
Abstract
There is currently no disease-modifying treatment for Huntington’s disease (HD), which is characterized by chorea motor impairment and cognitive decline. The zinc ionophore, PBT2, was previously shown to improve the phenotype of a HD mouse model and reported efficacy in certain cognitive tests in a phase II clinical trial in HD. Here we report that zinc deficiency is a feature of the hippocampus and cortex in the R6/1 mouse model of HD. Low cortical zinc has been shown to induce cognitive impairment, and indeed, dietary restriction of zinc in R6/1 mice was associated with cognitive impairment in the Y-maze, an exacerbated hippocampal long-term potentiation (LTP) deficit and reduction of AMPA receptors (and not other glutamatergic receptors). These data reveal the importance of zinc in maintaining brain function in HD.
Similar content being viewed by others

Introduction
Huntington’s disease (HD) is a fatal autosomal dominant neurodegenerative disorder resulting from an expanded polyglutamine tract in exon 1 of the gene encoding huntingtin (Htt) [1]. The mutant form of huntingtin (mHtt) elicits progressive motor, cognitive, and behavioral changes. R6/1 and R6/2 mice, which were originally generated with 116 and 144(+) CAG repeats in their human Htt transgene respectively, have often been used to model HD because they recapitulate many of the motor, cognitive, and affective symptoms of the disease, and since HD is a genetic disease, these mutant mice have strong construct and face validity [2]. While it is known that mHtt is the principal driver of neuronal toxicity in HD, several molecular pathways activated by mHtt to elicit cell dysfunction and death have been postulated, but there remains no disease-modifying treatment.
We previously showed that the zinc ionophore, PBT2 (Prana Biotechnology), improved lifespan and motor symptoms of the R6/2 mouse model of HD [3]. This drug was also recently shown to improve cognitive performance in the Trail Making Test Part B in a 26-week phase II clinical trial of 106 HD patients [4]. The brain, and in particular the hippocampus, contains the highest concentrations of zinc within the body [5], and loss of zinc compromises neuronal function. Zinc is an essential component of hundreds of enzymes and transcription factors, and we previously showed that low zinc levels in the brain result in age-dependent cognitive deterioration and neuronal loss [6]. Here, we investigated the status of zinc in R6/1 transgenic mice and whether lowering zinc with dietary restriction may contribute to the phenotype.
Methods
Mice and Mice Tissue Preparation
All mice were housed in a conventional animal facility according to standard animal care protocols and, unless otherwise stated, fed standard laboratory chow (Meat Free Rat and Mouse Diet, Specialty Feeds, Australia) and tap water ad libitum. R6/1 transgenic hemizygote males [7] were originally obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and bred with CBB6 (CBA~C57/B6) F1 females to establish the R6/1 colony at the Florey Institute of Neuroscience and Mental Health. To obtain the mouse brain, mice were euthanized with an overdose of sodium pentobarbitone (Lethabarb, 100 mg/kg) and perfused with phosphate-buffered saline. The brain was micro-dissected and stored at − 80 °C until required.
Mice Cohorts and Treatments
Natural History Study
The number of R6/1 and CBA~C57/B6 (wild-type (WT)) mice and their sex at each age are as follows: n[WT 8 weeks] = 16 (8 females); n[R6/1 8 weeks] = 16 (8 females); n[WT 12 weeks] = 12 (6 females); n[12 weeks R6/1] = 11 (5 females); n[16 weeks WT] = 10 (3 females); n[16 weeks R6/1] = 11 (5 females); n[20 weeks WT] = 32 (21 females); n[20 weeks R6/1] = 31 (20 females). Mice were euthanized at the given ages and brains dissected as described above.
Zinc Deficiency Study
A diet deficient in zinc (chow: Specialty Feeds [Product No. SF01-027], Glen Forest, Western Australia; water: Milli-Q) was administered to WT and R6/1 mice from age 6 weeks. Zinc-deficient animals were compared with mice on standard diet (chow: Speciality Feeds [Product No. AIN93G]; water: regular tap water). The n and sex of each group are as follows: n[WT control diet] = 16 (8 females); n[WT zinc-deficient diet] = 16 (8 female); n[R6/1 control diet] = 16 (8 females); n[R6/1 zinc-deficient diet] = 16 (9 females).
Y-Maze Test
The Y-maze test was performed as previously described [8]. Briefly, all mice were subjected to a 2-trial Y-maze test separated by a 1-h intertrial interval to assess spatial recognition memory, with all testing performed during the light phase of the circadian cycle. Behaviors were recorded on video during a 5-min trial and TopScan software (CleverSys Topscan lite) was used for analysis. Data are expressed as the percentage of frequency for novel arm entries made during the 5-min trial.
Accelerated Rotarod Test
A rotarod (Ugo Basile model 7650; Sandown Scientific, Hampton, UK) was used as a measure of motor performance. A detailed protocol has been described previously [9]. Mice were allowed to walk at the baseline speed for 2 min before allowing the rod to accelerate to 20 rpm over 150 s. If the mice fell off during this time, they were placed back on the rod until the acclimatization period was complete. During the test, acceleration of the revolving cylinder was set from 4 to 40 rpm and the latency to fall was recorded as a measure of motor coordination.
Clasping
Clasping severity was assessed using a previously described scoring system [10]. Mice were held by the tip of the tail against the edge of a table to prevent swinging motion. The mice were observed for a period of 30 s, with the highest clasping score occurring during this time being recorded. A four-point scale was used ranging from 0 to 3. If the hindlimbs are consistently splayed outward, away from the abdomen, it is assigned a score of 0. If one hindlimb is retracted toward the abdomen for more than 50% of the time suspended, it receives a score of 1. If both hindlimbs are partially retracted toward the abdomen for more than 50% of the time suspended, it receives a score of 2. If its hindlimbs are entirely retracted and touching the abdomen for more than 50% of the time suspended, it receives a score of 3. Scorers were blinded to both genotype and treatment. Scorers were also blinded to the purpose of the study.
Zinc Analysis
Zinc content was measured as previously described [8]. Briefly, samples from each experimental condition were freeze-dried and then resuspended in 69% nitric acid (ultraclean grade, Aristar) overnight. The samples were then heated for 20 min at 90 °C and an equivalent volume of hydrogen peroxide (30%, Merck) then added for a further 15-min incubation at 70 °C. The samples were diluted in double-distilled water and assayed by inductively coupled plasma mass spectrometer (ICP-MS; Ultramass 700, Varian). Each sample was measured in triplicate and the concentrations determined from the standard curve were normalized to wet tissue weight.
Western Blot
Samples from each experiment were homogenized in TBS (pH = 7.4) with EDTA-free protease inhibitor cocktail (1:50, Roche) + phosphatase inhibitors I and II (1:1000). Protein concentration was determined by BCA protein assay (Pierce). Aliquots of homogenate with equal protein concentrations were separated in 4–12% bis-Tris gels with NuPAGE MES running buffer (Invitrogen) and transferred to nitrocellulose membranes by iBlot (Invitrogen). The membranes were blocked with milk (10% v/v) and probed with appropriate primary and secondary IgG-HRP-conjugated antibodies (Dako). Enhanced chemiluminescence detection system (GE Healthcare) was used for developing and a Fujifilm LAS-3000 was used for visualization. Densitometry quantification of immunoreactive signals was performed by ImageJ (1.49b, NIH) and normalized to the relative amount of β-actin and expressed as percentage of the mean of the control group. The following antibodies were used: anti-β-actin (Sigma); anti-AMPAR (Cell Signaling); anti-NMDAR1 (Cell Signaling); anti-NMDAR2A (Cell Signaling); anti-NMDAR2B (Cell Signaling).
Acute Hippocampal Slice Preparations
Mice (WT or R6/1, either male or female aged from 18 to 20 weeks old) were anesthetized with isoflurane, and the whole brains were quickly removed and placed in ice-cold, carboxygenated (95% O2, 5% CO2) cutting solution (composition in mmol/L: sucrose, 206; KCl, 3; CaCl2, 0.5; MgCl2-H2O, 6; NaH2PO4, 1.25; NaHCO3, 25; D-glucose, 10.6). Transverse hippocampal slices (350 μm) were prepared with a VT 1200S tissue slicer (Leica) and quickly transferred to 34 °C artificial CSF (aCSF) (composition in mmol/L: NaCl, 126; KCl, 2.5; CaCl2, 2.4; 6 MgCl2-H2O, 1.3; NaH2PO4, 1.25; NaHCO3, 25; D-glucose, 10). Following 1 h of equilibration, slices were transferred to the multielectrode array (MEA) recording chamber.
Multielectrode Array Recordings
One acute hippocampal slice was placed on a 3D-MEA chip comprising 60 30-μm-high electrodes spaced by 200 μm (60 MEA 200/30 iR-Ti: MCS GmbH, Reutlingen, Germany). The slice was immobilized with a harp slice grid (ALA Scientific Instruments, New York, USA) to ensure optimal contact of the slices with the microelectrodes. The slice was continuously superfused with carboxygenated aCSF (3 ml/min at 32 °C) during the whole recording session. The Schäffer collateral was stimulated by injecting a biphasic current waveform (100 μs) through one selected electrode at 0.033 Hz. The amplitude of field excitatory postsynaptic potentials (fEPSPs) at the proximal stratum radiatum of CA1 were analyzed by using LTP-Analyzer (multichannel systems (MCS) GmbH, Reutlingen, Germany). Following a 10-min incubation on the MEA, CA1 fEPSPs were evoked by stimulating the Schäffer collateral pathway with a test stimulus (usually between ~ 1500 and 2500 mV). The basal stimulus was determined by an input-output (I-O) curve, which was obtained by stimulating the Schäffer collateral pathway with increasing stimulation intensities (at 30-s intervals) starting from 500 mV to a stimulation intensity that evoked the maximum fEPSP (determined by when the I-O curve became plateau). The basal stimulus was the stimulation intensity that evoked ~ 50% of the maximum fEPSP. The basal stimulus was then delivered at 20-ms interval (paired-pulse) to evoke paired-pulse facilitation (PPF), a common measure of healthy CA1 region wherein the second pulse evokes stronger fEPSP than the first pulse. Only slices showing PPF were chosen for LTP induction and recording. After 30 min of fEPSP baseline recording with the basal stimulus, LTP was induced by applying 3 trains (at 20-s intervals) of high-frequency stimulation (HFS) wherein each train consisted of the basal stimulus delivered at 100 Hz for a duration of 500 ms. LTP was calculated as the percentage of persistently enhanced fEPSP amplitude after HFS relative to the baseline. Only the last 10-min LTP was used for the statistical analysis.
Statistics
Statistical analysis was carried out in Prism 7 (GraphPad Software Inc). All tests were two-tailed, with the level of significance set at 0.05. The Tukey’s post hoc test was used with ANOVAs. A two-factor independent groups ANOVA was used for comparisons of zinc in different tissues at different ages and zinc treatments, Y maze test, LTP, and western blot results. Mixed model ANOVAs were used for analysis of weight, rotarod and clasping scores over time. The main effects of time and genotype, and their interaction, were included in mixed effects models of mice on low and normal zinc diets. The Benjamini, Krieger, and Yekutieli post hoc test was used for the mixed model ANOVAs.
Results
To investigate whether zinc content is altered in the R6/1 mouse model, we measured zinc in brain tissue during the ages of phenotypic presentation (Fig. 1). At 8 weeks of age, before onset of motor symptoms, zinc was not significantly changed in any region assessed, although there was a modest trend toward decreased zinc in the cortex and hippocampus of R6/1 mice. This trend was a prelude to manifest zinc loss (> 20%) in the cortex and hippocampus of R6/1 mice at 12 weeks of age, which was maintained through until 20 weeks (Fig. 1a, b). The striatum of R6/1 mice also exhibited loss of zinc content at 12 weeks of age, but this was not maintained at later ages (Fig. 1c). In exploratory analysis, we observed no significant difference in cortical zinc between males and females at all ages tested regardless of genotype (data not shown). This study was, however, not powered to test sex differences. Zinc content in the midbrain, cerebellum, and liver (comparatively less affected by HD) did not differ between genotypes at any age (Fig. 1d–f).

Low cortical zinc in R6/1 mice. WT and R6/1 mice were aged in different cohorts to 8, 12, 16, and 20 weeks. Zinc levels were measured in (A) cortex, (B) hippocampus, (C) striatum, (D) midbrain, (E) cerebellum, and (F) liver. Data are mean ± S.E. N is indicated in column bars. All data were analyzed with two-factor ANOVA. *P < 0.05; **P < 0.01; ***P < 0.001
To test whether brain zinc deficiency in R6/1 mice might be contributing to the behavioral phenotype, we used dietary restriction of zinc to determine whether this would exacerbate symptoms. The zinc-deficient diet was well tolerated, given that it did not affect weight in R6/1 mice or WT mice, although there was a nonsignificant trend toward lower weight in the WT mice on the zinc-deficient diet (Fig. 2a). As expected, R6/1 mice failed to accumulate weight like WT mice regardless of diet. Zinc deficiency did not affect motor performance of the WT mice as assessed by the rotarod (Fig. 2b). R6/1 mice on a zinc-deficient diet had impaired performance on rotarod by 8 weeks of age compared to R6/1 mice on a normal diet (ANOVA interaction P = 0.019; post hoc; 8 weeks P = 0.0038), which deteriorated compared to WT mice from week 9 onward. The zinc-deficient R6/1 mice did not have exacerbated motor impairment compared to WT mice thereafter, but these data may reveal that zinc deficiency brings forward the rotarod impairment onset, rather than worsens the severity of symptoms.

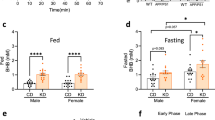
Zinc-deficient diet accelerates decline in R6/1 mice. WT and R6/1 mice were fed either a control or zinc-deficient diet from 6 weeks of age and their (A) weight, (B) motor performance on the rotarod, (C) clasping phenotype, and (D) cognitive performance on the Y-maze test were measured in the intervals recorded. n[WT control diet] = 16 (8 female); n[WT zinc-deficient diet] = 16 (8 female); n[R6/1 control diet] = 16 (8 female); n[R6/1 zinc-deficient diet] = 9 female). Data are mean ± S.E. *P < 0.05; **P < 0.01
This same trend was observed when monitoring the hindlimb clasping phenotype, which progresses in severity with age in R6/1 mice (Fig. 2c). By 19 weeks of age, R6/1 mice on both control and zinc-deficient diet had similar severity of clasping phenotype, but the clasping phenotype in mice on the zinc-deficient diet was more severe at weeks 14 and 15 compared to the mice on a normal diet (ANOVA interaction: P = 0.043; post hoc: 14 weeks P = 0.024; 15 weeks P = 0.001).
Similar to human HD, R6/1 mice develop cognitive impairment as the disease progresses. We assessed cognition using the Y-maze, which assesses the ability of the mice to use visuospatial cues to remember areas of the maze that they have been exposed to prior and to recognize whether an area is novel (Fig. 2d). At 10 weeks of age, WT and R6/1 mice were successful in recognizing the novel arm (because they investigated the novel arm more than what would be expected by chance), but by 16 weeks of age, when the test was repeated, R6/1 mice on the zinc-deficient diet had an early presentation of cognitive impairment compared to R6/1 mice on control diet, which had intact memory performance compared to WT mice (ANOVA, P = 0.047). Given the motor performance of R6/1 on control and zinc-deficient diet were equally impaired at 16 weeks of age, we do not think the changes in cognition on the Y-test are explained by motor impairment.
To explore cognitive changes further, we investigated long-term potentiation in a sub-group of mice (n = 4/group) using a multielectrode array. The maximal response to increasing stimulation of the tissue preparations was equivalent between groups (data not shown). WT mice had sustained LTP regardless of diet intervention (P < 0.001); however, R6/1 mice had reduced LTP performance that was impaired further in mice administered a zinc-deficient diet (P = 0.038; Fig. 3a, b).

Zinc-deficient diet worsens LTP impairment in R6/1 mice. (A) LTP trace of WT and R6/1 mice (aged 18–20 weeks) treated with normal or zinc-deficient diet. (B) LTP response (% over baseline). N = 4 mice (two males and two females) were used per group. Data are mean ± S.E. *P < 0.05; ***P < 0.001
Glutamatergic synapses degenerate in HD [11], so we investigated glutamate receptor levels in the hippocampus of WT and HD animals treated with a normal and zinc-deficient diet (n = 12/group) (Fig. 4). R6/1 mice exhibited decreased AMPA receptor (P < 0.001; Fig. 4a) and NMDA receptor subunits 1 (P = 0.004; Fig. 4b) and 2A (P < 0.001; Fig. 4c), while there was a decreased trend in NMDA receptor 2B (Fig. 4d), similar to our previous findings [12]. The zinc-deficient diet further lowered AMPA receptor levels, but not the NMDA receptor subunits.

Hippocampal glutamate receptors in R6/1 and WT mice treated on normal and zinc-deficient diet. (A) AMPA receptor, (B) NMDA receptor subunit 1, (C) NMDA receptor subunit 2A, and (D) NMDA receptor subunit 2B. Data are mean ± S.E. *P < 0.05; **P < 0.01; ***P < 0.001. Mice were euthanized at 20 weeks of age. N = 12 mice per group (6 males and 6 females)

Zinc levels in R6/1 and WT mice treated on normal and zinc-deficient diet. Mice were euthanized at 20 weeks of age, and zinc was measured in the regions indicated. Data are mean ± S.E. *P < 0.05; **P < 0.01; ***P < 0.001. The n and sex of each group are as follows: n[WT control diet] = 16 (8 females); n[WT zinc-deficient diet] = 16 (8 female); n[R6/1 control diet] = 15 (8 females); n[R6/1 zinc-deficient diet] = 16 (9 females)
Finally, we measured zinc levels in brain and liver of R6/1 and WT mice treated on normal and zinc-deficient diet. In R6/1 and not WT mice, the zinc-deficient diet lowered zinc levels in cortex (ANOVA; Tukey’s post hoc, P = 0.027; Fig. 5a), hippocampus (ANOVA; Tukey’s post hoc, P = 0.034; Fig. 5b), and midbrain (ANOVA; Tukey’s post hoc, P = 0.013; Fig. 5c). The restricted diet did not affect zinc levels in the striatum, cerebellum, or liver regardless of genotype (Fig. 5d–f).
Discussion
Here, we report that R6/1 mice have low levels of zinc in the hippocampus and cortex, areas which are normally highly concentrated zinc [13]. Synaptic zinc release plays a critical role in neurotransmission and the expression of NMDA receptor subunits [6, 14, 15]. Therefore, the conspicuous decrease in zinc levels in hippocampus in cortical regions of R6/1 mice that we observe here could contribute to functional impairments, and indeed, we showed behavioral, neurophysiological, and biochemical evidence that restricting zinc in R6/1 mice caused an exacerbation of the phenotype.
While the cause of the decrease of zinc content in hippocampus and cortex in R6/1 mice is uncertain, the fact that we observed zinc was decreased only in these structures, but not in other brain areas or peripheral tissues (such as liver) indicates that the drop was not due to malabsorption; otherwise, other deficits would be widespread. The decrease in zinc concentration is also not likely to be merely a result of neurodegeneration because the striatum undergoes the most severe degeneration in HD and this area did not show loss of zinc in the R6/1 mice.
Our observations indicate that mHtt might cause a selective impartment to zinc homeostatic machinery in cortical and not sub-cortical regions. There are currently 23 known zinc transporters (SLC30 and SLC39A families) [16], and it is possible that mHtt interreacts with one or more of these transporters to modulate zinc levels in the cortex. The zinc restriction challenge also revealed that the cortex, hippocampus, and possibly midbrain were particularly sensitive structures to loss of dietary zinc. In agreement with prior literature [13], in WT mice, we observed the highest concentrations of brain zinc in the hippocampus and cortex. Since these areas have the highest content of zinc, it is possible that these areas are the most vulnerable to lesions in the zinc regulatory pathways. Indeed, we observed reduction of zinc in R6/1 mice in these structures (and less so other brain regions), and it was these structures that also had further decline in zinc upon dietary zinc challenge. Although this severe zinc-deficient diet only caused a modest decrease in cortical zinc, it appears that these areas may be vulnerable to low zinc compared to other brain regions which were not generally as affected by the expression of the mHtt genotype or zinc-deficient diet.
The AMPA receptor is activated by glutamate and is crucial for the induction of LTP [17]. Repeated activation of the AMPA receptor cause a postsynaptic potential that removes a magnesium block in the NMDA receptor. Disinhibition of the NMDA receptor allows for glutamate to bind and contribute to signal transduction. The reduced level of AMPA receptor in the R6/1 mice as well as the reduced expression of NMDAR1 and NMDAR2A subunits might therefore explain the abnormality in LTP that we observed in the R6/1 mice. But the observation that zinc deficiency was only associated with a further reduction of AMPA receptors, and not NMDA receptors, suggests that the behavioral and electrophysiology abnormalities associated with zinc restriction were more strongly associated with AMPA receptors. In addition, within neurons, zinc uptake is stimulated by the activation of AMPA receptor [18,19,20], which we showed was decreased in the R6/1 mice, and declined further with zinc deficiency. Therefore, low level of AMPA receptor might contribute to both LTP and zinc abnormalities.
How could cortical zinc deficiency contribute to disease progression? There have been several toxic mechanisms proposed for mHtt, and the molecular mechanisms are not fully established. The nuclear localization of huntingtin protein has implicated the normal function of this protein in transcriptional regulation [21], and altered gene expression by mHtt is one of the major postulated pathogenic mechanisms [22]. Zinc activates metal response element-binding transcription factor 1 (MTF-1) which forms a complex with histone acetyltransferase p200/CBP and transcription factor SP1 to promote gene transcription, including zinc transporters [23]. Furthermore, SP1 is a promoter of ZIP8 [24] and ZIP9 [25], which both act to transport zinc into the cytoplasm. SP1 is a zinc finger transcription factor that has been shown to be disrupted in Huntington’s disease [26], so it is possible that zinc deficiency will prevent MTF-1-dependent recruitment of SP1, and the zinc-dependent SP1 gene transcription activity, resulting in an exacerbated phenotype. In addition, zinc has several important rules to maintain synaptic function, and deficiency has been shown to cause synaptic dysfunction, neurodegeneration, and cognitive impairment [6], so low zinc in cortical regions of HD may add to the toxicity of another primary effect of mHtt.
A limitation to our study is the lack of cognitive impairment in the 16-week-old R6/1 mice, which have previously been reported to be abnormal at this age [27]. One possible reason for this lack of memory deficit is that we performed this test twice, first at 10 weeks and then at 16 weeks. It is possible that a learning effect was the reason for this failure to observe any abnormality in the R6/1 mice treated with the normal diet. However, if this is the case, then the R6/1 mice on the zinc-deficient diet still had cognitive impairment, or possibly a failure of the learning response. While it would be important to replicate the cognitive impairment with other tasks, our behavioral data is supported by electrophysiology data which showed reduced hippocampal LTP (a neuronal correlate of learning and memory) response in the R6/1 mice and further decline with zinc restriction.
It was also not clear whether the early impairment on the rotarod in the R6/1 mice on the zinc-deficient diet represented an early presentation of the phenotype, or whether this result occurred by chance (since it was not replicated at older ages). This result on the rotarod was supported by the clasping behavioral analysis, where R6/1 mice on the zinc-deficient diet demonstrated a more severe phenotype at earlier ages, a trend that was possibly similar to what was observed with the rotarod. It would be important to replicate these findings, especially with additional motor tasks and/or with additional HD animal models before reaching a conclusion on the role of zinc in the motor symptoms of HD.
While it would be valuable to replicate in other animal models of HD, these results may still help interpret the prior findings with PBT2, where the drug was shown to improve aspects of cognitive function in mice [3] and in a phase 2 clinical trial [4]. PBT2 is a zinc ionophore, but it also could have other mechanisms, including by acting as a copper ionophore, and copper is also implicated in HD pathogenesis [28]. These results in the present study, which show loss of cortical zinc in R6/1 mice, support a proposed mechanism of action of PBT2 involving elevating brain zinc. Therefore, zinc ionophores warrant exploration as therapeutics for HD.
References
MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–983.
Pouladi MA, Morton AJ, Hayden MR. Choosing an animal model for the study of Huntington’s disease. Nature reviews Neuroscience. 2013;14(10):708–721.
Cherny RA, Ayton S, Finkelstein DI, Bush AI, McColl G, Massa SM. PBT2 Reduces Toxicity in a C. elegans Model of polyQ Aggregation and Extends Lifespan, Reduces Striatal Atrophy and Improves Motor Performance in the R6/2 Mouse Model of Huntington’s Disease. J Huntingtons Dis. 2012;1(2):211–219.
Huntington Study Group Reach HDI. Safety, tolerability, and efficacy of PBT2 in Huntington’s disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet neurology. 2015;14(1):39–47.
Frederickson CJ. Neurobiology of zinc and zinc-containing neurons. International review of neurobiology. 1989;31:145–238.
Adlard PA, Parncutt JM, Finkelstein DI, Bush AI. Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer’s disease? The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30(5):1631–1636.
Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87(3):493–506.
Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. 2012;18(2):291–295.
van Dellen A, Cordery PM, Spires TL, Blakemore C, Hannan AJ. Wheel running from a juvenile age delays onset of specific motor deficits but does not alter protein aggregate density in a mouse model of Huntington’s disease. BMC neuroscience. 2008;9:34.
Guyenet SJ, Furrer SA, Damian VM, Baughan TD, La Spada AR, Garden GA. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp. 2010(39). https://doi.org/10.3791/1787.
Dure LSt, Young AB, Penney JB. Excitatory amino acid binding sites in the caudate nucleus and frontal cortex of Huntington’s disease. Annals of neurology. 1991;30(6):785–793.
Wright DJ, Gray LJ, Finkelstein DI, Crouch PJ, Pow D, Pang TY, et al. N-acetylcysteine modulates glutamatergic dysfunction and depressive behavior in Huntington’s disease. Human molecular genetics. 2016;25(14):2923–2933.
Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nature reviews Neuroscience. 2005;6(6):449–462.
Sullivan JA, Zhang XL, Sullivan AP, Vose LR, Moghadam AA, Fried VA, et al. Zinc enhances hippocampal long-term potentiation at CA1 synapses through NR2B containing NMDA receptors. PloS one. 2018;13(11):e0205907.
Anderson CT, Radford RJ, Zastrow ML, Zhang DY, Apfel UP, Lippard SJ, et al. Modulation of extrasynaptic NMDA receptors by synaptic and tonic zinc. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(20):E2705–14.
Kambe T, Tsuji T, Hashimoto A, Itsumura N. The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism. Physiological reviews. 2015;95(3):749–784.
Bortolotto ZA, Fitzjohn SM, Collingridge GL. Roles of metabotropic glutamate receptors in LTP and LTD in the hippocampus. Current opinion in neurobiology. 1999;9(3):299–304.
Takeda A, Sakurada N, Ando M, Kanno S, Oku N. Facilitation of zinc influx via AMPA/kainate receptor activation in the hippocampus. Neurochemistry international. 2009;55(6):376–382.
Sensi SL, Yin HZ, Weiss JH. AMPA/kainate receptor-triggered Zn2+ entry into cortical neurons induces mitochondrial Zn2+ uptake and persistent mitochondrial dysfunction. The European journal of neuroscience. 2000;12(10):3813–3818.
Yin HZ, Ha DH, Carriedo SG, Weiss JH. Kainate-stimulated Zn2+ uptake labels cortical neurons with Ca2+-permeable AMPA/kainate channels. Brain research. 1998;781(1–2):45–56.
Kegel KB, Meloni AR, Yi Y, Kim YJ, Doyle E, Cuiffo BG, et al. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. The Journal of biological chemistry. 2002;277(9):7466–7476.
Thomson SB, Leavitt BR. Transcriptional Regulation of the Huntingtin Gene. Journal of Huntington's Disease. 2018;7(4):289–296.
Li Y, Kimura T, Huyck RW, Laity JH, Andrews GK. Zinc-induced formation of a coactivator complex containing the zinc-sensing transcription factor MTF-1, p300/CBP, and Sp1. Molecular and cellular biology. 2008;28(13):4275–4284.
Aiba I, Hossain A, Kuo MT. Elevated GSH level increases cadmium resistance through down-regulation of Sp1-dependent expression of the cadmium transporter ZIP8. Molecular pharmacology. 2008;74(3):823–833.
Qiu Y, Gao Y, Yu D, Zhong L, Cai W, Ji J, et al. Genome-wide analysis reveals zinc transporter ZIP9 regulated by DNA methylation promotes radiation-induced skin fibrosis via the TGF-β signaling pathway. Journal of Investigative Dermatology. 2019. https://doi.org/10.1016/j.jid.2019.04.027.
Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, et al. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science. 2002;296(5576):2238–2243.
Dowie MJ, Howard ML, Nicholson LF, Faull RL, Hannan AJ, Glass M. Behavioural and molecular consequences of chronic cannabinoid treatment in Huntington’s disease transgenic mice. Neuroscience. 2010;170(1):324–336.
Fox JH, Kama JA, Lieberman G, Chopra R, Dorsey K, Chopra V, et al. Mechanisms of copper ion mediated Huntington’s disease progression. PloS one. 2007;2(3):e334.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Funding
This study was supported by funds from the Australian National Health & Medical Research Council (NHMRC: APP1113686). The Florey Institute of Neuroscience and Mental Health recognizes support from the Victorian Government, in particular funding from the Operational Infrastructure Support Grant. No funder of this study had any role in the design and conduct of the study; collection, management, analysis, or interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All animal experiments were approved by the Howard Florey Animal Ethics Committee (permit # 13-110 and 14-056) and were conducted in accordance with state law and the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes as described by the National Health and Medical Research Council of Australia.
Disclosures
Dr. Bush is a shareholder in Prana Biotechnology Pty Ltd., Cogstate Pty Ltd., Eucalyptus Pty Ltd., Mesoblast Pty Ltd., Brighton Biotech LLC, Nextvet Ltd., Grunbiotics Pty Ltd., and Collaborative Medicinal Development LLC, and a paid consultant for Collaborative Medicinal Development. Dr. Adlard is a shareholder in, and consultant for, Prana Biotechnology Pty Ltd. The other authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PDF 514 kb)
Rights and permissions
About this article

Cite this article
Ayton, S., Lei, P., Appukuttan, A.T. et al. Brain Zinc Deficiency Exacerbates Cognitive Decline in the R6/1 Model of Huntington’s Disease. Neurotherapeutics 17, 243–251 (2020). https://doi.org/10.1007/s13311-019-00785-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-019-00785-6