synapse. 2008;62(8):574–81.
Wang L, Liu J, Harvey-White J, Zimmer A, Kunos G. Endocannabinoid signaling via cannabinoid receptor 1 is involved in ethanol preference and its age-dependent decline in mice. Proc Natl Acad Sci U S A 2003;100(3):1393–8.
Vinod KY, Yalamanchili R, Thanos PK, Vadasz C, Cooper TB, Volkow ND, et al. Genetic and pharmacological manipulations of the CB(1) receptor alter ethanol preference and dependence in ethanol preferring and nonpreferring mice. Synapse. 2008;62(8):574–81.
Ishiguro H, Iwasaki S, Teasenfitz L, Higuchi S, Horiuchi Y, Saito T, et al. Involvement of cannabinoid CB2 receptor in alcohol preference in mice and alcoholism in humans. Pharmacogenomics J 2007;7(6):380–5.
Onaivi ES, Ishiguro H, Gong J-P, Patel S, Meozzi PA, Myers L, et al. Brain neuronal CB2 cannabinoid receptors in drug abuse and depression: from mice to human subjects. PLoS One 2008;3(2):e1640.
Ortega-Alvaro A, Aracil-Fernández A, García-Gutiérrez MS, Navarrete F, Manzanares J. Deletion of CB2 Cannabinoid Receptor Induces Schizophrenia-Related Behaviors in Mice. Neuropsychopharmacology. 2011;36(7):1489–504.
Sanchis-Segura C, Spanagel R. Behavioural assessment of drug reinforcement and addictive features in rodents: an overview. Addict Biol 2006;11(1):2–38.
Pradier B, Erxlebe E, Markert A, Rácz I. Interaction of cannabinoid receptor 2 and social environment modulates chronic alcohol consumption. Behav Brain Res 2015;287:163–71.
Navarrete F, García-Gutiérrez MS, Manzanares J. Pharmacological regulation of cannabinoid CB2 receptor modulates the reinforcing and motivational actions of ethanol. Biochem Pharmacol 2018;157:227–34.
Blednov YA, Cravatt BF, Boehm SL, Walker D, Harris RA. Role of endocannabinoids in alcohol consumption and intoxication: studies of mice lacking fatty acid amide hydrolase. Neuropsychopharmacology 2007;32(7):1570–82.
Basavarajappa BS, Yalamanchili R, Cravatt BF, Cooper TB, Hungund BL. Increased ethanol consumption and preference and decreased ethanol sensitivity in female FAAH knockout mice. Neuropharmacology. 2006;50(7):834–44.
Perelló M, Zigman JM. The Role of Ghrelin in Reward-Based Eating. Biol Psychiatry 2012;72(5):347–53.
Müller TD, Nogueiras R, Andermann ML, Andrews ZB, Anker SD, Argente J, et al. Ghrelin. Mol Metab 2015;4(6):437–60.
Jerlhag E. Gut-brain axis and addictive disorders: A review with focus on alcohol and drugs of abuse. Pharmacol Ther 2018.
Al Massadi O, López M, Tschöp M, Diéguez C, Nogueiras R. Current Understanding of the Hypothalamic Ghrelin Pathways Inducing Appetite and Adiposity. Trends Neurosci 2017.
Yanagi S, Sato T, Kangawa K, Nakazato M. The Homeostatic Force of Ghrelin. Cell Metab 2018;27(4):786–804.
Zizzari P, Hassouna R, Grouselle D, Epelbaum J, Tolle V. Physiological roles of preproghrelin-derived peptides in GH secretion and feeding. Peptides. 2011;32(11):2274–82.
Labarthe A, Fiquet O, Hassouna R, Zizzari P, Lanfumey L, Ramoz N, et al. Ghrelin-Derived Peptides: A Link between Appetite/Reward, GH Axis, and Psychiatric Disorders? Front Endocrinol 2014;5.
Ge X, Yang H, Bednarek MA, Galon-Tilleman H, Chen P, Chen M, et al. LEAP2 Is an Endogenous Antagonist of the Ghrelin Receptor. Cell Metab 2017.
M’Kadmi C, Cabral A, Barrile F, Giribaldi J, Cantel S, Damian M, et al. N-terminal Liver-expressed antimicrobial peptide 2 (LEAP2) region exhibits inverse agonist activity toward the ghrelin receptor. J Med Chem 2018.
Damian M, Marie J, Leyris J-P, Fehrentz J-A, Verdié P, Martinez J, et al. High constitutive activity is an intrinsic feature of ghrelin receptor protein: a study with a functional monomeric GHS-R1a receptor reconstituted in lipid discs. J Biol Chem 2012;287(6):3630–41.
M’Kadmi C, Leyris J-P, Onfroy L, Galés C, Saulière A, Gagne D, et al. Agonism, Antagonism, and Inverse Agonism Bias at the Ghrelin Receptor Signaling. J Biol Chem 2015;290(45):27021–39.
Damian M, Mary S, Maingot M, M’Kadmi C, Gagne D, Leyris JP, et al. Ghrelin receptor conformational dynamics regulate the transition from a preassembled to an active receptor:Gq complex. Proc Natl Acad Sci U S A 2015;112(5):1601–6.
Els S, Schild E, Petersen PS, Kilian T-M, Mokrosinski J, Frimurer TM, et al. An Aromatic Region To Induce a Switch between Agonism and Inverse Agonism at the Ghrelin Receptor. J Med Chem 2012;55(17):7437–49.
Pantel J, Legendre M, Cabrol S, Hilal L, Hajaji Y, Morisset S, et al. Loss of constitutive activity of the growth hormone secretagogue receptor in familial short stature. J Clin Invest 2006;116(3):760–8.
Guan X-M, Yu H, Palyha OC, McKee KK, Feighner SD, Sirinathsinghji DJS, et al. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Mol Brain Res 1997;48(1):23–9.
Jerlhag E. Gut-brain axis and addictive disorders: A review with focus on alcohol and drugs of abuse. Pharmacol Ther 2019;196:1–14.
Cabral A, López Soto E, Epelbaum J, Perelló M. Is Ghrelin Synthesized in the Central Nervous System? Int J Mol Sci 2017;18(3):638.
Perello M, Cabral A, Cornejo MP, De Francesco PN, Fernandez G, Uriarte M. Brain accessibility delineates the central effects of circulating ghrelin. J Neuroendocrinol 2019;31(7):e12677.
Lee MR, Tapocik JD, Ghareeb M, Schwandt ML, Dias AA, Le AN, et al. The novel ghrelin receptor inverse agonist PF-5190457 administered with alcohol: preclinical safety experiments and a phase 1b human laboratory study. Mol Psychiatry 2018.
Farokhnia M, Faulkner ML, Piacentino D, Lee MR, Leggio L. Ghrelin: From a gut hormone to a potential therapeutic target for alcohol use disorder. Physiol Behav 2019;204:49–57.
Farokhnia M, Lee MR, Farinelli LA, Ramchandani VA, Akhlaghi F, Leggio L. Pharmacological manipulation of the ghrelin system and alcohol hangover symptoms in heavy drinking individuals: Is there a link? Pharmacol Biochem Behav 2018;172:39–49.
Haass-Koffler CL, Long VM, Farokhnia M, Magill M, Kenna GA, Swift RM, et al. Intravenous administration of ghrelin increases serum cortisol and aldosterone concentrations in heavy-drinking alcohol-dependent individuals: Results from a double-blind, placebo-controlled human laboratory study. Neuropharmacology. 2019;158:107711.
Haass-Koffler CL, Giovenco DE, Lee MR, Zywiak WH, de la Monte SM, Kenna GA, et al. Serum Insulin Levels Are Reduced by Intravenous Ghrelin Administration but Do Not Correlate with Alcohol Craving in Alcohol-Dependent Individuals. Int J Neuropsychopharmacol. 2016.
Haass-Koffler CL, Aoun EG, Swift RM, de la Monte SM, Kenna GA, Leggio L. Leptin levels are reduced by intravenous ghrelin administration and correlated with cue-induced alcohol craving. Transl Psychiatry 2015;5:e646.
Suchankova P, Nilsson S, von der Pahlen B, Santtila P, Sandnabba K, Johansson A, et al. Genetic variation of the growth hormone secretagogue receptor gene is associated with alcohol use disorders identification test scores and smoking. Addict Biol 2016;21(2):481–8.
Suchankova P, Yan J, Schwandt ML, Stangl BL, Jerlhag E, Engel JA, et al. The Leu72Met Polymorphism of the Prepro-ghrelin Gene is Associated With Alcohol Consumption and Subjective Responses to Alcohol: Preliminary Findings. Alcohol Alcohol 2017;52(4):425–30.
Landgren S, Jerlhag E, Zetterberg H, Gonzalez-Quintela A, Campos J, Olofsson U, et al. Association of pro-ghrelin and GHS-R1A gene polymorphisms and haplotypes with heavy alcohol use and body mass. Alcohol Clin Exp Res 2008;32(12):2054–61.
Zallar LJ, Farokhnia M, Tunstall BJ, Vendruscolo LF, Leggio L. The Role of the Ghrelin System in Drug Addiction. Int Rev Neurobiol. 136: Elsevier; 2017. p. 89–119.
Zallar LJ, Beurmann S, Tunstall BJ, Fraser CM, Koob GF, Vendruscolo LF, et al. Ghrelin receptor deletion reduces binge-like alcohol drinking in rats. J Neuroendocrinol 2019;31(7):e12663.
Simms JA, Steensland P, Medina B, Abernathy KE, Chandler LJ, Wise R, et al. Intermittent access to 20% ethanol induces high ethanol consumption in Long-Evans and Wistar rats. Alcohol Clin Exp Res 2008;32(10):1816–23.
Carnicella S, Kharazia V, Jeanblanc J, Janak PH, Ron D. GDNF is a fast-acting potent inhibitor of alcohol consumption and relapse. Proc Natl Acad Sci U S A 2008;105(23):8114–9.
Neasta J, Ben Hamida S, Yowell Q, Carnicella S, Ron D. Role for mammalian target of rapamycin complex 1 signaling in neuroadaptations underlying alcohol-related disorders. Proc Natl Acad Sci U S A 2010;107(46):20093–8.
Jeanblanc J, Rolland B, Gierski F, Martinetti MP, Naassila M. Animal models of binge drinking, current challenges to improve face validity. Neurosci Biobehav Rev 2018:S0149763418301234.
Al Massadi O, Nogueiras R, Dieguez C, Girault J-A. Ghrelin and food reward. Neuropharmacology. 2019;148:131–8.
Bhattacharya SK, Andrews K, Beveridge R, Cameron KO, Chen C, Dunn M, et al. Discovery of PF-5190457, a Potent, Selective, and Orally Bioavailable Ghrelin Receptor Inverse Agonist Clinical Candidate. ACS Med Chem Lett 2014;5(5):474–9.
Hasegawa Y, Higuchi S, Matsushita S, Miyaoka H. Association of a polymorphism of the serotonin 1B receptor gene and alcohol dependence with inactive aldehyde dehydrogenase-2. J Neural Transm (Vienna) 2002;109(4):513–21.
Cao JX, Hu J, Ye XM, Xia Y, Haile CA, Kosten TR, et al. Association between the 5-HTR1B gene polymorphisms and alcohol dependence in a Han Chinese population. Brain Res 2011;1376:1–9.
Zuo L, Zhang XY, Wang F, Li CS, Lu L, Ye L, et al. Genome-wide significant association signals in IPO11-HTR1A region specific for alcohol and nicotine codependence. Alcohol Clin Exp Res 2013;37(5):730–9.
Cao J, Liu X, Han S, Zhang CK, Liu Z, Li D. Association of the HTR2A gene with alcohol and heroin abuse. Hum Genet 2014;133(3):357–65.
Risinger FO, Doan AM, Vickrey AC. Oral operant ethanol self-administration in 5-HT1b knockout mice. Behav Brain Res 1999;102(1–2):211–5.
Crabbe JC, Wahlsten D, Dudek BC. Genetics of mouse behavior: interactions with laboratory environment. Science. 1999;284(5420):1670–2.
Gonzales RA, Job MO, Doyon WM. The role of mesolimbic dopamine in the development and maintenance of ethanol reinforcement. Pharmacol Ther 2004;103(2):121–46.
Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry 2016;3(8):760–73.
Le Foll B, Gallo A, Le Strat Y, Lu L, Gorwood P. Genetics of dopamine receptors and drug addiction: a comprehensive review. Behav Pharmacol 2009;20(1):1–17.
Parkitna JR, Sikora M, Golda S, Golembiowska K, Bystrowska B, Engblom D, et al. Novelty-seeking behaviors and the escalation of alcohol drinking after abstinence in mice are controlled by metabotropic glutamate receptor 5 on neurons expressing dopamine d1 receptors. Biol Psychiatry 2013;73(3):263–70.
Harrison SJ, Nobrega JN. Differential susceptibility to ethanol and amphetamine sensitization in dopamine D3 receptor-deficient mice. Psychopharmacology 2009;204(1):49–59.
Carnicella S, Drui G, Boulet S, Carcenac C, Favier M, Duran T, et al. Implication of dopamine D3 receptor activation in the reversion of Parkinson’s disease-related motivational deficits. Transl Psychiatry 2014;4:e401.
Beckley JT, Laguesse S, Phamluong K, Morisot N, Wegner SA, Ron D. The First Alcohol Drink Triggers mTORC1-Dependent Synaptic Plasticity in Nucleus Accumbens Dopamine D1 Receptor Neurons. J Neurosci 2016;36(3):701–13.
Elliot EE, Sibley DR, Katz JL. Locomotor and discriminative-stimulus effects of cocaine in dopamine D5 receptor knockout mice. Psychopharmacology 2003;169(2):161–8.
Newman TK, Parker CC, Suomi SJ, Goldman D, Barr CS, Higley JD. DRD1 5′UTR variation, sex and early infant stress influence ethanol consumption in rhesus macaques. Genes Brain Behav 2009;8(6):626–30.
Cunningham CL, Howard MA, Gill SJ, Rubinstein M, Low MJ, Grandy DK. Ethanol-conditioned place preference is reduced in dopamine D2 receptor-deficient mice. Pharmacol Biochem Behav 2000;67(4):693–9.
Gallo EF, Salling MC, Feng B, Moron JA, Harrison NL, Javitch JA, et al. Upregulation of dopamine D2 receptors in the nucleus accumbens indirect pathway increases locomotion but does not reduce alcohol consumption. Neuropsychopharmacology. 2015;40(7):1609–18.
Rubinstein M, Phillips TJ, Bunzow JR, Falzone TL, Dziewczapolski G, Zhang G, et al. Mice lacking dopamine D4 receptors are supersensitive to ethanol, cocaine, and methamphetamine. Cell. 1997;90(6):991–1001.
Dick DM, Wang JC, Plunkett J, Aliev F, Hinrichs A, Bertelsen S, et al. Family-based association analyses of alcohol dependence phenotypes across DRD2 and neighboring gene ANKK1. Alcohol Clin Exp Res 2007;31(10):1645–53.
Franke P, Nothen MM, Wang T, Knapp M, Lichtermann D, Neidt H, et al. DRD4 exon III VNTR polymorphism-susceptibility factor for heroin dependence? Results of a case-control and a family-based association approach. Mol Psychiatry 2000;5(1):101–4.
Moyer RA, Wang D, Papp AC, Smith RM, Duque L, Mash DC, et al. Intronic polymorphisms affecting alternative splicing of human dopamine D2 receptor are associated with cocaine abuse. Neuropsychopharmacology. 2011;36(4):753–62.
Berggren U, Fahlke C, Aronsson E, Karanti A, Eriksson M, Blennow K, et al. The taqI DRD2 A1 allele is associated with alcohol-dependence although its effect size is small. Alcohol Alcohol 2006;41(5):479–85.
Sasabe T, Furukawa A, Matsusita S, Higuchi S, Ishiura S. Association analysis of the dopamine receptor D2 (DRD2) SNP rs1076560 in alcoholic patients. Neurosci Lett 2007;412(2):139–42.
Bulwa ZB, Sharlin JA, Clark PJ, Bhattacharya TK, Kilby CN, Wang Y, et al. Increased consumption of ethanol and sugar water in mice lacking the dopamine D2 long receptor. Alcohol. 2011;45(7):631–9.
McGeary J. The DRD4 exon 3 VNTR polymorphism and addiction-related phenotypes: a review. Pharmacol Biochem Behav 2009;93(3):222–9.
Larsen H, van der Zwaluw CS, Overbeek G, Granic I, Franke B, Engels RC. A variable-number-of-tandem-repeats polymorphism in the dopamine D4 receptor gene affects social adaptation of alcohol use: investigation of a gene-environment interaction. Psychol Sci 2010;21(8):1064–8.
Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 2010;50:295–322.
Haass-Koffler CL, Goodyear K, Loche A, Long VM, Lobina C, Tran HH, et al. Administration of the metabotropic glutamate receptor subtype 5 allosteric modulator GET 73 with alcohol: A translational study in rats and humans. J Psychopharmacol 2018;32(2):163–73.
Haass-Koffler CL, Goodyear K, Long VM, Tran HH, Loche A, Cacciaglia R, et al. A Phase I randomized clinical trial testing the safety, tolerability and preliminary pharmacokinetics of the mGluR5 negative allosteric modulator GET 73 following single and repeated doses in healthy volunteers. Eur J Pharm Sci 2017;109:78–85.
Cleva RM, Olive MF. mGlu receptors and drug addiction. Wiley Interdiscip Rev Membr Transp Signal 2012;1(3):281–95.
Joffe ME, Centanni SW, Jaramillo AA, Winder DG, Conn PJ. Metabotropic Glutamate Receptors in Alcohol Use Disorder: Physiology, Plasticity, and Promising Pharmacotherapies. ACS Chem Neurosci 2018;9(9):2188–204.
Domart MC, Benyamina A, Lemoine A, Bourgain C, Blecha L, Debuire B, et al. Association between a polymorphism in the promoter of a glutamate receptor subunit gene (GRIN2A) and alcoholism. Addict Biol 2012;17(4):783–5.
Schumann G, Johann M, Frank J, Preuss U, Dahmen N, Laucht M, et al. Systematic analysis of glutamatergic neurotransmission genes in alcohol dependence and adolescent risky drinking behavior. Arch Gen Psychiatry 2008;65(7):826–38.
Meyers JL, Salling MC, Almli LM, Ratanatharathorn A, Uddin M, Galea S, et al. Frequency of alcohol consumption in humans; the role of metabotropic glutamate receptors and downstream signaling pathways. Transl Psychiatry 2015;5:e586.
Lovinger DM, Kash TL. Mechanisms of Neuroplasticity and Ethanol’s Effects on Plasticity in the Striatum and Bed Nucleus of the Stria Terminalis. Alcohol Res 2015;37(1):109–24.
Erb S, Shaham Y, Stewart J. Stress-induced relapse to drug seeking in the rat: role of the bed nucleus of the stria terminalis and amygdala. Stress. 2001;4(4):289–303.
Heaney CF, Kinney JW. Role of GABA(B) receptors in learning and memory and neurological disorders. Neurosci Biobehav Rev 2016;63:1–28.
Kumar K, Sharma S, Kumar P, Deshmukh R. Therapeutic potential of GABA(B) receptor ligands in drug addiction, anxiety, depression and other CNS disorders. Pharmacol Biochem Behav 2013;110:174–84.
Jacobson LH, Vlachou S, Slattery DA, Li X, Cryan JF. The Gamma-Aminobutyric Acid B Receptor in Depression and Reward. Biol Psychiatry 2018;83(11):963–76.
Agabio R, Maccioni P, Carai MA, Gessa GL, Froestl W, Colombo G. The development of medications for alcohol-use disorders targeting the GABAB receptor system. Recent Pat CNS Drug Discov 2012;7(2):113–28.
Kasten CR, Boehm SL, 2nd. Identifying the role of pre-and postsynaptic GABA(B) receptors in behavior. Neurosci Biobehav Rev 2015;57:70–87.
Pin JP, Kniazeff J, Binet V, Liu J, Maurel D, Galvez T, et al. Activation mechanism of the heterodimeric GABA(B) receptor. Biochem Pharmacol 2004;68(8):1565–72.
Pinard A, Seddik R, Bettler B. GABAB receptors: physiological functions and mechanisms of diversity. Adv Pharmacol 2010;58:231–55.
Xu C, Zhang W, Rondard P, Pin JP, Liu J. Complex GABAB receptor complexes: how to generate multiple functionally distinct units from a single receptor. Front Pharmacol 2014;5:12.
Vigot R, Barbieri S, Brauner-Osborne H, Turecek R, Shigemoto R, Zhang YP, et al. Differential compartmentalization and distinct functions of GABAB receptor variants. Neuron. 2006;50(4):589–601.
Phillips TJ, Reed C. Targeting GABAB receptors for anti-abuse drug discovery. Expert Opin Drug Discovery 2014;9(11):1307–17.
Augier E, Dulman RS, Damadzic R, Pilling A, Hamilton JP, Heilig M. The GABAB Positive Allosteric Modulator ADX71441 Attenuates Alcohol Self-Administration and Relapse to Alcohol Seeking in Rats. Neuropsychopharmacology. 2017;42(9):1789–99.
Vengeliene V, Takahashi TT, Dravolina OA, Belozertseva I, Zvartau E, Bespalov AY, et al. Efficacy and side effects of baclofen and the novel GABAB receptor positive allosteric modulator CMPPE in animal models for alcohol and cocaine addiction. Psychopharmacology 2018;235(7):1955–65.
Lee C, Mayfield RD, Harris RA. Altered gamma-aminobutyric acid type B receptor subunit 1 splicing in alcoholics. Biol Psychiatry 2014;75(10):765–73.
Ribeiro AF, Correia D, Torres AA, Boas GR, Rueda AV, Camarini R, et al. A transcriptional study in mice with different ethanol-drinking profiles: possible involvement of the GABA(B) receptor. Pharmacol Biochem Behav 2012;102(2):224–32.
Jacobson LH, Sweeney FF, Kaupmann K, O’Leary OF, Gassmann M, Bettler B, et al. Differential roles of GABAB1 subunit isoforms on locomotor responses to acute and repeated administration of cocaine. Behav Brain Res. 2016;298(Pt B):12–6.
Caputo F, Ciminelli BM, Jodice C, Blasi P, Vignoli T, Cibin M, et al. Alcohol use disorder and GABAB receptor gene polymorphisms in an Italian sample: haplotype frequencies, linkage disequilibrium and association studies. Ann Hum Biol 2017;44(4):384–8.
Terranova C, Tucci M, Di Pietra L, Ferrara SD. GABA Receptors Genes Polymorphisms and Alcohol Dependence: No Evidence of an Association in an Italian Male Population. Clin Psychopharmacol Neurosci 2014;12(2):142–8.
Lorrai I, Maccioni P, Gessa GL, Colombo G. R(+)-Baclofen, but Not S(-)-Baclofen, Alters Alcohol Self-Administration in Alcohol-Preferring Rats. Front Psychiatry. 2016;7:68.
Kasten CR, Blasingame SN, Boehm SL, 2nd. Bidirectional enantioselective effects of the GABAB receptor agonist baclofen in two mouse models of excessive ethanol consumption. Alcohol. 2015;49(1):37–46.
Kasten CR, Boehm SL, 2nd. Intra-nucleus accumbens shell injections of R(+)- and S(-)-baclofen bidirectionally alter binge-like ethanol, but not saccharin, intake in C57Bl/6J mice. Behav Brain Res 2014;272:238–47.
Loi B, Maccioni P, Lobina C, Carai MA, Gessa GL, Thomas AW, et al. Reduction of alcohol intake by the positive allosteric modulator of the GABA(B) receptor, rac-BHFF, in alcohol-preferring rats. Alcohol. 2013;47(1):69–73.
Maccioni P, Fara F, Lorrai I, Acciaro C, Mugnaini C, Corelli F, et al. Suppressing effect of CMPPE, a new positive allosteric modulator of the GABAB receptor, on alcohol self-administration and reinstatement of alcohol seeking in rats. Alcohol. 2019;75:79–87.
Maccioni P, Lorrai I, Contini A, Leite-Morris K, Colombo G. Microinjection of baclofen and CGP7930 into the ventral tegmental area suppresses alcohol self-administration in alcohol-preferring rats. Neuropharmacology. 2018;136(Pt A):146–58.
Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol 2004;44:525–57.
Hauger RL, Risbrough V, Brauns O, Dautzenberg FM. Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: new molecular targets. CNS Neurol Disord Drug Targets 2006;5(4):453–v79.
Justice NJ, Yuan ZF, Sawchenko PE, Vale W. Type 1 corticotropin-releasing factor receptor expression reported in BAC transgenic mice: implications for reconciling ligand-receptor mismatch in the central corticotropin-releasing factor system. J Comp Neurol 2008;511(4):479–96.
Korosi A, Veening JG, Kozicz T, Henckens M, Dederen J, Groenink L, et al. Distribution and expression of CRF receptor 1 and 2 mRNAs in the CRF over-expressing mouse brain. Brain Res 2006;1072(1):46–54.
Kuhne C, Puk O, Graw J, Hrabe de Angelis M, Schutz G, Wurst W, et al. Visualizing corticotropin-releasing hormone receptor type 1 expression and neuronal connectivities in the mouse using a novel multifunctional allele. J Comp Neurol 2012;520(14):3150–80.
Lemos JC, Wanat MJ, Smith JS, Reyes BA, Hollon NG, Van Bockstaele EJ, et al. Severe stress switches CRF action in the nucleus accumbens from appetitive to aversive. Nature. 2012;490(7420):402–6.
Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, et al. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol 2000;428(2):191–212.
Reyes TM, Lewis K, Perrin MH, Kunitake KS, Vaughan J, Arias CA, et al. Urocortin II: a member of the corticotropin-releasing factor (CRF) neuropeptide family that is selectively bound by type 2 CRF receptors. Proc Natl Acad Sci U S A 2001;98(5):2843–8.
Ryabinin AE, Tsoory MM, Kozicz T, Thiele TE, Neufeld-Cohen A, Chen A, et al. Urocortins: CRF’s siblings and their potential role in anxiety, depression and alcohol drinking behavior. Alcohol. 2012;46(4):349–57.
Phillips TJ, Reed C, Pastor R. Preclinical evidence implicating corticotropin-releasing factor signaling in ethanol consumption and neuroadaptation. Genes Brain Behav 2015;14(1):98–135.
Henckens MJ, Deussing JM, Chen A. Region-specific roles of the corticotropin-releasing factor-urocortin system in stress. Nat Rev Neurosci 2016;17(10):636–51.
Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci 2007;30(8):399–406.
Zorrilla EP, Heilig M, de Wit H, Shaham Y. Behavioral, biological, and chemical perspectives on targeting CRF(1) receptor antagonists to treat alcoholism. Drug Alcohol Depend 2013;128(3):175–86.
Schreiber AL, Gilpin NW. Corticotropin-Releasing Factor (CRF) Neurocircuitry and Neuropharmacology in Alcohol Drinking. Handb Exp Pharmacol 2018;248:435–71.
Shalev U, Erb S, Shaham Y. Role of CRF and other neuropeptides in stress-induced reinstatement of drug seeking. Brain Res 2010;1314:15–28.
Blomeyer D, Treutlein J, Esser G, Schmidt MH, Schumann G, Laucht M. Interaction between CRHR1 gene and stressful life events predicts adolescent heavy alcohol use. Biol Psychiatry 2008;63(2):146–51.
Ray LA. Stress-induced and cue-induced craving for alcohol in heavy drinkers: Preliminary evidence of genetic moderation by the OPRM1 and CRH-BP genes. Alcohol Clin Exp Res 2011;35(1):166–74.
Ribbe K, Ackermann V, Schwitulla J, Begemann M, Papiol S, Grube S, et al. Prediction of the risk of comorbid alcoholism in schizophrenia by interaction of common genetic variants in the corticotropin-releasing factor system. Arch Gen Psychiatry 2011;68(12):1247–56.
Treutlein J, Kissling C, Frank J, Wiemann S, Dong L, Depner M, et al. Genetic association of the human corticotropin releasing hormone receptor 1 (CRHR1) with binge drinking and alcohol intake patterns in two independent samples. Mol Psychiatry 2006;11(6):594–602.
Olive MF, Mehmert KK, Koenig HN, Camarini R, Kim JA, Nannini MA, et al. A role for corticotropin releasing factor (CRF) in ethanol consumption, sensitivity, and reward as revealed by CRF-deficient mice. Psychopharmacology 2003;165(2):181–7.
Palmer AA, Sharpe AL, Burkhart-Kasch S, McKinnon CS, Coste SC, Stenzel-Poore MP, et al. Corticotropin-releasing factor overexpression decreases ethanol drinking and increases sensitivity to the sedative effects of ethanol. Psychopharmacology 2004;176(3–4):386–97.
Pastor R, McKinnon CS, Scibelli AC, Burkhart-Kasch S, Reed C, Ryabinin AE, et al. Corticotropin-releasing factor-1 receptor involvement in behavioral neuroadaptation to ethanol: a urocortin1-independent mechanism. Proc Natl Acad Sci U S A 2008;105(26):9070–5.
Giardino WJ, Cocking DL, Kaur S, Cunningham CL, Ryabinin AE. Urocortin-1 within the centrally-projecting Edinger-Westphal nucleus is critical for ethanol preference. PLoS One 2011;6(10):e26997.
Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci 2005;8(5):571–8.
Ellacott KL, Cone RD. The role of the central melanocortin system in the regulation of food intake and energy homeostasis: lessons from mouse models. Philos Trans R Soc Lond Ser B Biol Sci 2006;361(1471):1265–74.
Hadley ME, Haskell-Luevano C. The proopiomelanocortin system. Ann N Y Acad Sci 1999;885:1–21.
Rodrigues AR, Almeida H, Gouveia AM. Intracellular signaling mechanisms of the melanocortin receptors: current state of the art. Cell Mol Life Sci 2015;72(7):1331–45.
Buckley DI, Ramachandran J. Characterization of corticotropin receptors on adrenocortical cells. Proc Natl Acad Sci U S A 1981;78(12):7431–5.
Mountjoy KG. Distribution and function of melanocortin receptors within the brain. Adv Exp Med Biol 2010;681:29–48.
Koch M, Horvath TL. Molecular and cellular regulation of hypothalamic melanocortin neurons controlling food intake and energy metabolism. Mol Psychiatry 2014;19(7):752–61.
Kokare DM, Singru PS, Dandekar MP, Chopde CT, Subhedar NK. Involvement of alpha-melanocyte stimulating hormone (alpha-MSH) in differential ethanol exposure and withdrawal related depression in rat: neuroanatomical-behavioral correlates. Brain Res 2008;1216:53–67.
Navarro M, Cubero I, Knapp DJ, Breese GR, Thiele TE. Decreased immunoreactivity of the melanocortin neuropeptide alpha-melanocyte-stimulating hormone (alpha-MSH) after chronic ethanol exposure in Sprague-Dawley rats. Alcohol Clin Exp Res 2008;32(2):266–76.
Navarro M, Cubero I, Thiele TE. Decreased immunoreactivity of the polypeptide precursor pro-opiomelanocortin (POMC) and the prohormone convertase pc1/3 after chronic ethanol exposure in Sprague-Dawley rats. Alcohol Clin Exp Res 2013;37(3):399–406.
Lindblom J, Wikberg JE, Bergstrom L. Alcohol-preferring AA rats show a derangement in their central melanocortin signalling system. Pharmacol Biochem Behav 2002;72(1–2):491–6.
Navarro M. The Role of the Melanocortin System in Drug and Alcohol Abuse. Int Rev Neurobiol. 136: Elsevier; 2017. p. 121–50.
Gerlai R. Gene targeting: technical confounds and potential solutions in behavioral brain research. Behav Brain Res 2001;125(1–2):13–21.
Navarro M, Carvajal F, Lerma-Cabrera JM, Cubero I, Picker MJ, Thiele TE. Evidence that Melanocortin Receptor Agonist Melanotan-II Synergistically Augments the Ability of Naltrexone to Blunt Binge-Like Ethanol Intake in Male C57BL/6J Mice. Alcohol Clin Exp Res 2015;39(8):1425–33.
Ercil NE, Galici R, Kesterson RA. HS014, a selective melanocortin-4 (MC4) receptor antagonist, modulates the behavioral effects of morphine in mice. Psychopharmacology 2005;180(2):279–85.
Civelli O. Orphan GPCRs and neuromodulation. Neuron. 2012;76(1):12–21.
Kononoff J, Kallupi M, Kimbrough A, Conlisk D, de Guglielmo G, George O. Systemic and Intra-Habenular Activation of the Orphan G Protein-Coupled Receptor GPR139 Decreases Compulsive-Like Alcohol Drinking and Hyperalgesia in Alcohol-Dependent Rats. eNeuro. 2018;5(3).
Zhang LL, Wang JJ, Liu Y, Lu XB, Kuang Y, Wan YH, et al. GPR26-deficient mice display increased anxiety- and depression-like behaviors accompanied by reduced phosphorylated cyclic AMP responsive element-binding protein level in central amygdala. Neuroscience. 2011;196:203–14.
Mizushima K, Miyamoto Y, Tsukahara F, Hirai M, Sakaki Y, Ito T. A novel G-protein-coupled receptor gene expressed in striatum. Genomics. 2000;69(3):314–21.
Van Waes V, Tseng KY, Steiner H. GPR88—a putative signaling molecule predominantly expressed in the striatum: Cellular localization and developmental regulation. Basal Ganglia 2011;1(2):83–9.
Massart R, Mignon V, Stanic J, Munoz-Tello P, Becker JA, Kieffer BL, et al. Developmental and adult expression patterns of the G-protein-coupled receptor GPR88 in the rat: Establishment of a dual nuclear-cytoplasmic localization. J Comp Neurol 2016;524(14):2776–802.
Ghate A, Befort K, Becker JA, Filliol D, Bole-Feysot C, Demebele D, et al. Identification of novel striatal genes by expression profiling in adult mouse brain. Neuroscience. 2007;146(3):1182–92.
Becker JA, Befort K, Blad C, Filliol D, Ghate A, Dembele D, et al. Transcriptome analysis identifies genes with enriched expression in the mouse central extended amygdala. Neuroscience. 2008;156(4):950–65.
Massart R, Guilloux JP, Mignon V, Sokoloff P, Diaz J. Striatal GPR88 expression is confined to the whole projection neuron population and is regulated by dopaminergic and glutamatergic afferents. Eur J Neurosci 2009;30(3):397–414.
Le Merrer J, Befort K, Gardon O, Filliol D, Darcq E, Dembele D, et al. Protracted abstinence from distinct drugs of abuse shows regulation of a common gene network. Addict Biol 2012;17(1):1–12.
Quintana A, Sanz E, Wang W, Storey GP, Guler AD, Wanat MJ, et al. Lack of GPR88 enhances medium spiny neuron activity and alters motor- and cue-dependent behaviors. Nat Neurosci 2012;15(11):1547–55.
Lovinger DM. New twist on orphan receptor GPR88 function. Nat Neurosci 2012;15(11):1469–70.
Meirsman AC, Le Merrer J, Pellissier LP, Diaz J, Clesse D, Kieffer BL, et al. Mice Lacking GPR88 Show Motor Deficit, Improved Spatial Learning, and Low Anxiety Reversed by Delta Opioid Antagonist. Biol Psychiatry 2016;79(11):917–27.
Harris JA, Hirokawa KE, Sorensen SA, Gu H, Mills M, Ng LL, et al. Anatomical characterization of Cre driver mice for neural circuit mapping and manipulation. Front Neural Circuits 2014;8:76.
Smith JS, Lefkowitz RJ, Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov 2018;17(4):243–60.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 568 kb)
Rights and permissions
About this article

Cite this article
Neasta, J., Darcq, E., Jeanblanc, J. et al. GPCR and Alcohol-Related Behaviors in Genetically Modified Mice. Neurotherapeutics 17, 17–42 (2020). https://doi.org/10.1007/s13311-019-00828-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-019-00828-y