
Abstract
Antimicrotubulin chemotherapeutic agents, including plant-derived vincaalkaloids such as vincristine, can cause peripheral neuropathic pain. Exogenously activated heme oxygenase 1 (HO-1) is a potential therapy for chemotherapy-induced neuroinflammation. In this study, we investigated a role for Nrf2/HO-1/CO in mediating vincristine-induced neuroinflammation by inhibiting connexin 43 (Cx43) production in the spinal cord following the intrathecal application of the HO-1 inducer protoporphyrin IX cobalt chloride (CoPP) or inhibitor protoporphyrin IX zinc (ZnPP), and we analyzed the underlying mechanisms by which levo-corydalmine (l-CDL, a tetrahydroprotoberberine) attenuates vincristine-induced pain. Treatment with levo-corydalmine or oxycodone hydrochloride (a semisynthetic opioid analgesic, used as a positive control) attenuated vincristine-induced persistent pain hypersensitivity and degeneration of the sciatic nerve. In addition, the increased prevalence of atypical mitochondria induced by vincristine was ameliorated by l-CDL in both A-fibers and C-fibers. Next, we evaluated whether nuclear factor E2-related factor 2 (Nrf2), an upstream activator of HO-1, directly bound to the HO-1 promoter sequence and degraded heme to produce carbon monoxide (CO) following stimulation with vincristine. Notably, l-CDL dose-dependently increased HO-1/CO expression by activating Nrf2 to inhibit Cx43 expression in both the spinal cord and in cultured astrocytes stimulated with TNF-α, corresponding to decreased Cx43-mediated hemichannel. Furthermore, l-CDL had no effect on Cx43 following the silencing of the HO-1 gene. Taken together, our findings reveal a novel mechanism by which Nrf2/HO-1/CO mediates Cx43 expression in vincristine-induced neuropathic pain. In addition, the present findings suggest that l-CDL likely protects against nerve damage and attenuates vincristine-induced neuroinflammation by upregulating Nrf2/HO-1/CO to inhibit Cx43 expression.
Similar content being viewed by others


Introduction
Chemotherapy-induced neuropathic pain (CINP) is characterized by mixed sensory motor neuropathy, depending on the drug type [1]. Vincristine (VCR), a commonly prescribed chemotherapeutic agent, has long been used to control the proliferation of solid tumors and brain tumors and to treat lymphomas and leukemias [2, 3]. Because of its low permeability through the blood-brain barrier, its neurotoxic effects are mainly observed on the peripheral nervous system [4]. The severity and incidence of VCR-induced neuropathic pain is positively correlated with the duration of the dose and treatment, which limit continued treatment [1, 5, 6]. The possible cause of peripheral neuropathy is axonal damage subsequent to microtubule destruction [7]. Vincristine possesses neurotoxic and anticancer effects due to its ability to induce mitochondrial dysfunction and a subsequent energy deficiency [8]. Thus, based on the current clinical situation, a key goal is to identify novel, potentially effective drugs for future treatment of chemotherapy-induced neuropathic pain.
Levo-corydalmine (l-CDL) is a new compound that was obtained by replacing the methoxy group at the C-10 position of levo-tetrahydropalmatine (l-THP) with a phenolic hydroxyl group. l-THP, a tetrahydroprotoberberine isoquinoline alkaloid, has been identified as a primary active constituent of the genera Stephania and Corydalis [9]. Importantly, l-THP induces robust anti-hyperalgesic effects on a mouse model of chemotherapy-induced neuropathic pain [9]. Oxycodone, a morphine-benzyltetrahydroisoquinoline alkaloid, is a semisynthetic opioid analgesic, and among the clinically used opioids, oxycodone exerts excellent anti-allodynic and anti-hyperalgesic effects on neuropathic pain [10, 11]. In addition, l-CDL and oxycodone have a similar structure to benzyltetrahydroisoquinoline alkaloid. Considering these factors, we chose oxycodone as a positive control in vivo to investigate whether the drug l-CDL is superior to the currently clinically used drug oxycodone in attenuating vincristine-induced pain hypersensitivity and biochemical alterations.
Heme oxygenase 1 (HO-1, encoded by HMOX1) is a rate-limiting enzyme that catalyzes the oxidative degradation of heme into biliverdin, iron, and carbon monoxide (CO) [12]. A recent study confirmed that exogenously induced HO-1 represents a potential therapeutic agent for chemotherapy-induced neuropathic pain [13]. Protoporphyrin IX cobalt chloride (CoPP) is a substrate for the inducible isoform HO-1 [12, 14]. Repeated systemic injection of CoPP attenuates vincristine-induced pain hypersensitivity [13], and we compared the effects of l-CDL and CoPP on reducing vincristine-induced pain in the present study. The nuclear factor E2-related factor 2 (Nrf2)-antioxidant response element (ARE) signalling pathway functions by regulating the expression cell survival-related genes, antioxidants, and anti-inflammatory factors [15]. The Nrf2/HO-1 signalling pathway also regulates mitochondrial oxidative damage [16]. According to a recent, interesting study, the release of CO molecules to exogenously deliver CO or increase endogenous CO production represents a potentially novel approach to the treatment of neuropathic pain [17]. In addition, CO is a potential novel inhibitor of connexin hemichannels, including connexin 43 and connexin 46, in HeLa and MCF-7 cells [18]. Connexin 43 (Cx43) constitutes the main gap junction protein in astrocytes [19]. Spinal cord injury and nerve stimulation upregulate Cx43 expression [20, 21]. In particular, continuous upregulation of Cx43 maintains late-phase neuropathic pain [22]. In the current study, we aimed to identify a novel mechanism and determine whether Nrf2/HO-1/CO-mediated Cx43 expression is involved in vincristine-induced neuropathic pain. Strategies targeting this pathway with selective antagonists represent potentially effective interventions. We also explored whether pharmacological increases in Nrf2/HO-1/CO induced by l-CDL exerts an inhibitory effect on Cx43. l-CDL conferred neuroprotection and reduced the prevalence of atypical mitochondria, supporting the potential clinical applications of l-CDL to prevent vincristine-induced neuropathic pain.
Materials and Methods
Animals
Animals (adult male ICR mice weighing 22–25 g; Qinglongshan Animal Farm of Nanjing, China; Production Licence No. SCXK (Su) 2017-0001) were housed in plastic cages and maintained under controlled conditions (a fixed 12-h light/dark cycle with free access to food and water, controlled temperature of 22–26 °C, and 60 ± 10% relative humidity). All animal procedures were performed between 9:00 am and 5:00 pm. All animal experiments complied with the ARRIVE guidelines and were performed in accordance with the International Association for the Study of Pain. Furthermore, all experiments were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals and approved by China Pharmaceutical University (Nanjing, China; licence number: SYXK (Su) 2016-0011). Mice were randomly allocated to each experiment. All efforts were made to minimize animal suffering and the number of animals used.
Chemotherapy-Related Pain Induction and Drug Administration Protocols
Mice were intraperitoneally injected with vincristine sulfate (Lingnan Pharmaceuticals Incorporated, Guangzhou, China; dissolved in normal saline) at a dose of 0.1 mg/kg (injection volume, 0.1 ml/10 g) once a day or with the same volume of normal saline (by the same schedule) for 5 consecutive days as described previously [13, 23].
l-CDL (98% purity) was prepared by the School of Traditional Chinese Medicine, China Pharmaceutical University (Nanjing, China). l-CDL (administered to three groups at doses of 5, 10, and 20 mg/kg) was dissolved in sodium carboxyl methyl cellulose (CMC-Na) and administered intragastrically once daily for 9 consecutive days, with the first administration beginning on the first day after the last vincristine injection. Compound doses were based on the results of previous laboratory studies [23, 24]. The vehicle (CMC-Na) was administered to the control group according to the same schedule. Oxycodone (oxycodone hydrochloride prolonged-release tablets) was purchased from Bard Pharmaceuticals Limited (United Kingdom, package distributed in China). Oxycodone (1.5 mg/kg, dissolved in CMC-Na according to specified conversion in the drug instructions) and the HO-1 inducer CoPP (St. Louis, Sigma-Aldrich, USA; 5 mg/kg) were also administered intragastrically according to the same schedule as l-CDL for 9 consecutive days, based on a previous study [13].
For intrathecal injection, mice in the CoPP (8 μg) group, the HO-1 inhibitor protoporphyrin IX zinc (ZnPP, 8 μg, OKA, Beijing, China) group, or the vehicle (saline + 1% DMSO) group were intrathecally injected with 20 μl of the appropriate formulation using a 30-G needle between the L5 and L6 intervertebral space to deliver the reagents to the cerebrospinal fluid [25], whereas l-CDL (20 mg/kg) was administered intragastrically alone or administered following the intrathecal ZnPP (8 μg) injection as the control group.
Behavioral Assessments of Mechanical Allodynia and Heat Hyperalgesia in Mice
All manipulations were performed in a test room by the same experimenter under quiet conditions to avoid stress. For the assessment of mechanical sensitivity, animals were placed on a wire mesh bottom suspended in cages and allowed 30 min for habituation before the examination. The plantar surface of each hind paw was stimulated with a series of von Frey hairs with logarithmically increasing stiffness (0.02–1.4 g, Woodland Hills, Los Angeles, CA). When the animals were resting, von Frey hairs were presented perpendicularly to the plantar surface (2–3 s for each hair), and a sharp withdrawal of the paw was considered a positive response. The 50% paw withdrawal threshold (PWT) was determined using Dixon’s up-down method [26]. Heat sensitivity was measured as the duration of immersion and was assessed using the tail-flick latency test (TFL). The tail of the mouse was immersed in hot water maintained at 48 °C until withdrawal, with a cutoff time of 15 s to prevent tissue damage [27].
Mechanical allodynia and heat hyperalgesia were evaluated before the intrathecal injection of vehicle, CoPP or ZnPP and at 2, 4, 7, and 24 h after the intrathecal injections. For the groups receiving intrathecal injection of ZnPP followed by l-CDL intragastric administration or l-CDL administered intragastrically alone, mechanical allodynia and heat hyperalgesia were simultaneously evaluated.
Assessment of Sciatic Nerve Degradation and Mitochondrial Abnormalities Using Transmission Electron Microscopy
For histological examinations, mice were sacrificed after behavioral testing on day 14. The sciatic nerve was exposed, cut into 3-mm-long segments, and transferred to 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M phosphate buffer overnight at 4 °C, pH 7.4. The fixed nerves were postfixed with osmium tetroxide at 25 °C for 1 h, dehydrated in a graded series of alcohol solutions, and embedded in Epon812. Ultrathin sections (70 nm) were cut with an ultramicrotome (Leica, German) and stained with uranyl acetate and lead citrate for 30 min and 5 min [28], respectively. Electron photomicrographs were captured at a × 5000 magnification to assess sciatic nerve degradation and count atypical mitochondria, whereas electron photomicrographs at a × 30,000 magnification were captured for further evaluation of mitochondrial abnormalities. The observer was blinded to the group assignment in each analysis and quantification. Photographs of all A-fibers and C-fibers were captured at a × 30,000 magnification. Each myelinated or unmyelinated fiber and axon was randomly selected for imaging. Swelling and vacuolar mitochondria were identified as previously described [29].
Primary Astrocyte Cultures
Primary astrocyte cultures were prepared using previously described methods [30]. Briefly, astrocyte cultures were prepared from the cerebral cortexes of neonatal mice (P2). The meninges were carefully removed from the isolated cerebral hemispheres and then minced before triturating, filtering through a 200 μm nylon screen, and collection by centrifugation at ~ 1500 rpm for 10 min. The dissociated cells were suspended in DMEM/F12 (KeyGEN BioTECH, Nanjing, China) supplemented with 10% (v/v) foetal bovine serum (FBS), penicillin (100 U/ml), and streptomycin (100 μg/ml). After trituration, the cell suspension was plated in 75 cm2 tissue culture flasks at a density of 1 × 107 cells/flask. The medium was replaced twice per week. After culture for 8–10 days, astrocytes were prepared by shaking the flasks for 3 h and then incubating the cells with 0.05% trypsin in a cell incubator for 15 min to separate microglial cells and oligodendrocytes from the astrocytes. Prepared astrocytes exhibited a purity of 80–90%, as determined by glial fibrillary acidic protein (GFAP) immunoreactivity, and showed a star-shaped morphology with processes extending from the soma. Prior to stimulation with TNF-α (PeproTech, Rocky Hill, USA), the media was replaced with Opti-MEM (Gibco, Grand Island, USA). Cells were incubated with l-CDL (3, 10, or 30 μM), CoPP (10 μM) or Gap 27 (30 μM) treatments for 24 h after the TNF-α treatment (10 ng/ml, 60 min at 37 °C). Following treatment with the HO-1 siRNA (100 nM, 36 h, CAAGGAGGTACACATCCAA; Ribobio, Guangzhou, China) and before the TNF-α- incubation (10 ng/ml, 60 min), astrocytes were incubated with l-CDL (3, 10, or 30 μM) and CoPP (10 μM) for 24 h. After the treatments, astrocytes were collected for enzyme-linked immunosorbent assays (ELISA), real-time quantitative PCR, and Western blotting.
Immunohistochemical Staining
The spinal cords were harvested from the mice in each group 14 days after the vincristine injection, and a standard immunohistochemistry methodology was used. Briefly, paraffin-embedded spinal cord sections were prepared, deparaffinized, rehydrated, and pre-treated with citrate buffer (pH 6.0) in a microwave before cooling to room temperature. Sections were washed with 0.01 M phosphate-buffered saline (PBS), blocked with 10% goat serum for 15 min to reduce non-specific antibody binding, and then incubated with the primary antibody (HO-1, 1:100, rabbit; Proteintech, Rosemont, USA) at 4 °C overnight. Sections were subsequently rinsed with PBS and then reacted with secondary antibodies and a polymer helper for 30 min at 37 °C, followed by PBS and polyclonal HRP-conjugated goat-anti-rabbit IgG for 1 h at 37 °C. Finally, the slides were washed with PBS and developed with 3,3′-diaminobenzidine (DBA), followed by counterstaining with haematoxylin. The specimens were examined under a positive biological microscope (BX53, Olympus, USA) and assessed using ImageJ 1.50i software (National Institutes of Health, USA).
Immunofluorescence Staining
After appropriate survival times, mice were transcardially perfused through the ascending aorta with 0.1 M PBS (pH 7.4), followed by 4% paraformaldehyde in 0.16 M phosphate buffer containing 1.5% picric acid. After perfusion, the L4-L5 spinal cord segments were removed and postfixed with the same fixative overnight; the fixative was then replaced with 30% sucrose until sectioning. The embedded blocks were sectioned at a thickness of 30 μm using a cryostat (Leica CM3050S; Germany) and stored (free-floating) until immunofluorescence staining as previously described [31]. Briefly, the sections were blocked with 10% normal goat serum and 0.3% Triton X-100 in 0.01 M PBS (pH 7.4) for 2 h at room temperature and then incubated overnight at 4 °C with the following primary antibodies: Cx43 antibody (1:100, rabbit; Cell Signaling Technology, Danvers, USA), GFAP antibody (1:300, mouse; Cell Signaling Technology, Danvers, USA), Iba-1 antibody (1:200, mouse; Santa Cruz Biotechnology, CA, USA), and RBFOX3/NeuN antibody (1:300, mouse; OriGene Technologies, Rockville, USA). Sections were rinsed with PBS three times and then incubated with FITC- or Cy3 (cyanine 3)-conjugated secondary antibodies (1:100; Bioss, Beijing, China) for 2 h at room temperature. For double immunofluorescence staining, sections were incubated with a mixture of polyclonal and monoclonal primary antibodies, followed by a mixture of FITC- and Cy3-conjugated secondary antibodies (Bioss, Beijing, China). Sections were cover-slipped using a water-based mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; Beyotime, Shanghai, China). Finally, the stained sections were examined under a Carl Zeiss fluorescence microscope, and images were captured with a CCD Spot camera equipped with image acquisition software (Axio Vision; Carl Zeiss). Image acquisition was conducted with fixed settings for exposure, camera gain, and laser intensity. Data were quantified in a completely blinded manner using ImageJ 1.50i software (National Institutes of Health, USA) at the appropriate intensity to reveal all positive cells.
For the immunofluorescence staining of cultured astrocytes, after an incubation with TNF-α followed by l-CDL or CoPP, astrocytes were fixed with 4% paraformaldehyde for 30 min and processed for immunofluorescence staining with the Cx43 antibody (1:100, rabbit; Cell Signaling Technology, Danvers, USA), HO-1 antibody (1:100, rabbit; Proteintech, Rosemont, USA), GFAP antibody (1:300, mouse; Cell Signaling Technology, Danvers, USA), Iba-1 antibody (1:200, mouse; Santa Cruz Biotechnology, CA, USA), and RBFOX3/NeuN antibody (1:300, mouse; OriGene Technologies, Rockville, USA), as indicated above. We also performed double staining with a mixture of polyclonal and monoclonal primary antibodies. After immunostaining, DAPI (Bioss, Beijing, China) was added for 20 min at room temperature to stain all the nuclei of cells in the cultures.
Analysis of the Hemichannel Function Analysis in Cultured Astrocytes
Ethidium bromide uptake was used to measure the function of hemichannels due to its permeability for hemichannels. Astrocytes were stimulated with TNF-α for 1 h followed by l-CDL (3, 10, or 30 μM), CoPP (10 μM), or Gap 27 (30 μM) and then exposed to 0.5 μM ethidium bromide (Sigma-Aldrich; St. Louis, USA) for 10 min at 37 °C [22]. Next, the cells were washed with Hank’s balanced salt solution (HBSS, Gibco, Grand Island, USA). Astrocytes were examined with a fluorescence microscope (Nikon, Ts2R), and images were captured with a CCD Spot camera. Data were quantified in a completely blinded manner using ImageJ 1.50i software (National Institutes of Health, USA) at the appropriate intensity to identify all cells displaying positive staining for ethidium bromide.
Western Blot
The L4-L5 spinal segments were homogenized in a radio immunoprecipitation assay (RIPA) lysis buffer containing proteinase inhibitors (Applygen Technologies, Beijing, China). The supernatants were collected and protein concentrations were determined using the bicinchoninic acid (BCA) protein assay method. Equivalent amounts of protein (50 μg) were loaded in each lane, separated on 10% SDS-PAGE gels, and transferred to nitrocellulose membranes. After transfer, the blots were first saturated with 10% skim milk (in 10 mM Tris-HCl containing 150 mM sodium chloride and 0.5% Tween 20), incubated for 60 min, and then incubated overnight at 4 °C with antibodies against Nrf2 (1:1000, rabbit; Abcam, Shanghai, China), HO-1 (1:500, rabbit; Proteintech, Rosemont, USA), and Cx43 (1:1000, rabbit; Cell Signaling Technology, Danvers, USA). As a loading control, the blots were probed with a GAPDH antibody (1:1000, rabbit; Bioss, Beijing, China), β-actin antibody (1:2000, rabbit; Bioss, Beijing, China), or histone H3 antibody (1:1000, rabbit; Abcam, Shanghai, China). These blots were further incubated with a horseradish peroxidase-conjugated secondary antibody for 2 h and developed using an electrochemiluminescence (ECL) solution. The experiment was repeated in triplicate, and GAPDH, β-actin, or histone H3 served as an internal control. Specific bands were evaluated based on their apparent molecular size. The intensity of the selected bands was analyzed using ImageJ 1.50i software (National Institutes of Health, USA). Boxes of the same size were used to select positive bands.
Real-Time Quantitative PCR
Total RNA was extracted from the L4-L5 spinal segments and treated astrocytes using Trizol reagent (Takara, Shiga, Japan). RNA quantity and purity were determined using an ultraviolet spectrophotometer (Eppendorf biophotometer). The cDNA templates were amplified using the following primers: m-HO-1 forward, 5′-TAG CTC ATC CCA GAC ACC GC-3′; m-HO-1 reverse, 5′-CAG GCA AGA TTC TCC CTT ACA GA-3′; m-Cx43 forward, 5′-CCT TGG TGT CTC TCG CTC TGA-3′; m-Cx43 reverse, 5′-GAG CAG CCA TTG AAG TAA GCA TA-3′; m-GAPDH forward, 5′-GAA CGG GAA GCT CAC TGG-3′; and m-GAPDH reverse, 5′-GCC TGC TTC ACC ACC TTC T-3′. All quantitative PCR experiments were performed using a Real-time Detection System (Eppendorf AG, Hamburg, Germany) with an SYBR Premix Ex Taq™ II kit (Yifeixue Bio Tech, Nanjing, China). PCR amplifications were performed at 95 °C for 30 s, followed by 45 cycles at 95 °C for 5 s, 56 °C for 30 s, and 72 °C for 30 s. Levels of specific mRNAs were calculated after the normalization of the results from each sample to the expression of the GAPDH mRNA. The data are presented as relative mRNA units with respect to control values (expressed as fold increase compared with the sham value or vehicle value). Melting curves were obtained upon completion of the cycles to ensure that nonspecific products were absent. Quantification was performed using the comparative CT method (2 –ΔΔCt: normalizing cycle threshold (Ct) values to the GAPDH Ct).
ELISA
After the animals were sacrificed, L4–L6 spinal cord segments were collected and homogenized for the CO analysis. For primary cultures of astrocytes, cells were collected after treatment for analysis using a mouse CO ELISA kit (Enzyme-linked Biotechnology, Shanghai, China). ELISAs were performed in triplicate according to the manufacturer’s instructions. The standard curve was included in the experiment.
Transfection and Luciferase Assays
The wild-type HO-1 (HO-1-wt) promoter (− 1 to − 2000 bp) and HO-1 mutant (HO-1-mut) promoter (− 1 to 2000 bp) were ligated into a pGL3 basic luciferase reporter vector (KeyGEN BioTECH, Jiangsu, China). 293 T cells were grown to 75–85% confluence and transfected with or without Nrf2 DNA-Lipofectamine 2000 Transfection Reagent (Biosharp, Hefei, China) in a 24-well plate according to the manufacturer’s instructions. As an internal control, the reporter plasmids described above containing wild-type Renilla luciferase (Yeasen, Shanghai, China) were co-transfected into cells. At 24 h after transfection, cellular extracts were prepared. The Dual-Luciferase reporter assay system (Yeasen, Shanghai, China) was employed to evaluate luciferase activity according to the manufacturer’s protocol using a multi-function microplate reader (POLARstar Omega, BMG LABTECH, Germany).
Statistical Analysis
The experimental results are presented as means±S.D. Adult male ICR mice (weighing 22–25 g) were randomly allocated for behavioral studies (n = 6), histological studies (n = 5), or biochemical studies (n = 3), and n refers to the number of animals or cells in each group. Differences between groups were compared using Student’s t test, one-way analysis of variance (ANOVA), or two-way ANOVA. Data were analyzed using SPSS (IBM SPSS Statistics v19.0), and P < 0.05 was considered statistically significant. All statistical figures were created using GraphPad Prism 5.0 software.
Results
l-CDL Attenuates Vincristine-Induced Pain Hypersensitivity in Mice
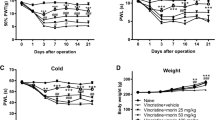
As shown in Fig. 1A, mice were injected with l-CDL, oxycodone, or CoPP daily for 9 consecutive days (from 6 to 14 days), and pain behavior was assessed on days 0, 4, 6, 8, 10, 12, and 14. On day 14, we analyzed the expression of Nrf2/HO-1/CO and Cx43 and performed a histological analysis of the sciatic nerve. Regarding mechanical sensitivity, the PWT was significantly decreased at day 6 in response to von Frey hair stimulation and decreased from 0.99 ± 0.06 g before vincristine injection to 0.06 ± 0.04 g at day 14 (Fig. 1B). Regarding heat sensitivity, the TFL in response to the heat stimulation in the vincristine-treated group was obviously decreased compared with the sham group at day 6 and decreased from 12.46 ± 1.36 s before vincristine administration to 4.63 ± 0.74 s at day 14 after the vincristine treatment (Fig. 1C), indicating the development of heat hyperalgesia.

l-CDL attenuates vincristine-induced pain hypersensitivity in mice. (A) Timeline of vincristine, l-CDL, CoPP, and oxycodone treatments, behavioral tests, histological analysis, and the detection of various factors. (B, C) The PWT and the TFL were both significantly decreased at day 6 and maintained at day 14 in the vincristine-treated group. l-CDL and the CoPP and oxycodone positive control groups exhibited a reversal of vincristine-induced mechanical allodynia and thermal hyperalgesia after the first injection, and the effect was maintained at day 14. ###P < 0.001 compared with the sham control; **P < 0.01 and ***P < 0.001 compared with the vincristine-treated group, two-way ANOVA followed by Bonferroni’s post hoc tests, n = 6 mice/group. All data are presented as means±S.D.
Notably, l-CDL reversed both heat hyperalgesia and mechanical allodynia after the first injection, and this effect was maintained at day 14 (Fig. 1B, C). The pain threshold values of the l-CDL-treated, oxycodone-treated, and CoPP-treated groups also increased with an increasing duration of administration, approaching the value of the sham group. Moreover, the group that received a high dose of l-CDL showed a higher mechanical pain threshold than the positive group treated with oxycodone after 10 days and was similar to the positive group treated with the HO-1 inducer CoPP.
l-CDL Attenuates the Vincristine-Induced Degeneration of the Sciatic Nerve and Reduces the Prevalence of Atypical Mitochondria
As shown in Fig. 2A, normal myelinated (m) and unmyelinated fibers (n) were observed in the sham group. An examination of the sciatic nerve at 14 days after the vincristine injection revealed that the myelinated fibers, particularly the large fibers, were severely damaged, whereas the myelin sheath and axons were severely degraded (p). Some of the unmyelinated fibers appeared to be in looser bundles. Some damage to myelin was observed, but many myelinated fibers appeared normal in the l-CDL and oxycodone groups, particularly the group treated with the high dose of l-CDL, for which the degree of damage to myelinated fibers was still far less severe than the damage observed in the vincristine-treated group. On the other hand, the protective effects of l-CDL and oxycodone on damage to unmyelinated fibers were consistent with the effects on myelinated fibers.

l-CDL attenuates vincristine-induced degeneration of the sciatic nerve and the prevalence of atypical mitochondria. (A) All six micrographs are shown at the same magnification (× 5000, bar = 2 μm). Normal myelinated (m) and unmyelinated fibers (n) were observed in the sham group. Severe degeneration of myelin and axons in large myelinated fibers was observed in the vincristine-treated group (day 14), whereas unmyelinated fibers appeared in looser bundles and were swollen. Shrinkage of the axons of myelinated fibers was observed on day 14 (p). l-CDL, particularly the high dose of l-CDL, and oxycodone reduced the degeneration of myelinated and unmyelinated fibers compared with the vincristine-treated group. n = 5 mice/group. (B–E) Representative ultrastructural images show the prevalence of atypical mitochondria in A-fibers (B) and C-fibers (C) in the six different groups (× 30,000, bar = 0.5 μm). The application of all three doses of l-CDL, especially the high dose of l-CDL, significantly decreased the prevalence of atypical mitochondria (indicated by arrows) compared with the vincristine-treated group. ###P < 0.001 compared with the sham control; **P < 0.01 and ***P < 0.001, compared with the vincristine-treated group, ANOVA followed by Tukey’s post hoc test, n = 5 mice/group. All data are presented as means±S.D.
Both l-CDL and oxycodone protected mitochondria. According to the histological examination, the prevalence of atypical mitochondria (swollen and vacuolated) in myelinated A-fibers and C-fibers in the vincristine-treated group was 42.04 ± 5.92% and 30.66 ± 2.75%, respectively, whereas 7.61 ± 2.47% of the mitochondria in A-fibers and 8.04 ± 4.55% of the mitochondria in C-fibers were atypical in saline-treated rats (Fig. 2D, E). The application of all three doses of l-CDL significantly decreased the prevalence of atypical mitochondria (indicated by arrows) compared with the vincristine-treated group at 14 days. In particular, the high dose of l-CDL (20 mg/kg) significantly decreased the prevalence of atypical mitochondria in A-fibers (10.48 ± 3.56%) and C-fibers (10.22 ± 2.35%) and had a better effect than oxycodone on both the prevalence of mitochondrial abnormalities in A-fibers (17.65 ± 5.14%) and C-fiber (14.20 ± 2.09%).
l-CDL Upregulates Nrf2/HO-1/CO in the Spinal Cord After the Vincristine Treatment
We harvested the spinal cord 14 days after the vincristine injection to determine whether l-CDL increased HO-1 expression in the spinal dorsal horn. Compared with the sham group, the vincristine injection induced a marked increase in HO-1 immunoreactivity, whereas l-CDL and oxycodone induced overexpression of HO-1 compared with the vincristine group according to samples taken from the same region of the superficial dorsal horn (laminae I–III) (Fig. 3A–C). As shown in Fig. 3D, the levels of the HO-1 protein were also significantly increased in the l-CDL and oxycodone groups at 14 days, and the high dose of l-CDL was the most effective treatment. Compared with the vincristine-treated group, the l-CDL- and oxycodone-treated groups exhibited lower cytoplasmic Nrf2 levels but higher nuclear Nrf2 levels (Fig. 3E, F); thus, activated Nrf2 was transported from the cytoplasm to the nucleus. Consistent with the trends of increased HO-1 and Nrf2 levels, l-CDL and oxycodone increased CO release in the spinal cord (Fig. 3G). At a concentration of 20 mg/kg, l-CDL increased CO levels to a greater extent than oxycodone (1.5 mg/kg).

l-CDL induces Nrf2/HO-1/CO upregulation in the spinal cord after the vincristine treatment. (A–C) l-CDL and oxycodone induced HO-1-overexpression in the same region of the superficial dorsal horn (laminae I–III) compared with the vincristine group, as assessed using immunostaining. Scale bar = 100 μm. (D) Western blots also showed that l-CDL increased HO-1 expression. (E–F) The l-CDL and oxycodone groups exhibited lower cytoplasmic Nrf2 levels and higher nuclear Nrf2 levels. (G) The l-CDL and oxycodone groups also exhibited increased CO release in the spinal cord, and the high dose of l-CDL (20 mg/kg) was the most effective at increasing CO levels. ###P < 0.001 compared with the sham control; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the vincristine-treated group, ANOVA followed by Tukey’s post hoc test, n = 3 mice/group. All data are presented as means±S.D.
l-CDL Attenuates Cx43 Expression by Regulating the HO-1/CO Pathway in the Spinal Cord after the Vincristine Treatment
To investigate the role of Cx43 in the maintenance of vincristine-induced neuropathic pain and whether HO-1/CO regulates Cx43 expression, we injected animals with CoPP (8 μg), ZnPP (8 μg), l-CDL (20 mg/kg), or ZnPP (8 μg) + l-CDL (20 mg/kg) 6 days after the vincristine injection and assessed pain behavior to investigate the role of Cx43 in the maintenance of vincristine-induced neuropathic pain and determine whether HO-1/CO regulates Cx43 expression. Treatments with 8 μg of CoPP and 20 mg/kg l-CDL almost completely reversed the vincristine-induced mechanical allodynia and heat hyperalgesia. This reversal began at 2 h and was maintained at 7 h and pain hypersensitivity was attenuated for more than 24 h in the l-CDL group, whereas the reversal of CoPP group diminished at 24 h following the intrathecal injection (Fig. 4A, B). At a dose of 8 μg, ZnPP had no effect on pain hypersensitivity, but the ZnPP (8 μg) + l-CDL (20 mg/kg) group exhibited significantly attenuated pain hypersensitivity for more than 24 h. Next, the CoPP, l-CDL, and ZnPP + l-CDL groups displayed increased HO-1 and CO levels in the spinal cord compared with the vehicle group at 7 h, whereas ZnPP alone had no effect on the HO-1 and CO levels (Fig. 4C, D). In contrast, the Western blot results showed that CoPP, l-CDL, and ZnPP + l-CDL significantly decreased Cx43 levels compared with vehicle administration (Fig. 4E).

l-CDL decreases Cx43 expression by the regulating HO-1/CO pathway in the spinal cord after the vincristine treatment. (A, B) CoPP (8 μg), l-CDL (20 mg/kg), and ZnPP (8 μg) + l-CDL (20 mg/kg) treatments attenuated vincristine-induced mechanical allodynia and heat hyperalgesia. **P < 0.01 and ***P < 0.001 compared with the vehicle-treated group, two-way ANOVA followed by Bonferroni’s post hoc tests, n = 6 mice/group. (C–E) The CoPP group, the l-CDL group and the ZnPP + l-CDL group exhibited increased HO-1 and CO expression at 7 h after the intrathecal injection, but significantly attenuated Cx43 expression compared with the vehicle group. **P < 0.01 and ***P < 0.001 compared with the vehicle-treated group, ANOVA followed by Tukey’s post hoc test, n = 3 mice/group. (F–G) l-CDL inhibited the vincristine-induced increase in Cx43 immunoreactivity in the spinal cord 14 days after the vincristine injection. Confocal images of spinal sections showed the co-localization of Cx43 with the astrocytic marker GFAP but not with neuronal marker NeuN or microglial marker Iba-1. Scale bar = 50 μm. ###P < 0.001 compared with the sham control; **P < 0.01 and ***P < 0.001 compared with the vincristine-treated group, ANOVA followed by Tukey’s post hoc test, n = 3 mice/group. (H, I) l-CDL inhibited vincristine-induced increases in the levels of the Cx43 protein and mRNA at 14 days. ###P < 0.001, compared with the sham control; *P < 0.05 and ***P < 0.001, compared with the vincristine-treated group, ANOVA followed by Tukey’s post hoc test, n = 3 mice/group. All data are presented as the means±S.D.
Furthermore, immunostaining showed that all three doses of l-CDL inhibited the vincristine-induced increase in Cx43 immunoreactivity in the spinal cord (Fig. 4F, G). In particular, the high dose of l-CDL produced effects similar to the sham group and better effects than the oxycodone control. Double immunostaining for Cx43/GFAP, Cx43/NeuN, and Cx43/Iba-1 on day 14 indicated that Cx43 was localized in spinal cord astrocytes (Fig. 4F). Based on the Western blot results, l-CDL inhibited vincristine-induced Cx43 expression at 14 days (Fig. 4H). The expression of the Cx43 mRNA was not changed in saline-treated animals but was significantly increased at day 14 in vincristine-treated animals. l-CDL and oxycodone attenuated the increase in the expression of the CXCL1 mRNA induced by the vincristine injection (Fig. 3I).
l-CDL Increases Nrf2-Induced HO-1/CO Expression in Cultured Astrocytes Treated with TNF-α
We explored whether Nrf2 binds to the HO-1 promoter and induces HO-1 expression to further determine whether the regulation of Nrf2/HO-1/CO expression by l-CDL in primary astrocytes was consistent with the pattern observed in the spinal cord. The HO-1-wt promoter (− 1 to 2000 bp) and the HO-1-mut promoter (− 1 to 2000 bp) were ligated into a luciferase reporter vector and transfected into 293 T cells with or without Nrf2 DNA using Lipofectamine 2000. As shown in Fig. 5A, the Nrf2-HO-1-wt group exhibited significantly reduced HO-1 promoter activity, indicating that Nrf2 bound to the HO-1-wt promoter in the luciferase reporter vector. However, Nrf2 had no effect on the HO-1-mut promoter, in which the binding sites in the HO-1 promoter sequence were mutated, confirming that Nrf2 indeed binds to the HO-1 promoter. In addition, the HO-1-wt promoter or the HO-1-mut promoter alone did not influence basic activity (Fig. 5A). Next, 3 nM VCR induced the release of 10 ng/ml TNF-a (Supplementary Fig. 1A and B) and compared with the TNF-α-treated group (10 ng/ml, 60 min at 37 °C), and l-CDL and CoPP evoked a significant increase in nuclear Nrf2 levels in astrocyte cultures (Fig. 5C). However, due to the transfer of Nrf2 from the cytoplasm to the nucleus, the l-CDL and CoPP groups exhibited lower cytoplasmic Nrf2 levels (Fig. 5B). Treatment with l-CDL (30 μM) increased nuclear Nrf2 levels and decreased cytoplasmic Nrf2 levels in astrocyte cultures after an incubation with VCR (3 nM, 60 min at 37 °C), although it had no effect on the nuclear transfer of Nrf2 in cells treated with the TNF-a inhibitor (lenalidomide, 3 μM) (Supplementary Fig. 2A and B). Consistent with the activation of Nrf2 transcription, l-CDL and CoPP subsequently promoted the mRNA expression of the downstream target HO-1 (Fig. 5D).

l-CDL increases Nrf2-activated HO-1 expression in cultured astrocytes. (A) The Nrf2-HO-1-wt group exhibited significantly reduced HO-1 promoter activity, whereas the Nrf2-HO-1-mut promoter group, HO-1-wt promoter group, and HO-1-mut promoter group did not exhibit changes in basic activity. The promoter activity is represented by the level of luciferase activity indicated by relative light units (RLU). Wild-type Renilla luciferase plasmids were also transfected into cells and used as an internal control. ***P < 0.001 compared with the Nrf2-HO-1-wt promoter; N.S., not significant, ANOVA followed by Tukey’s post hoc test. n = 3 cultures/group. (B, C) l-CDL and CoPP evoked lower cytoplasmic Nrf2 levels and higher nuclear Nrf2 levels in astrocyte cultures at 1 h after TNF-α incubation. #P < 0.05 compared with the sham control; **P < 0.01 and ***P < 0.001 compared with the TNF-α group, ANOVA followed by Tukey’s post hoc test, n = 3 cultures/group. (D) Real-time quantitative PCR results showed that l-CDL and CoPP increased the expression of the HO-1 mRNA. **P < 0.01 and ***P < 0.001 compared with the TNF-α group, ANOVA followed by Tukey’s post hoc test, n = 3 cultures/group. All data are presented as means±S.D.
A TNF-α treatment (10 ng/ml, 60 min at 37 °C) followed by l-CDL and CoPP (24 h at 37 °C) evoked a significant increase in the HO-1 fluorescence intensity (Fig. 6A, B). Double immunostaining for HO-1/GFAP in cultured astrocytes revealed the localization of HO-1 in astrocytes (Fig. 6C). Both l-CDL and CoPP substantially increased CO levels in cultured astrocytes (Fig. 6D). Based on these data, l-CDL increased HO-1/CO expression in cultured astrocytes by activating Nrf2 in response to TNF-α stimulation.

l-CDL increased HO-1/CO levels in cultured astrocytes after the TNF-α treatment. (A, B) l-CDL and CoPP significantly increased the HO-1 fluorescence intensity at 24 h after the TNF-α incubation (10 ng/ml, 60 min at 37 °C). Scale bar = 100 μm. *P < 0.05 and ***P < 0.001 compared with the TNF-α group, ANOVA followed by Tukey’s post hoc test, n = 3 cultures/group. (C) Double staining for HO-1 and GFAP showed the expression of HO-1 in astrocytes. Scale bar = 100 μm. (D) ELISA results showed that both l-CDL and CoPP substantially increased CO levels in cultured astrocytes. #P < 0.05 compared with the sham control; **P < 0.01 and ***P < 0.001 compared with TNF-α group, ANOVA followed by Tukey’s post hoc test, n = 3 cultures/group. All data are presented as means±S.D.
l-CDL Reduces the TNF-α-Induced Increase in Cx43 Levels and Hemichannel Activity in Cultured Astrocytes
We performed immunohistochemical staining for Cx43 and GFAP to verify that treatment with a known inflammatory mediator, TNF-α, directly reproduced pathological increases in Cx43 expression. The TNF-α treatment (10 ng/ml, 60 min at 37 °C) evoked a significant increase in Cx43 and GFAP expression in astrocyte cultures (Fig. 7A, B). Notably, these increases were abolished by an l-CDL, CoPP, or Gap 27 treatment for 24 h. l-CDL dose-dependently suppressed TNF-α-induced Cx43 expression in astrocytes, whereas a high dose of l-CDL exerted a greater inhibitory effect on Cx43 expression than CoPP and Gap 27 (Fig. 7C). Under the control conditions and in the presence of external calcium, only a few astrocytes exhibited ethidium bromide absorption in the sham group (3.51 ± 0.47% intensity/arbitrary units). However, at 1 h after, the TNF-α treatment (10 ng/ml), the fluorescence intensity of ethidium bromide uptake was significantly increased (18.63 ± 2.06% intensity/arbitrary units) (Fig. 7D, E). Notably, the l-CDL and Gap 27 groups exhibited a significant decrease in the TNF-α-induced uptake of ethidium bromide, particularly the high-dose l-CDL group and the Gap 27 group (5.37 ± 0.94% and 4.46 ± 0.63% intensity/arbitrary units, respectively). Interestingly, CoPP also suppressed basal ethidium bromide uptake by 57%. Taken together, l-CDL reduced the TNF-α-induced upregulation of astrocytic Cx43 expression, which corresponded to reduced Cx43-mediated hemichannel activity.

l-CDL reduced TNF-α-induced increases in Cx43 levels and hemichannel activity in cultured astrocytes. (A, B) Cx43 was expressed at very low levels in sham control astrocytes and increased 1 h after TNF-α incubation. Double staining for Cx43 and GFAP revealed the expression of Cx43 in astrocytes. Scale bar = 100 μm. ##P < 0.01 and ###P < 0.001 compared with the sham control, Student’s t test, n = 3 cultures/group. (C) Cx43 expression in cultured astrocytes stimulated with TNF-α (10 ng/ml, 60 min) was suppressed by treatment with l-CDL, CoPP, and Gap 27. ###P < 0.001, compared with the sham control and ***P < 0.001 compared with the TNF-α group, ANOVA followed by Tukey’s post hoc test, n = 3 cultures/group. (D, E) The TNF-α treatment (10 ng/ml, 60 min) increased hemichannel function, as revealed by ethidium bromide uptake in astrocytes. This increase was suppressed by l-CDL, Gap 27, and CoPP. Scale bar = 50 μm. ###P < 0.001 compared with the sham control and ***P < 0.001 compared with the TNF-α group, ANOVA followed by Tukey’s post hoc test, n = 3 cultures/group. All data are presented as means±S.D.
l-CDL Has No Effect on Cx43 Following HO-1 Gene Silencing
We pre-treated cultured astrocytes with a specific small interfering RNA that targets the HO-1 messenger RNA and display high transfection efficiency to confirm the possible mechanism by which l-CDL mediated HO-1/CO-dependent Cx43 expression. Significantly lower expression of the HO-1 mRNA was observed in the 3 l-CDL dose groups following treatment with the HO-1 small interfering RNA (100 nM, 36 h) before the TNF-α incubation (10 ng/ml, 1 h) and drug treatment than in the vehicle-treated group, whereas HO-1 siRNA-scramble had no effect on HO-1 expression in cultured astrocytes (Supplementary Fig. 3A and B). In contrast, the HO-1 mRNA was expressed at significantly higher levels in the CoPP-treatment control group without a pre-incubation with the HO-1 small interfering RNA, than in the vehicle-treated group (Fig. 8A). After transfection, all l-CDL-treatment groups incubated with the HO-1 siRNA expressed the HO-1 protein at very low levels, indicating that HO-1 gene silencing was stable (Fig. 8B). Notably, this small interfering RNA pretreatment also inhibited the TNF-α-induced increase in CO levels, whereas l-CDL decreased CO levels in response to HO-1 silencing (Fig. 8C). Furthermore, following the TNF-α incubation and HO-1 gene silencing, l-CDL and CoPP did not affect the levels of the Cx43 mRNA or protein in cultured astrocytes (Fig. 8D, E), whereas Gap 27 decreased the expression of the Cx43 mRNA compared with the vehicle-treated group without the HO-1 small interfering RNA pre-incubation. Thus, l-CDL did not alter the expression of Cx43 following HO-1 gene silencing and a TNF-α incubation. Furthermore, l-CDL also had no effect on Cx43 expression following HO-1 gene silencing and the VCR treatment (Supplementary Fig. 4A and B).

l-CDL has no effect on Cx43 expression following HO-1 gene silencing. (A, B) Low levels of the HO-1 mRNA and protein were detected expression after a TNF-α incubation in the presence of l-CDL with an HO-1 small interfering RNA pretreatment, indicating that HO-1 gene silencing was stable. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the vehicle-treated group, ANOVA followed by Tukey’s post hoc test, n = 3 cultures/group. (C) l-CDL induced lower CO production in response to HO-1 silencing. ***P < 0.001 compared with the vehicle-treated group, ANOVA followed by Tukey’s post hoc test, n = 3 cultures/group. (D, E) l-CDL had no effect on Cx43 expression following HO-1 silencing. ***P < 0.001 compared with the vehicle-treated group; N.S., not significant compared with the vehicle-treated group, ANOVA followed by Tukey’s post hoc test, n = 3 cultures/group. All data are presented as means±S.D.
Discussion
In this study, we reported a novel mechanism by which Nrf2-activated HO-1/CO transcription mediates Cx43 expression to promote vincristine-induced neuropathic pain, and we investigated the anti-nociceptive and neuroprotective effects of l-CDL. Oxycodone hydrochloride, CoPP, ZnPP, and Gap 27 were used as positive controls in this study. Vincristine was injected for 5 consecutive days, and the resulting behavioral alterations and histopathological changes were ameliorated by l-CDL. Moreover, l-CDL pharmacologically increased the levels of Nrf2/HO-1/CO to exert an inhibitory effect on Cx43 expression and prevent vincristine-induced neuropathic pain.
l-CDL, a tetrahydroberberine (THPB), attenuates tibia bone cavity tumor cell implantation (TCI)-induced bone cancer pain by co-inhibiting N-methyl-D-aspartate (NMDA) and mGlu1/5 receptors [32] and alleviating tumor compression-induced pain (TCIP) by suppressing the CCL2/CCR2 pathway [24]. In our previous study, l-CDL alleviated vincristine-induced neuropathic pain by inhibiting an NF-kappa B-dependent CXCL1/CXCR2 signalling pathway [23]. l-CDL is the structural analogue of l-tetrahydropalmatine (l-THP), which induces a dose-dependent anti-hyperalgesic effect on neuropathic pain caused by the chemotherapeutic agent oxaliplatin [9]. In the present study, we selected oxycodone as a positive standard in vivo based on the factors listed below. According to the configurations of the compounds, l-CDL is a member of the class of protoberberine-benzyltetrahydroisoquinoline alkaloids, whereas oxycodone belongs to the class of morphine-benzyltetrahydroisoquinoline alkaloids. Chemotherapy-induced peripheral neuropathy (CIPN)-related pain predominantly has a neuropathic aetiology. Among the opioids used in the clinic, oxycodone has shown excellent anti-hyperalgesic and anti-allodynic effects [33] and is efficacious and well-tolerated when administered to relieve chemotherapy-induced neuropathic pain [34]. However, the mechanism by which oxycodone modulates the expression of HO-1 and Cx43 in the spinal cord has not yet been studied. Thus, our study provides new insights for subsequent drug research. Notably, peak behavioral alterations in vincristine-induced neuropathic pain occurred in the 2nd and 3rd weeks in previous well-established research findings [35, 36]. The attenuation of pain hypersensitivity by l-CDL may reflect the protective responses of the spinal cord after peripheral nerve injury.
The transmission electron microscopy evaluation performed in our study revealed myelinated fiber damage and unmyelinated fiber loosening and swelling following vincristine administration, consistent with previously reported results [37, 38]. l-CDL and oxycodone protected myelinated fibers and unmyelinated fibers after vincristine administration. Spontaneous discharges of A-fiber and C-fiber nociceptors engage central mechanisms that create and amplify neuropathic pain abnormalities [39, 40]. In the present study, the vincristine-induced increased in the prevalence of atypical mitochondria in both A-fibers and C-fibers was ameliorated by l-CDL. Furthermore, intraepidermal nerve fiber (IENF) degeneration and abnormal spontaneous discharges of primary afferent nerve fibers may be enhanced by mitochondrial dysfunction and the subsequent energy deficiency [8]. A loss of IENF is directly related to pain behaviors and thus is considered a key chemotherapy-induced neuropathic pain mechanism [41, 42]. Although l-CDL reduced the vincristine-induced increase in the prevalence of atypical mitochondria, this study has some limitations. We did not explore whether and how l-CDL modulates the loss of IENFs induced by vincristine by protecting the mitochondria; therefore, the preventative effect of l-CDL on chemotherapy-induced neuropathic pain requires further investigation.
A wide variety of in vivo and in vitro studies have shown that HO-1 is an inducible enzyme, and pharmacological upregulation of HO-1 leads to the profound inhibition of behavioral hypersensitivity development, further generating robust antioxidant and anti-inflammatory responses [12, 43]. According to Hervera et al. [44], repeated treatment with CoPP increases HO-1 expression in dorsal root ganglia (DRG) and the spinal cord and alleviates CCI-induced neuropathic inflammation, reduces pain hypersensitivity, and reduces the expression of nitric oxide synthase-1 (NOS1) and NOS2 induced by sciatic nerve injury [17]. A systemic injection of CoPP or lentiviruses encoding HO-1 persistently alleviate vincristine-induced pain hypersensitivity for more than 1 week [13]. We confirmed that l-CDL plays an important role in increasing the expression of HO-1; thus, the l-CDL treatment exerted antinociceptive effects on vincristine-induced neuropathic pain.
HO-1 expression is induced by different transcription factors, such as nuclear factor kB (NF-κB), hypoxia-inducible factor-1 (HIF-1), activating protein-1 (AP-1), and Nrf2 [45]. A striking finding of this study is that Nrf2 directly binds to the upstream sequence in the HO-1 promoter in vincristine-induced neuropathic pain, inducing the degradation of heme to produce carbon monoxide (CO). Moreover, l-CDL and CoPP activated the transfer of Nrf2 from the cytoplasm to the nucleus, consistent with the findings reported by Shan et al. [46] who showed that high concentrations of CoPP enhanced Nrf2 activity. Nrf2 activation is known to inhibit NF-κB activation, whereas NF-κB is also known to inhibit Nrf2 [47, 48], indicating that cross-talk between NF-κB activation and Nrf-2 inhibition leads to neuroinflammation. These findings also confirm that l-CDL inhibits NF-κB and simultaneously increases Nrf2 expression to inhibit the neuroinflammatory reaction caused by vincristine. Activation of the HO-1/carbon monoxide pathway attenuates acute and chronic inflammatory pain [49], painful diabetic neuropathy, and CCI-induced neuropathic pain [50, 51]. Based on our results, l-CDL dose-dependently increased the release of CO in the spinal cord and cultured astrocytes.
An increase in the levels of the HO-1 protein is usually accompanied by higher total levels of the gap junction protein Cx43 [52]. Additionally, abundant CO, which functions as a novel inhibitor of Cx43-hemichannels, crosses the astrocyte membrane and induce Cx43-hemichannel closure after a CORM-2 injection [18]. Furthermore, upregulation of the endogenous CO producer HO-1 and the administration of exogenous CORM-2 both inhibit SNL-induced Cx43 expression, gap junction function and hemichannel function [53]. Therefore, our study evaluated whether Cx43 is involved in HO-1/CO-mediated analgesia after the vincristine injection. Recently, the inhibition of Cx43 has been proposed as a potential therapeutic target for the prevention and/or treatment of bone cancer pain (BCP) [54], CCI [22], paclitaxel-induced and oxaliplatin-induced peripheral neuropathy [55, 56], among other types of pain. Based on previous findings, the absolute change in astrocyte Cx43 expression that appears to cause neuropathy is critical, and the direction of change appears to dependent on the aetiology. We are the first to show that Cx43 participated in vincristine-induced neuropathic pain (VINP). Nevertheless, according to current data, restoring normal astrocyte Cx43 expression is an important component of the treatment of neuropathic pain [57]. The present study is the first to support a role for Nrf-2/HO-1/CO in mediating vincristine-induced neuroinflammation by inhibiting Cx43 production in the spinal cord and cultured astrocytes. Although the exact mechanism of VCR-induced neuropathy is not clearly understood, a few studies have reported that the upregulation of proinflammatory interleukins and TNF-α in the injured region of the spinal cord plays a key role in neuropathic pain [58]. However, following a TNF-α incubation and HO-1 gene silencing, l-CDL did not affect the levels of the Cx43 protein or mRNA in cultured astrocytes, indicating that l-CDL was indeed acting on HO-1/CO to modulate Cx43 expression.
In addition to the formation of gap junctions, Cx43 also forms an unopposed hemichannel, providing a pathway for molecular exchange between the cytoplasmic and extracellular compartments [59]. TNF-α upregulates astrocytic Cx43 expression, which corresponds to increased Cx43-mediated hemichannel activity but not gap junction communication [22]. Our previous study also verified the same conclusion that TNF-α does not affect Cx43-mediated gap junction channels (data not shown). In the present study, l-CDL reduced the TNF-α-induced upregulation of Cx43 expression in astrocytes, which corresponded to a decrease Cx43-mediated hemichannel activity.
In conclusion, we provide evidence that a vincristine injection caused obvious pain hypersensitivity and a high dose of l-CDL (20 mg/kg) exerted a greater antinociceptive effect than oxycodone, similar to the effect on the positive control group treated with the HO-1 inducer CoPP. Meanwhile, l-CDL attenuated vincristine-induced degeneration of the sciatic nerve and the prevalence of atypical mitochondria. Our data provide the first evidence that Nrf2/HO-1/CO-mediated Cx43 expression is involved in VINP. Based on these findings, l-CDL represents a potential anti-inflammatory drug that targets the Nrf2/HO-1/CO axis to modulate the Cx43 signalling pathway, thereby relieving VINP.
Abbreviations
- l-CDL:
-
Levo-corydalmine
- l-THP:
-
Levo-tetrahydropalmatine
- Oxy:
-
Oxycodone
- VCR:
-
Vincristine
- HO-1:
-
Heme oxygenase 1
- Nrf2:
-
Nuclear factor E2-related factor 2
- CO:
-
Carbon monoxide
- Cx43:
-
Connexin 43
- CoPP:
-
Protoporphyrin IX cobalt chloride
- ZnPP:
-
Protoporphyrin IX zinc
- GFAP:
-
Glial fibrillary acidic protein
- PBS:
-
Phosphate-buffered saline
- DMSO:
-
Dimethyl sulfoxide
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- VINP:
-
Vincristine-induced neuropathic pain
- CINP:
-
Chemotherapy-induced neuropathic pain
- siRNA:
-
Small interfering RNA
- DMEM:
-
Dulbecco’s Modified Eagle’s medium
- RT-qPCR:
-
Real-time quantitative polymerase chain reaction
- ELISA:
-
Enzyme-linked immunosorbent assay
References
Bessaguet F, Danigo A, Bouchenaki H et al. Neuroprotective effect of angiotensin II type 2 receptor stimulation in vincristine-induced mechanical allodynia. Pain 2018;159(12):2538–46.
Wang Y, Cao SE, Tian J et al. Auraptenol attenuates vincristine-induced mechanical hyperalgesia through serotonin 5-HT1A receptors. Sci Rep 2013;3:3377.
Mora E, Smith EM, Donohoe C, Hertz DL. Vincristine-induced peripheral neuropathy in pediatric cancer patients. Am J Cancer Res 2016;6(11):2416–30.
Jackson DV, Jr., Castle MC, Poplack DG, Bender RA. Pharmacokinetics of vincristine in the cerebrospinal fluid of subhuman primates. Cancer Res 1980;40(3):722–4.
Geisler S, Doan RA, Strickland A et al. Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 2016;139(Pt 12):3092–108.
Miltenburg NC, Boogerd W, . Chemotherapy-induced neuropathy: A comprehensive survey. Cancer Treat Rev 2014;40(7):872–82.
Djaldetti R, Hart J, Alexandrova S et al. Vincristine-induced alterations in Schwann cells of mouse peripheral nerve. Am J Hematol 1996;52(4):254–7.
Xiao WH, Bennett GJ. Effects of mitochondrial poisons on the neuropathic pain produced by the chemotherapeutic agents, paclitaxel and oxaliplatin. Pain 2012;153(3):704–9.
Guo Z, Man Y, Wang X et al. Levo-tetrahydropalmatine attenuates oxaliplatin-induced mechanical hyperalgesia in mice. Sci Rep 2014;4:3905.
Watson CP, Babul N. Efficacy of oxycodone in neuropathic pain: a randomized trial in postherpetic neuralgia. Neurology 1998;50(6):1837–41.
Watson CP, Moulin D, Watt-Watson J, Gordon A, Eisenhoffer J. Controlled-release oxycodone relieves neuropathic pain: a randomized controlled trial in painful diabetic neuropathy. Pain 2003;105(1–2):71–8.
Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev 2008;60(1):79–127.
Shen Y, Zhang ZJ, Zhu MD et al. Exogenous induction of HO-1 alleviates vincristine-induced neuropathic pain by reducing spinal glial activation in mice. Neurobiol Dis 2015;79:100–10.
Syapin PJ. Regulation of haeme oxygenase-1 for treatment of neuroinflammation and brain disorders. Br J Pharmacol 2008;155(5):623–40.
Ferrandiz ML, Nacher-Juan J, Alcaraz MJ. Nrf2 as a therapeutic target for rheumatic diseases. Biochem Pharmacol 2018;152:338–46.
Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res 2008;103(11):1232–40.
Hervera A, Leanez S, Motterlini R, Pol O. Treatment with carbon monoxide-releasing molecules and an HO-1 inducer enhances the effects and expression of micro-opioid receptors during neuropathic pain. Anesthesiology 2013;118(5):1180–97.
León-Paravic CG, Figueroa VA, Guzmán DJ et al. Carbon Monoxide (CO) Is a Novel Inhibitor of Connexin Hemichannels. J Biol Chem 2014;289(52):36150–57.
Bennett MV, Garre JM, Orellana JA et al. Connexin and pannexin hemichannels in inflammatory responses of glia and neurons. Brain Res 2012;1487:3–15.
Morioka N, Fujii S, Kondo S et al. Downregulation of spinal astrocytic connexin43 leads to upregulation of interleukin-6 and cyclooxygenase-2 and mechanical hypersensitivity in mice. Glia 2018;66(2):428–44.
Chen MJ, Kress B, Han X et al. Astrocytic CX43 hemichannels and gap junctions play a crucial role in development of chronic neuropathic pain following spinal cord injury. Glia 2012;60(11):1660–70.
Chen G, Park CK, Xie RG et al. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain 2014;137(Pt 8):2193–209.
Zhou L, Hu Y, Li C et al. Levo-corydalmine alleviates vincristine-induced neuropathic pain in mice by inhibiting an NF-kappa B-dependent CXCL1/CXCR2 signaling pathway. Neuropharmacology 2018;135:34–47.
Hu Y, Kodithuwakku ND, Zhou L et al. Levo-Corydalmine Alleviates Neuropathic Cancer Pain Induced by Tumor Compression via the CCL2/CCR2 Pathway. Molecules 2017;22(6)
Wu XF, Liu WT, Liu YP et al. Reopening of ATP-sensitive potassium channels reduces neuropathic pain and regulates astroglial gap junctions in the rat spinal cord. Pain 2011;152(11):2605–15.
Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 1980;20:441–62.
McRoberts JA, Ennes HS, Marvizon JC et al. Selective knockdown of NMDA receptors in primary afferent neurons decreases pain during phase 2 of the formalin test. Neuroscience 2011;172:474–82.
Wei XH, Yang T, Wu Q et al. Peri-sciatic administration of recombinant rat IL-1beta induces mechanical allodynia by activation of src-family kinases in spinal microglia in rats. Exp Neurol 2012;234(2):389–97.
Jin HW, Flatters SJ, Xiao WH, Mulhern HL, Bennett GJ. Prevention of paclitaxel-evoked painful peripheral neuropathy by acetyl-L-carnitine: effects on axonal mitochondria, sensory nerve fiber terminal arbors, and cutaneous Langerhans cells. Exp Neurol 2008;210(1):229–37.
Gao YJ, Zhang L, Samad OA et al. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci 2009;29(13):4096–108.
Xu ZZ, Liu XJ, Berta T et al. Neuroprotectin/protectin D1 protects against neuropathic pain in mice after nerve trauma. Ann Neurol 2013;74(3):490–5.
Dai WL, Yan B, Jiang N et al. Simultaneous inhibition of NMDA and mGlu1/5 receptors by levo-corydalmine in rat spinal cord attenuates bone cancer pain. Int J Cancer 2017;141(4):805–15.
Thibault K, Calvino B, Rivals I et al. Molecular mechanisms underlying the enhanced analgesic effect of oxycodone compared to morphine in chemotherapy-induced neuropathic pain. PLoS One 2014;9(3):e91297.
Cartoni C, Brunetti GA, Federico V et al. Controlled-release oxycodone for the treatment of bortezomib-induced neuropathic pain in patients with multiple myeloma. Support Care Cancer 2012;20(10):2621–6.
Jaggi AS, Singh N. Analgesic potential of intrathecal farnesyl thiosalicylic acid and GW 5074 in vincristine-induced neuropathic pain in rats. Food Chem Toxicol 2012;50(5):1295–301.
Boehmerle W, Huehnchen P, Peruzzaro S, Balkaya M, Endres M. Electrophysiological, behavioral and histological characterization of paclitaxel, cisplatin, vincristine and bortezomib-induced neuropathy in C57Bl/6 mice. Sci Rep 2014;4:6370.
Topp KS, Tanner KD, Levine JD. Damage to the cytoskeleton of large diameter sensory neurons and myelinated axons in vincristine-induced painful peripheral neuropathy in the rat. J Comp Neurol 2000;424(4):563–76.
Tanner KD, Levine JD, Topp KS. Microtubule disorientation and axonal swelling in unmyelinated sensory axons during vincristine-induced painful neuropathy in rat. J Comp Neurol 1998;395(4):481–92.
Xiao WH, Bennett GJ. Chemotherapy-evoked neuropathic pain: Abnormal spontaneous discharge in A-fiber and C-fiber primary afferent neurons and its suppression by acetyl-L-carnitine. Pain 2008;135(3):262–70.
Cata JP, Weng HR, Chen JH, Dougherty PM. Altered discharges of spinal wide dynamic range neurons and down-regulation of glutamate transporter expression in rats with paclitaxel-induced hyperalgesia. Neuroscience 2006;138(1):329–38.
Zheng H, Xiao WH, Bennett GJ. Mitotoxicity and bortezomib-induced chronic painful peripheral neuropathy. Exp Neurol 2012;238(2):225–34.
Flatters SJ, Bennett GJ. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain 2006;122(3):245–57.
Bijjem KR, Padi SS, lal Sharma P. Pharmacological activation of heme oxygenase (HO)-1/carbon monoxide pathway prevents the development of peripheral neuropathic pain in Wistar rats. Naunyn Schmiedeberg's Arch Pharmacol 2013;386(1):79–90.
Hervera A, Leanez S, Negrete R, Motterlini R, Pol O. Carbon monoxide reduces neuropathic pain and spinal microglial activation by inhibiting nitric oxide synthesis in mice. PLoS One 2012;7(8):e43693.
Tian Y, Li Z, Shen B, Zhang Q, Feng H. Protective effects of morin on lipopolysaccharide/d-galactosamine-induced acute liver injury by inhibiting TLR4/NF-kappaB and activating Nrf2/HO-1 signaling pathways. Int Immunopharmacol 2017;45:148–55.
Shan Y, Lambrecht RW, Donohue SE, Bonkovsky HL. Role of Bach1 and Nrf2 in up-regulation of the heme oxygenase-1 gene by cobalt protoporphyrin. FASEB J 2006;20(14):2651–3.
Negi G, Kumar A, Sharma SS. Nrf2 and NF-kappaB modulation by sulforaphane counteracts multiple manifestations of diabetic neuropathy in rats and high glucose-induced changes. Curr Neurovasc Res 2011;8(4):294–304.
Ganesh Yerra V, Negi G, Sharma SS, Kumar A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-kappaB pathways in diabetic neuropathy. Redox Biol 2013;1:394–7.
Negrete R, Hervera A, Leanez S, Pol O. Treatment with a carbon monoxide-releasing molecule inhibits chronic inflammatory pain in mice: nitric oxide contribution. Psychopharmacology 2014;231(5):853–61.
Castany S, Carcole M, Leanez S, Pol O. The role of carbon monoxide on the anti-nociceptive effects and expression of cannabinoid 2 receptors during painful diabetic neuropathy in mice. Psychopharmacology 2016;233(11):2209–19.
Chen Y, Chen H, Xie K et al. H2 Treatment Attenuated Pain Behavior and Cytokine Release Through the HO-1/CO Pathway in a Rat Model of Neuropathic Pain. Inflammation 2015;38(5):1835–46.
Lakkisto P, Csonka C, Fodor G et al. The heme oxygenase inducer hemin protects against cardiac dysfunction and ventricular fibrillation in ischaemic/reperfused rat hearts: role of connexin 43. Scand J Clin Lab Invest 2009;69(2):209–18.
Wang H, Sun X. Carbon Monoxide-Releasing Molecule-2 Inhibits Connexin 43-Hemichannel Activity in Spinal Cord Astrocytes to Attenuate Neuropathic Pain. J Mol Neurosci 2017;63(1):58–69.
Yang H, Yan H, Li X et al. Inhibition of Connexin 43 and Phosphorylated NR2B in Spinal Astrocytes Attenuates Bone Cancer Pain in Mice. Front Cell Neurosci 2018;12:129.
Tonkin RS, Bowles C, Perera CJ et al. Attenuation of mechanical pain hypersensitivity by treatment with Peptide5, a connexin-43 mimetic peptide, involves inhibition of NLRP3 inflammasome in nerve-injured mice. Exp Neurol 2018;300:1–12.
Yoon SY, Robinson CR, Zhang H, Dougherty PM. Spinal astrocyte gap junctions contribute to oxaliplatin-induced mechanical hypersensitivity. J Pain 2013;14(2):205–14.
Morioka N, Zhang FF, Nakamura Y et al. Tumor necrosis factor-mediated downregulation of spinal astrocytic connexin43 leads to increased glutamatergic neurotransmission and neuropathic pain in mice. Brain Behav Immun 2015;49:293–310.
Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci 2007;10(11):1361–8.
Retamal MA, Froger N, Palacios-Prado N et al. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J Neurosci 2007;27(50):13781–92.
Acknowledgments
This study was supported by the National Major Scientific and Technological Special Project for “Significant New Drugs Development” during the Thirteenth Five-year Plan Period (No. 2018ZX09301043-001 and No. 2016ZX09101031, respectively), National Natural Science Foundation of China (No. 81803642), the “Double First-Class” Construction Technology Innovation Team Project of China Pharmaceutical University (No. CPU2018GY23), and Postgraduate Research & Practice Innovation Program of Jiangsu Province (No. KYCX18_0804). We thank the State Key Laboratory of Natural Medicines (China Pharmaceutical University) for providing the large-scale instrument sharing platform.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhou, L., Ao, L., Yan, Y. et al. Levo-corydalmine Attenuates Vincristine-Induced Neuropathic Pain in Mice by Upregulating the Nrf2/HO-1/CO Pathway to Inhibit Connexin 43 Expression. Neurotherapeutics 17, 340–355 (2020). https://doi.org/10.1007/s13311-019-00784-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-019-00784-7