
Abstract
Cerebral edema is commonly associated with cerebral pathology, and the clinical manifestation is largely related to the underlying lesioned tissue. Brain edema usually amplifies the dysfunction of the lesioned tissue and the burden of cerebral edema correlates with increased morbidity and mortality across diseases. Our modern-day approach to the medical management of cerebral edema has largely revolved around, an increasingly artificial distinction between cytotoxic and vasogenic cerebral edema. These nontargeted interventions such as hyperosmolar agents and sedation have been the mainstay in clinical practice and offer noneloquent solutions to a dire problem. Our current understanding of the underlying molecular mechanisms driving cerebral edema is becoming much more advanced, with differences being identified across diseases and populations. As our understanding of the underlying molecular mechanisms in neuronal injury continues to expand, so too is the list of targeted therapies in the pipeline. Here we present a brief review of the molecular mechanisms driving cerebral edema and a current overview of our understanding of the molecular targets being investigated.
Similar content being viewed by others

Introduction
Cerebral edema is the pathologic accumulation of intracellular and interstitial brain tissue water [1, 2] that is commonly associated with neurologic pathology. The clinical manifestation is largely related to the primary brain pathology and the associated brain edema usually amplifies the dysfunction of the lesioned tissue. The clinical presentation of cerebral edema varies widely depending on the primary brain pathology from asymptomatic, mild worsening of deficits, coma, and brain death. As a potential complication of both primary neurologic and nonneurologic disorders [1], increased burden of cerebral edema correlates with increased morbidity and mortality across diseases [3,4,5,6]. A growing understanding of the cellular mechanisms underlying cerebral edema have re-invigorated the scientific community to identify better targeted therapeutics for the management of cerebral edema [2, 7,8,9]. The focus of this review will be to discuss the distinct cellular pathways leading to cerebral edema, discuss the current and investigational pharmacologic treatment strategies for management of cerebral edema, and briefly discuss the future therapeutic strategies currently being investigated.
Pathophysiology of Cerebral Edema
The cellular mechanisms resulting in cerebral edema are divided into three distinct categories: cytotoxic edema, vasogenic edema, and hydrostatic edema [1, 10].
Cytotoxic Edema and Ionic Pump Failure
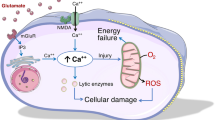
Cytotoxic edema results from an influx of ions, primarily Na+ and Cl−, from the interstitial space into the intracellular compartment, carrying with it water that results in oncotic cellular swelling [7]. The formation of cytotoxic edema has been documented in all central nervous system cell types [7, 11]; however, astrocytes appear to be the most sensitive [12]. Cytotoxic edema does not on its own generate tissue swelling, as it simply leads to the intracellular movement of interstitial water [11]. However, this disruption of ionic transport across cellular membranes leads to alteration in the primary and secondary drivers of active and secondarily active transport cellular transport [7]; rearrangement of one ion (i.e., Na+) requires the movement of additional ions (i.e., Cl−) to maintain electrical and osmotic neutrality. This attempt to maintain homeostasis rapidly becomes maladaptive and leads to cellular swelling and depletion of intracellular adenosine triphosphate (ATP) and it is this maladaptive maintenance of ionic transport that characterizes cytotoxic edema [7, 11].
There are however, a few particularly important cellular targets integral to cytotoxic edema. The constitutively expressed NKCC1 transporter is located primarily on astrocytes and found in all regions of the adult brain [13] and activated in the setting of ischemia, trauma, and acute liver failure [14]. Although molecular regulation of this transporter is complex, there is growing in vivo and in vitro data suggesting that modulation of this transporter may regulate cytotoxic edema [11, 13, 14]. The pathologic role of the NKCC1 transporter in the injured brain is not the only example of maladaptive transmembrane channels. Aquaporins particularly aquaporin-4 (AQP-4), the major AQP expressed on astrocytes [15], are gaining recognition for their role in cytotoxic edema [16,17,18]. When ionic transport is abnormal in pathologic states, AQPs facilitate transport of water across the blood–brain barrier (BBB) and therefore worsen cerebral edema in the setting of an intact BBB [17, 18]. Although the precise contribution of AQP physiology to cerebral edema remains unclear, AQP-4 knockout animal models have demonstrated reduced astrocyte swelling under cytotoxic conditions [18]. The last main constitutively expressed membrane transporter that contributed to cytotoxic edema is the sodium–hydrogen antiporter (NHE) family [19]. This luminal family of transporters, the NHE proteins facilitate Na+ influx across the BBB membranes in hypoxic and hypoglycemic states following ischemia, leading to the development of cytotoxic cerebral edema [19].
Cytotoxic edema is not simply the result of maladaptive normal cellular mechanism. Aberrant cellular transporters additionally form under specific pathologic states. One such transporter, transient receptor potential melastatin 4 (Trpm4), is a naturally existing cation that is activated in the presence of depleted cytosolic ATP [20]. Alone, Trmp4 is activated by intracellular Ca2+ [21]. However, in the presence of cerebral pathology such as ischemia, astrocytes exhibit expression of an additional transporter, sulfonylurea receptor 1 (Sur1) [21]. These transports Trpm4 and Sur1 physically associate to form Sur1Trpm4, which has furthermore been implicated in astrocyte and brain edema [22, 23].
Cytotoxic edema additionally results from exposure to endogenous toxins. One such endogenous toxin is glutamate. Following neuronal injury such as ischemia or trauma [24, 25], glutamate accumulates and can lead to neuronal lysis and astrocyte swelling [26]. One potential mechanism for astrocyte swelling could be via the metabotropic glutamate receptor 5 (mGluR5) complex with Na+/K+-ATPase and AQP-4 [27]. When mGluR5 is minimally expressed in the normal healthy brain, if glutamate uptake is inhibited in the pathologic brain, neurotoxicity and cytotoxic edema may be worsened [28].
Vasogenic Edema, BBB Disruption, and the Ensuing Inflammatory Response
Vasogenic edema results from edema formation in the absence of an intact BBB [29]. In the absence of an intact BBB, protein and water communicate between the vascular compartment and the interstitial compartment [30]. Contrary to cytotoxic edema, in which ionic forces are the primary driver [7], hydrostatic forces drive the formation of vasogenic edema [31]. During cerebral injury, inflammation and ischemia contribute to endothelial injury and increase permeability [32]. One such mechanism by which BBB permeability is increased in cerebral injury is via increased expression of vascular endothelial growth factor (VEGF) [33, 34].VEGF decouples tight junctions, leading to permeability of cerebral vasculature and increased edema [33]. This is not simply a maladaptive mechanism; however, it is believed to potentially promote angiogenesis to at risk brain in at risk territories [9]. VEGF is not the only molecular signaling that degrades the BBB and contributes to vasogenic edema; matrix metalloproteinase (MMP) activity is increased in territories of cerebral injury [35]. MMP activity however actively degrades the integrity of the microvascular matrix leading to increase edema formation [36].
This destruction of the BBB free communication of the intravascular space with the cerebral interstitial space leads to extravasation of hemorrhage [7]. Local hemorrhage and the degradation of these blood products are quite toxic to the surrounding cells [9, 37, 38]. Once such mechanism is via the secretion of tumor necrosis factor (TNF) and interleukin-1β, Toll receptors, and other inflammatory cytokines that inhibit the BBB’s integrity [7, 35, 39]. The ensuing inflammatory response can continue for multiple days and contributes to the formation of reactive oxygen species, MMP activation, and further BBB breakdown [38, 39].
Hydrostatic Edema
Hydrostatic edema is the result of unfavorable hydrostatic pressure vectors between intracranial vasculature, ventricular system, and brain parenchyma [40], leading to transependymal movement of cerebral spinal fluid (CSF) into the brain parenchyma [10]. The mainstay of treatment for this pathology is primarily surgical (CSF diversion, surgical decompression) [10] and is largely outside the scope of this discussion.
Targeted and Nontargeted Therapeutics for Cerebral Edema
Historically, identifying the underlying mechanism of cerebral edema directly influenced the primary treatment of choice. Traditionally, vasogenic edema was treated with agents such as hyperosmolar therapies and corticosteroids, while the efficacy of treatment options for cytotoxic edema remain limited [10]. That being said, the use of nontargeted therapies such as osmolar agents, sedation, neuromuscular blockade, and temperature management have been used in the acute management of cytotoxic edema [1]. Based on a growing understanding of the cellular mechanisms driving cytotoxic and vasogenic edema, a growing list of targeted therapies are in the pipeline for the medical management of cytotoxic and vasogenic edema [8].
Nontargeted Therapies
Nontargeted therapies are the current clinical mainstay for the medical management of cerebral edema. At this point in time, there are five main classes of nontargeted therapies of the management of cerebral edema. These agents have undergone various stages of preclinical and clinical research (see Table 1).
Osmolar therapies
The use of hyperosmolar therapies (e.g., urea, mannitol, hypertonic saline) leverages the selective permeability of an intact blood–brain barrier. The goal of hyperosmolar therapy is to create an osmotic gradient within the cerebral vasculature, where the dominant diffusion vectors are from the parenchyma to the vascular space, thereby decreasing the intracranial free water volume and decreasing intracerebral pressure [42]. This osmotic gradient requires an intact blood–brain barrier, where solute permeability across the BBB depends largely on the agent’s reflection coefficient [43]. Of note, there are cellular transport channels and carrier proteins that facilitate movement of these molecules (saline and urea) across the BBB into the parenchyma; however, the greatest component of transport remains intravascularly due to concentration gradients [42]. Therefore, the reflection coefficient of each agent represents the degree to which each molecule is able to diffuse passively across an intact BBB membrane and defines the selectivity of the BBB for this solute [43]. Reflection coefficients, inherent to the molecular properties of each solute, are measured on a 0 to 1 scale, with 0 representing complete permeability and 1 representing complete exclusion to passive movement across a membrane [43]. Those agents that have been identified to be most effective hyperosmolar agents are those whose reflection coefficients approach 1 [44].
Urea
The use of hyperosmolar therapies, such as urea, is first described as early as the mid-1950s [45]. The use of hyperosmolar solutions of urea rapidly fell out of favor given the many negative side effects attributed to their infusion. Gastrointestinal disturbances, electrocardiographic complications, coagulopathy, and, perhaps most importantly, rebound cerebral edema, due to the relatively low reflection coefficient of urea (0.59) [46], were only some of the reason’s urea fell out of favor [47]. This however allowed for the use of mannitol solutions to take hold in the medical management of cerebral edema.
No randomized clinical trials are available for the use of urea in the management of cerebral edema. Clinical experience has been reported in by Javid et al. in the 1950s [45, 48]. The majority of the physiologic evidence is obtained from animal models [47, 49].
Mannitol
First described in the early 1960s [50], mannitol remains part of the routine medical management of cerebral edema. Mannitol is most widely available as a 20% solution and administered on a weight-based bolus dosing schedule: 0.5 to 2 mg/kg [10]. Continuous infusions of mannitol are no longer used clinically, as rebound cerebral edema due to mannitol deposition in the area of compromised cerebral blood–brain barriers are now clearly described [51]. The proposed mechanism of action for mannitol is through direct osmotic effects on astrocytes and neurons with intact blood–brain barriers, as well as opening tight junctions within the blood–brain barrier to enhance osmotic pull of intracellular cerebral fluid into the intravascular space [52]. Mannitol has a reflection coefficient of 0.9 [47] which facilitates its ability to remain intravascular and provide this osmotic pull. Although the osmotic diuresis seen with mannitol does facilitate reduction in cerebral edema, redistribution of the cerebral plasma volume ultimately leads to increased cerebral blood flow and a concomitant vasoconstrictive response of the intracerebral vasculature that decreases intracerebral pressure [53]. Although effective in ICP control, mannitol has potentially adverse unintended consequences [54, 55]. Mannitol initially and fairly rapidly induces increased intravascular volume, which has the potential to precipitate acute hypervolemia intravascularly and lead to acute pulmonary edema or heart failure in patients predisposed to these conditions [55]. After the initial increase in intravascular volume, mannitol exerts its effect as a potent osmotic diuretic; the diuresis which follows mannitol administration can lead to intravascular volume loss, hypotension that may lead to acute kidney injury and worst outcomes particularly in patients with TBI [54, 55]. Clinicians need to be aware of these effects and address them proactively to ensure optimal outcome in patients.
Hypertonic Saline
It was not until the 1990s when the use of hypertonic saline fluids gained widespread acceptance as a first-line agent for the management of cerebral edema [56]. Now a mainstay in clinical practice for the management of cerebral edema hypertonic saline fluids are available in varying concentrations based on institutional practices, from 2 to 23.4% [10]. Similar to the mechanism of action of mannitol, hypertonic saline exerts osmotic properties on cerebral tissues with intact blood–brain barriers, drawing intracellular fluid intravascularly to reduce cerebral volume [46]. Due to the higher reflection coefficient of sodium chloride (1.0) [46], it is believed that hypertonic saline may be even more effective than mannitol and have a lower rate of rebound cerebral edema. In contrast to mannitol, hypertonic saline does not cause diuresis and hypovolemia, but instead increases intravascular volume. Complications of hypertonic saline administration include metabolic derangements, volume overload, phlebitis, hypotension (particularly with rapid infusions of high concentrations of hypertonic solutions), and coagulopathy to name a few [57]. Hypertonic saline is administered in bolus of 250 to 500 ml volumes [42], typically in 3% concentrations, or as a salvage technique with 30 ml bolus of 23.4% saline concentrations [58]. Continuous infusions of hypertonic saline are also used to maintain osmotic gradients [59]; however, when doing so, targeted sodium levels should be identified and serum sodium levels should remain below 160 mmol/l [60]. Hypertonic saline has additional, potentially beneficial effects beyond its role in mitigating cerebral edema. Hypertonic saline, when administered, can improve mean arterial pressure, cardiac output, and stroke volume, potentially lifesaving in traumatic injuries or septic shock [55, 61].
Up until this point, there have been small randomized clinical trials demonstrating the beneficial effects of hypertonic saline in the management of cerebral edema; however, the demonstration of improvement in patient outcome had remained elusive. However, recently announced is the COBI trial, continuous hyperosmolar therapy for traumatic brain injured patients’ trial, which is a phase 3 clinical trial that has been designed to test whether continuous infusions of hypertonic 20% saline may improve clinical outcomes in moderate to severe traumatic brain injury, with planned completion in February of 2020 (Clinicaltrial.gov; NCT03143751).
Diuretics
Loop diuretics, such as furosemide, have been described and used independently for the management of cerebral edema; however, the results of clinical studies have been inconsistent [62]. Specifically, the administration of diuresis in acute intracranial hypertension and cerebral edema can lead to acute hypovolemia, drop in mean arterial pressure, and potentially worst cerebral perfusion and poor clinical outcomes. Such was seen in a recent trial of administration of furosemide for brain relaxation (Clinicaltrials.gov; NCT01054404). Because of this, the routine use of loop diuretics as a sole management of cerebral edema is not routine clinical practice. There is however data to suggest that furosemide, dosed at 0.7 mg/kg, can prolong the reversal of the blood–brain barrier osmotic gradient established with hypertonic saline and mannitol by primarily excreting free water [62, 63].
Anesthesia and Sedation
Anesthetic agents are part of the medical management of elevated intracerebral pressure. Although not directly affecting cerebral edema, per se, pharmacologically induced coma lowers intracerebral pressure through lowering the cerebral metabolic demand of neurons and thus lowering the intracranial arterial volume [10, 46]. Although there is no clear direct effect on cerebral edema, barbiturates, such as thiopental, phenobarbital, and pentobarbital [46, 64], have long been instituted in the treatment of traumatic brain injury and elevated intracerebral pressure [65]. The most recent traumatic brain injury guidelines include the use of barbiturate coma to management of refractory elevated intracerebral pressure, however, not as a prophylactic measure [66]. There is no clear evidence supporting the use of barbiturate induced coma in the management of cerebral edema for tumor, intracerebral hemorrhage, or ischemic stroke [46]. Barbiturate induced coma should be used with caution; however, given the lack of demonstrable improvement in clinical outcomes [65], the use of this strategy remains somewhat controversial in the USA [64]. Additionally, many complications from high-dose barbiturate therapy including hypotension, immunosuppression, and hypothermia further limit the benefits from this intervention [46, 65]. When employed pentobarbital is, in general, the agent of choice for treatment given its intermediate half-life of ~20 h. Pentobarbital dosing is typically titrated to sustained intracerebral pressure control or burst suppression on electroencephalogram [46].
Propofol, 2,6-dilsopropylphenol, a sedative used routinely in intensive care units, also is employed in the medical management of elevated intracranial pressure. The Brain Trauma Foundation TBI Guidelines, however, reports caution when using propofol infusion for the management of elevated ICP in TBI patients, as similar to barbiturates, clinically beneficial outcomes with propofol are not clearly established [66]. The hypothesized mechanism of propofol on reduction of intracerebral pressure is via reduction in cerebral metabolic rate and thus reducing cerebral blood volume [60]. There is, however, growing evidence to suggest a more direct effect of propofol on cerebral edema via modulation of aquaporin-4 [67, 68]. This modulation in aquaporin-4 is believed to decreased the subsequent neuroinflammatory response by attenuating the expression of IL-1β and TNF-α expression [68]. Although the exact mechanism of propofol’s effects on cerebral edema and intracranial pressure remain unclear, the clinical utilization of propofol for the management of cerebral edema has not been extensively studied in human populations [69]. However, when used in clinical practice, propofol bolus and continuous infusion is considered second line therapy for the medical management of elevated intracerebral pressure by the Neurocritical Care Society [60]. It is important to bear in mind that a subset of patients who are treated with propofol infusions, particularly those of younger ages and treated with higher doses [70], may develop propofol infusion syndrome [71], metabolic acidosis, rhabdomyolysis, cardiac dysfunction, and hypertriglyceridemia which may be fatal [60, 66, 70].
Midazolam, a benzodiazepine; fentanyl, an opiate; and dexmedetomidine, a centrally acting α2 adrenergic receptor agonist, all may be used as adjunctive interventions for the management of elevated cerebral pressures [69]. The mechanism of each is indirect and believed to be through systemic effects on pain management and systemic vascular resistance. These agents are not mainstays in the medical management of cerebral edema.
Hypothermia
Induced hypothermia or targeted temperature management, to 32–35 °C, for the management of elevated intracerebral pressure is well-known [72]. The mechanisms by which hypothermia treats elevated intracerebral pressure and cerebral edema is thought to be multifactorial. Firstly, hypothermia is thought to lower the cerebral metabolic rate and thus limit the risk of sodium pump failure, calcium influx, and cellular death [73]. Hypothermia is additionally thought to attenuate free radical production and pro-inflammatory cytokines and maintain cerebral perfusion which collectively mitigates the formation of cytotoxic edema formation [73,74,75]. Finally, a direct effect on cerebral vasogenic edema within human clinical studies has additionally been described [76]. Although there are clinical data to suggest a mechanistic effect on cerebral edema and elevated intracranial pressure, one of the most recent clinical trials does not suggest improved clinical outcomes with induced hypothermia plus standard management in TBI patients with elevated intracerebral pressure [77].
Glucocorticoids
Glucocorticoids have been the clinical mainstay for management of perilesional vasogenic edema [78]. The mechanisms through which glucocorticoids act on vasogenic edema are many. Glucocorticoids bind to the glucocorticoid receptor releasing heat-shock proteins and translocating to the nucleus mediating transcriptional changes [79]. Some transcriptional mediators are upregulated, such as glucocorticoid-induced leucine zipper [80] that are responsible for the transcription of proteins that suppress inflammatory mediators such as AP-1 and NF-κB [81]. There are additional direct interactions between glucocorticoid bound, glucocorticoid receptor homodimer and AP-1 and NF-κB, which result in decreased pro-inflammatory cytokine release and direct reduction in peritumoral vasogenic cerebral edema [82]. Although these are the postulated primary mechanisms for glucocorticoids on vasogenic cerebral edema, many preclinical studies suggest that they may mediate many additional pathways including VEGF, multiple aquaporin pathways, multiple claudin pathways, and via MMP/TIMP mechanisms [78]. Although many of these targets appear to be important in in vivo models, the specific pathways mediating vasogenic edema in brain neoplasms in humans remains less clear.
Glucocorticoids are not recommended for the management of cytotoxic cerebral edema in traumatic brain injury, intracerebral hemorrhage, or stroke [66, 83, 84], although the role of glucocorticoids in aneurysmal subarachnoid hemorrhage remains unclear [85].
Potential Targeted Therapies
Our approach to the medical management of cerebral edema has largely revolved around an increasingly artificial distinction between cytotoxic and vasogenic cerebral edema [7, 35, 86]. A recent and growing understanding of the molecular mechanisms underlying cerebral edema in neurologic injury has provided a framework within which to identify novel therapeutic targets and potential promise for new clinical interventions [87]. These potential molecular targets have been identified based on a growing understanding of the cell–cell signaling that occurs amongst the neurovascular unit [88,89,90]. The neurovascular unit is the conceptual paradigm describing the cellular signaling that occurs between glial, vascular, and neuronal partitions within the central nervous system (CNS) [90]. The dynamic interplay of cellular signals within the neurovascular unit is not only important for trophic support but also increasingly recognized as active during and after CNS injury, releasing neurotoxic inflammatory mediators, coordinating cellular remodeling, and organizing recovery [89, 90]. The role of these molecular signals in the clinical management of cerebral edema remains unclear; many serve as investigational therapeutic targets. However, this review hopes to detail the current landscape of these promising therapeutic interventions (see Table 2).
Bumetanide: a Potential NKCCl Target
Bumetanide is a loop diuretic approved for the management of fluid retention in the management of chronic heart failure [91]. A specific antagonist of the NKCCl transporter [92], bumetanide offers promise for management of cerebral edema through the potential inhibition of this transporter and reduced swelling of astrocytes after injury [13, 14]. Under normal environment conditions and the presence of ATP, NKCCl cotransports ions and water volume to maintain plasma membrane homeostasis [13, 14, 93]. Sodium is transported into endothelial cells and expelled into the interstitial via a sodium potassium ATPase [93]. However, under pathologic stress resulting from ischemia, the blood–brain barrier becomes permeable to an increased concentration of sodium and potassium resulting in increased astrocyte uptake leading to edema [94]. Furthermore, in vitro evidence suggests cerebral tissues express increased concentrations of NKCCl [94] and that under conditions of hypoxia, NKCCl appears to be upregulated, further facilitating transport of sodium, potassium, and water interstitially potentiating cerebral edema [95]. There is additional evidence that although NKCCl may play a direct effect on cerebral edema formation, there are perhaps downstream inflammatory effects such as IL-1 that may potentiate cerebral injury [96]. Collectively, this has made bumetanide a potential pharmacologic target for the management of cerebral edema. Animal studies have demonstrated that in the management of cerebral edema in diabetic ketoacidosis [97], ischemic stroke [94], and traumatic brain injury [96, 98] that bumetanide offers promise in lowering the degree of edema seen. Additionally, in the management of perihematomal edema in intracerebral hemorrhage, there is the suggestion that early administration of bumetanide may attenuate perihematomal edema in rats, but may not improve outcomes [92]. There are no reports of bumetanide in the management of cerebral edema in humans.
AER-270, AER-271, and Curcumin: Targeting Aquaporins
Aquaporins are ubiquitous within the human body; however, two main isoforms are primarily prevalent in the central nervous systems of mammals: aquaporin-1 and aquaporin-4 [99]. Within the brain, aquaporin-4 is the most abundant [100] and are localized primarily to the astrocyte foot processes [99, 101]. Aquaporin-4 transporter, although molecularly connected with other neuroinflammatory mediators [87], facilitates intracellular water movement in cytotoxic edema [102] and decreased water clearance from astrocyte foot processes in cytotoxic edema [103, 104]. Given that aquaporin-4 does not appear to have one universal role, the facilitation of water movement, understanding the temporal relationship of aquaporin-4 with cerebral injury, appears paramount. In murine models of traumatic brain injury, there are conflicting reports of the role and regulation of aquaporin-4 within the first 48 h after injury [105, 106]. These observations may however be a manifestation generalized aquaporin dysregulation seen after traumatic brain injury, as seen in an aquaporin knockout mouse model [107] and potentially serve as a compensatory mechanism to counteract cytotoxic edema in favor of vasogenic edema [108]. However, available animal model evidence suggests that in general, aquaporin deletion, or by extension inhibition, may worsen vasogenic edema and limit cytotoxic edema [99, 100, 109].
Although human data is limited, the role of aquaporin-4 upregulation has been reported in traumatic brain injury [110], intracranial neoplasms [111], cerebral infarction [112], and subarachnoid hemorrhage [113], with the degree of upregulation correlating to the degree of cerebral edema [111, 114]. As such, inhibition of aquaporin-4 offers potential promise in the management of cerebral edema. AER-270 and AER-271 (a phosphorylated pro-drug of AER-270) are potentially theraputic small molecules [115]. Demonstrating promise in the management of cerebral edema in animal models [115, 116], the selective aquaporin-4 inhibitor, AER-271, is currently under investigation in phase 1 clinical trial for eventual use in humans for the treatment of acute ischemic stroke (ClinicalTrials.gov Identifier: NCT03804476).Footnote 1
Curcumin, a derivative curry spice rhizome Curcuma longa is known for its anti-inflammatory effects and a potent inhibitor of NF-κB [117]. It has more recently been demonstrated to have additionally anti-inflammatory effects through reduction of IL-1β, which has been demonstrated to directly correlate with aquaporin-4 expression [118]. It therefore is postulated that through reduction in aquaporin-4, curcumin may attenuate cerebral edema [119]. This hypothesis has been supported by growing evidence within rodent models of TBI [117], ischemic stroke [120], hemorrhagic stroke [121], diffuse cerebral hypoxia [119], and cerebral neoplasms [122].
Glibenclamide: Targeting Sur1-Trpm4
Sulfonylurea receptor 1 (Sur1) is a cation channel that is upregulated along capillary beds, astrocytes, and neurons after neurotrauma or ischemia [123]. Once Sur1 is upregulated in the setting of cerebral injury, transient receptor potential melastatin 4 (Trpm4) [124] associates forming a nonselective cation channel ultimately leading to intracellular water influx, edema, and cellular death [123, 124]. With Sur1-Trmp4 not present under normal homeostatic pressure [87], this protein channel offers a promising target for therapeutic development. Sur1 in domains that bind sulfonylurea drugs such as glibenclamide or glyburide, which inhibit the channel activity [124]. Animal models in TBI and hemorrhagic shock have demonstrated promising results in improved functional outcomes and decreased volume of edema [125,126,127]. This has provided the enthusiasm with which human studies have been pursued. Two small human trials, both in TBI, have demonstrated that with administration of glyburide, decreased contusion expansion rates and improvement in short-term functional outcomes were observed [128, 129]. The culmination of these data largely leads to the creation of a prospective randomized controlled trial investigating the efficacy of glyburide IV administration in the management of large territory ischemic infarcts: GAMES-RP [130]. GAMES-RP recently reported results suggesting improvement in midline shift, levels of alertness, NIH stroke scale scores, and decreased mortality in patients treated with glyburide [131]. This work has largely been the reason glyburide appears to be a very promising intervention in the management of cerebral edema.
Glibenclamide and SB-3CT: Targeting Matrix Metalloproteinases
Glibenclamide is additionally an indirect matrix metalloproteinase (MMP) inhibitor, specifically of MMP-9, which further contributes to its potential as a therapeutic target [130, 131]. There is also growing evidence that MMP-9 plays an important role in stroke recovery, with tissue remodeling, angiogenesis, and neuronal plasticity [132]. The mechanism through which MMP contributes to cerebral edema is via proteolytic cleavage of the basement membrane and disruption of the blood–brain barrier [133]. In cerebral injury MMP is upregulated and thereby reduced the integrity of the underlying BBB, contributing to cerebral edema [133]. Clinically, this has been demonstrated in murine models, in which MMP knockout mice have improved functional outcomes and decreased cerebral edema in traumatic brain injury [134], hepatic encephalopathy [135], ischemic stroke [132], and subarachnoid hemorrhage [136]. SB-3CT, a selective inhibitor of MMP, has been investigated in multiple preclinical models of cerebral injury and has demonstrated promise as a potential translatable target [137,138,139,140]. Yet given the ubiquitous nature of MMPs and the prior failed trials utilizing MMP inhibitors for cancer, secondary to undesired side effects should encourage discretion with translation to clinical populations [138]. There is however, an ongoing large clinical trial investigating the use of glibenclamide injection for the management of severe cerebral edema post large hemispheric infarct (ClinicalTrials.gov Identifier: NCT02864953),Footnote 2 with results pending.
Amiloride, SM-20220, and HOE-642: Targeting NHE1 and NHE2
Multiple sodium–hydrogen exchange (NHE) protein isoforms exist in humans [19, 141]; however, two isoforms, NHE1 and NHE2, appear to have a particularly important role in the permeability of blood–brain barrier in acute ischemic stroke [19]. NHE1 and NHE2 are both present on luminal blood–brain barrier, stimulated by hypoxia and hypoglycemia, both conditions present in the setting of acute ischemia [142, 143]. Murine model suggests that with small molecule NHE inhibitors, such as SM-20220 and HOE-642, perilesional edema secondary to ischemia can be limited [142, 143]. Amiloride, a thiazide diuretic [144], has additionally been described to decrease cerebral edema via decreased brain water content; however, this precise mechanism has not been completely elucidated [145]. No human studies have been completed to verify these results in clinical populations [2].
SR 49059, V1880, Conivaptan, and Vasopressin: Targeting V1a Receptors
Arginine vasopressin is a nanopeptide that is produced in the hypothalamus and released into the blood stream via the neurohypophysis and is heavily responsible for cerebral water homeostasis [146]. Furthermore, there is growing evidence that arginine vasopressin expression may be related to aquaporin expression in the brain as well [147]. Specifically, V1a receptors facilitate aquaporin-4-mediated water flow in the rat cortices [148]. This observation coupled with results from animal studies identifying V1a as a potential therapeutic target to mediate cerebral edema post TBI and ischemia [148,149,150] has made small molecules inhibitors of V1a, such as SR 49059 and V1880, of particular interest [2]. Mice models of ischemic and hemorrhagic stroke have demonstrated decreased cerebral edema and improved functional outcomes with pretreatment of SR 49059 [151, 152]. TBI mouse models demonstrate that treatment with V1880 decreases cerebral edema observed post injury [153]. Similar results in reducing cerebral edema have been seen with the mixed V1a and V2a inhibitor, conivaptan, when administered to mice post ischemic stroke [154, 155]. Collectively, these data have supported the development of a single center phase 1 trial of conivaptan for the treatment of intracerebral hemorrhage (ClinicalTrials.gov Identifier: NCT03000283).Footnote 3 Vasopressin, an arginine vasopressin agonist, has not demonstrated the expected increase in cerebral edema when administered to patients with TBI [156], calling into question the exact mechanism through which V1a antagonism decreases cerebral edema.
ML-7: Targeting MLCK inhibition
Myosin light chain kinase has been demonstrated to increase cerebral edema [157]. The proposed mechanism via which myosin light chain kinase augments cerebral edema is via contraction of endothelial cells thereby increasing the permeability of the endothelial blood–brain barrier [157, 158]. ML-7 is a small molecule inhibitor of myosin light chain kinase and in preclinical animal studies have demonstrated decreased rates of edema and improved functional outcomes in TBI models [157, 158]. ML-7 has not been translated to human studies as of yet. Endothelial myosin light chain kinase is ubiquitous throughout human endothelial layers, potentially inhibiting the translatability of this target into clinical practice [159].
Vascular Endothelial Growth Inhibitor and Bevacizumab: Targeting Vascular Endothelial Growth Inhibitor and Anti-vascular Endothelial Growth Factor Antibody
Vascular endothelial growth factor (VEGF) is most well-recognized for its role in angiogenesis and vascular homeostasis in the human brain [160, 161]. However, over the past several years, many additional angiogenic independent functions of VEGF have been identified and are likely the reason VEGF expression in the adult brain tends to localize in geographically specific regions in the cerebral tissues [160]. VEGF’s role in BBB permeability is clearly delineated, and furthermore, VEGF has been demonstrated to be upregulated in neuronal injury and inducible by hypoxia-inducible factor 1 and hypoxia-inducible factor 2 [161,162,163,164]. Once hypoxia-inducible factors are released, MMPs then facilitate cleavage of the native basement membrane, resulting in increased permeability of the native vessel and facilitate extravasation of intraluminal components into the extracellular space [164] The role of VEGF and more specifically VEGF inhibition is best described and most widely used in the management of cerebral neoplasms [165]. One commonly used inhibitor is bevacizumab, a humanized monoclonal antibody against VEGF [166], has demonstrated efficacious effects in the treatment of neoplasms, particularly when combined with chemotherapeutic regiments; with one phase II trial further demonstrated decreased cerebral edema in the treatment arm [167]. Rebound cerebral edema has also been described in patient being treated with bevacizumab for CNS neoplasms who undergo rapid cessation of their treatment [166].
The role of VEGF and VEGF inhibition outside the management of CNS neoplasms is unclear. Within the management of ischemic stroke, it appears that VEGF inhibition reduces the edema formation and decreases the risk of ischemia related reperfusion injury [168]. Although VEGF upregulation may offer some neuroprotective effects, the corresponding increase risk of CNS microbleeds and worsening edema suggests the role VEGF serves for edema regulation may be more important in ischemic injury [169]. TBI models suggest that post traumatic injury, regional upregulation of VEGF is seen [33] and that these regional differences contribute to the formation of post TBI cerebral edema [170]. The importance of VEGF in mediating cerebral edema in TBI has been demonstrated in rats with VEGF receptor antagonism [171]. The role of VEGF upregulation in ischemia and trauma remains unclear in humans. Although we do know that TBI augments VEGF expression in the CSF of humans [172], no human trials have been completed.
NK1 Receptor Antagonists: Targeting NK1
Neurokinin-1 (NK1) receptor antagonists, such as tachykinin receptor antagonist N-acetyl-l-tryptophan [173], have been demonstrated to inhibit substance P [174,175,176]. One of many neuropeptides released in neuronal injury, substance P [177] has been implicated for its role in breaking down the blood–brain barrier leading to increased permeability and the development of cerebral edema [174, 178]. This coupled with the evidence from both animal and human studies that substance P levels in the cortex are elevated in the setting of TBI [178] and ischemia [179, 180]and have made substance P antagonists promising targets for the medical management of cerebral edema. One downstream inhibitor of substance P was the NK1 receptor antagonist SR140333, which demonstrated reduction in infarct volume size in animal models [181]. Additional models in rodents for cerebral and spinal ischemia have replicated these findings with molecular inhibition of NK1 [176, 180]. Similar findings in animal models of TBI have demonstrated similarly promising findings: decreased cerebral edema and improved functional outcomes [174, 175]. Although the efficacy of substance P inhibition has yet to be replicated in human populations, serum substance P levels have been associated with TBI severity and mortality in humans as well [182].
Fenofibrate, Pioglitazone, and Rosiglitazone: Targeting PPAR Agonist
Fenofibrate, pioglitazone, and rosiglitazone are commonly used peroxisome proliferator-activated receptors (PPAR) agonists in clinical practice [183]. PPARs, membrane associated transcription factors, are present in three different isotypes and are all present in the brain [184]. Through a combination of downstream effects, agonism of these PPAR receptors leads to down-transcription of pro-inflammatory mediators [185]. This has led to the application of clinically available PPAR agonists to the management of cerebral edema and neuronal injury. In TBI, fenofibrate [186], pioglitazone [187], and rosiglitazone [188] have demonstrated decreased neuronal inflammation and subsequent decreases in edema when administered in rodent models. Similarly, the PPAR agonists have demonstrated promise in the management of neuronal injury post ischemic stroke models [189,190,191]. This has led to the development of novel, neuronally designed PPAR agonists, GW0742, for the management of acute stroke, with similarly promising results [192]. Interestingly, PPAR has additionally been demonstrated to be a potential target in the management of cerebral edema post neurosurgical interventions [193], a novel population in which targeted therapy could be very beneficial. Although these findings have also not been translated into human studies, the proposed role of PPAR in neuroinflammation and cerebral edema in intracerebral hemorrhage [194, 195] has led to the design and completion of a phase 2 clinical trial which has recently been completed (ClinicalTrials.gov Identifier: NCT00827892)Footnote 4; results of which are still pending.
HMGB1: Targeting Toll Receptors
High mobility group box protein 1 (HMGB1) is a DNA binding protein that translocate to the extracellular space following cerebral injury and activates Toll-like receptors [196]. These Toll-like receptors are then responsible for cell signaling which is prompts a secondary neuroinflammatory cascade seen in TBI and ischemic stroke [197]. There appears to be a dose–response relationship between neuronal injury, cerebral edema, and HMGB1 as well in animal models, further making HMGB1 a potential therapeutic target [197]. HMGB1 a-Box, a molecular fragment which competitively inhibits HMGB1 at its receptor binding site, has demonstrated efficacy at reversing edema and improving neurologic function in TBI models [198]. Similar results with small molecule inhibitors or antibodies to HMGB1 have demonstrated promise in the management of intracerebral hemorrhage [199] and subarachnoid hemorrhage [200] models as well. However, when translating these results to human populations, HMGB1 inhibition, either through small molecules or antibodies, has been rife with complications [201]. However probenecid, initially developed to be administered with penicillin and increase serum levels [202], has recently demonstrated efficacious effects at inhibiting HMGB1 levels and decreasing infarct volume size in animals [203]. Given its relatively safe pharmacokinetic profile, it offers a potential early translatable target for clinical populations.
Fingolimod, RPC1063, and RP101075: Targeting Sphingosine-1-Phosphate Receptors
Fingolimod is an established oral medication for the management of multiple sclerosis and targets sphingosine-1-phosphate receptors (S1PR) [9, 204]. Preclinical rodent models have demonstrated improved clinical outcomes of intracerebral hemorrhage by mitigating post injury inflammatory response by inhibiting migration of lymphocytes to the site of injury [9]. Cerebral edema is reduced by lymphocyte suppression and decreased expression of secondary inflammatory cytokines that are believed to exacerbate edema progression [204,205,206]. S1PRs have multiple subunits, with fingolimod primarily mediating inflammation via S1PR1 binding, although having affinity for additional subunits as in S1PR3 [204]. The importance of S1PR1 selectivity in the mediation of cerebral edema has been tested with the use of RPC1063, a selective S1PR1 modulator, and its active metabolite RP101075 [204, 206]. This active metabolite, RP101075, has been shown to limit cerebral edema in rodent models of intracerebral hemorrhage, enhance BBB integrity, and improve the microvasculature circulation post injury, further suggesting the importance of S1PR1 modulation in cerebral injury [204, 206]. There are no active human clinical trials investigating the effects of S1PR1 on cerebral edema, however, given the promising preclinical models, making sphingomyelin-1-phosphate receptor modulators potentially viable for the management of cerebral edema.
Conclusions and Future Directions
Despite the rapid modernization and development of specialized neurocritical care units and staffed by a subspecialty trained population of neurointensivists and staff [207,208,209], the medical management of cerebral edema developed within this clinical practice is independent of targeting the underlying cellular and molecular mechanisms that drive it [7]. As a result, the nontargeted therapies that are employed in-practice remain the standard of clinical practice [60] and focus on the downstream effects of cerebral edema formation without modulation of the neuroinflammatory cascade that is paramount in all neuronal injury [29, 210, 211]. However, in the recent years, a growing understanding in the molecular mechanisms of cerebral inflammation, blood–brain barrier permeability, and cerebral edema in all modalities of neuronal injury have led to an inspiring number of potential therapeutic targets [7]. The complicated nature of these pathways is just now becoming clear through animal models in TBI, ischemic stroke, subarachnoid hemorrhage, and cerebral neoplasm. And although a lack of the detailed understanding of many of these pathways have limited the clinical utility targeting many of these molecular targets in clinical practice, our growing understanding of these mechanisms will surely aid in further clinical development of future therapeutics. Few large-scale human trials have been designed around these targeted interventions; however, those that have been completed demonstrated promising results [130, 131]. With several human trials in development (Clinical Trials Identifier: NCT03000283 and NCT03804476), and more on the way, it is clear that our future understanding of targeted medical management of cerebral edema will rapidly evolve, offering promise for a currently pressing clinical problem.
Notes
Phase 1 study to assess AER-271. Clinical Trials.Gov. https://clinicaltrials.gov/ct2/show/NCT03804476 Accessed 3/16/2019
Phase 3 study to evaluate the efficacy and safety of intravenous BIIB093 (glibenclamide) for severe cerebral edema following large hemispheric infarction (CHARM). Clinical Trials.gov. https://clinicaltrials.gov/ct2/show/NCT02864953. Accessed 3/18/2019
Conivaptan for the reduction of cerebral edema in intracerebral hemorrhage—a safety and tolerability study. Clinical Trials.Gov. https://clinicaltrials.gov/ct2/show/study/NCT03000283. Accessed 3/16/2019
Safety of Pioglitazone for Hematoma Resolution In Intracerebral Hemorrhage (SHRINC). Clinical Trials.gov. https://clinicaltrials.gov/ct2/show/NCT00827892. Accessed 3/17/2019
References
Shah S, Kimberly WT. Today’s approach to treating brain swelling in the neuro intensive care unit. Semin Neurol 2016;36(6):502–7.
Jha RM, Kochanek PM, Simard JM. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology 2019;145:230–46.
Battey TW, Karki M, Singhal AB, et al. Brain edema predicts outcome after nonlacunar ischemic stroke. Stroke 2014;45(12):3643–8.
Urday S, Beslow LA, Dai F, et al. Rate of perihematomal edema expansion predicts outcome after intracerebral hemorrhage. Crit Care Med 2016;44(4):790–7.
Tucker B, Aston J, Dines M, et al. Early brain edema is a predictor of in-hospital mortality in traumatic brain injury. J Emerg Med 2017;53(1):18–29.
Kok B, Karvellas CJ. Management of cerebral edema in acute liver failure. Semin Respir Crit Care Med 2017;38(6):821–9.
Stokum JA, Gerzanich V, Simard JM. Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab 2016;36(3):513–38.
Winkler EA, Minter D, Yue JK, Manley GT. Cerebral edema in traumatic brain injury: pathophysiology and prospective therapeutic targets. Neurosurg Clin N Am 2016;27(4):473–88.
Zheng H, Chen C, Zhang J, Hu Z. Mechanism and therapy of brain edema after intracerebral hemorrhage. Cerebrovasc Dis 2016;42(3–4):155–69.
Koenig MA. Cerebral edema and elevated intracranial pressure. Continuum (Minneap Minn) 2018;24(6):1588–602.
Rungta RL, Choi HB, Tyson JR, et al. The cellular mechanisms of neuronal swelling underlying cytotoxic edema. Cell 2015;161(3):610–21.
Norenberg MD. Astrocyte responses to CNS injury. J Neuropathol Exp Neurol 1994;53(3):213–20.
Jayakumar AR, Liu M, Moriyama M, et al. Na-K-Cl cotransporter-1 in the mechanism of ammonia-induced astrocyte swelling. J Biol Chem 2008;283(49):33874–82.
Jayakumar AR, Norenberg MD. The Na-K-Cl co-transporter in astrocyte swelling. Metab Brain Dis 2010;25(1):31–8.
Saadoun S, Papadopoulos MC, Watanabe H, Yan D, Manley GT, Verkman AS. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J Cell Sci 2005;118(Pt 24):5691–8.
Haj-Yasein NN, Vindedal GF, Eilert-Olsen M, et al. Glial-conditional deletion of aquaporin-4 (Aqp4) reduces blood-brain water uptake and confers barrier function on perivascular astrocyte endfeet. Proc Natl Acad Sci U S A 2011;108(43):17815–20.
Higashida T, Kreipke CW, Rafols JA, et al. The role of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J Neurosurg 2011;114(1):92–101.
Manley GT, Fujimura M, Ma T, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 2000;6(2):159–63.
Lam TI, Wise PM, O'Donnell ME. Cerebral microvascular endothelial cell Na/H exchange: evidence for the presence of NHE1 and NHE2 isoforms and regulation by arginine vasopressin. Am J Phys Cell Physiol 2009;297(2):C278–89.
Mehta RI, Tosun C, Ivanova S, et al. Sur1-Trpm4 cation channel expression in human cerebral infarcts. J Neuropathol Exp Neurol 2015;74(8):835–49.
Stokum JA, Kwon MS, Woo SK, et al. SUR1-TRPM4 and AQP4 form a heteromultimeric complex that amplifies ion/water osmotic coupling and drives astrocyte swelling. Glia 2018;66(1):108–25.
Simard JM, Chen M, Tarasov KV, et al. Newly expressed SUR1-regulated NC(Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nat Med 2006;12(4):433–40.
Chen M, Simard JM. Cell swelling and a nonselective cation channel regulated by internal Ca2+ and ATP in native reactive astrocytes from adult rat brain. J Neurosci 2001;21(17):6512.
Obrenovitch TP, Urenjak J. Is high extracellular glutamate the key to excitotoxicity in traumatic brain injury? J Neurotrauma 1997;14(10):677–98.
Guyot LL, Diaz FG, O'Regan MH, McLeod S, Park H, Phillis JW. Real-time measurement of glutamate release from the ischemic penumbra of the rat cerebral cortex using a focal middle cerebral artery occlusion model. Neurosci Lett 2001;299(1–2):37–40.
Schneider GH, Baethmann A, Kempski O. Mechanisms of glial swelling induced by glutamate. Can J Physiol Pharmacol 1992;70(Suppl):S334–43.
Illarionova NB, Gunnarson E, Li Y, et al. Functional and molecular interactions between aquaporins and Na,K-ATPase. Neuroscience 2010;168(4):915–25.
Izumi Y, Kirby CO, Benz AM, Olney JW, Zorumski CF. Muller cell swelling, glutamate uptake, and excitotoxic neurodegeneration in the isolated rat retina. Glia 1999;25(4):379–89.
Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience 2004;129(4):1021–9.
Vorbrodt AW, Lossinsky AS, Wisniewski HM, et al. Ultrastructural observations on the transvascular route of protein removal in vasogenic brain edema. Acta Neuropathol 1985;66(4):265–73.
Durward QJ, Del Maestro RF, Amacher AL, Farrar JK. The influence of systemic arterial pressure and intracranial pressure on the development of cerebral vasogenic edema. J Neurosurg 1983;59(5):803–9.
Garcia JG, Siflinger-Birnboim A, Bizios R, Del Vecchio PJ, Fenton JW 2nd, Malik AB. Thrombin-induced increase in albumin permeability across the endothelium. J Cell Physiol 1986;128(1):96–104.
Dore-Duffy P, Wang X, Mehedi A, Kreipke CW, Rafols JA. Differential expression of capillary VEGF isoforms following traumatic brain injury. Neurol Res 2007;29(4):395–403.
Fischer S, Wobben M, Marti HH, Renz D, Schaper W. Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc Res 2002;63(1):70–80.
Simard JM, Kent TA, Chen M, Tarasov KV, Gerzanich V. Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol 2007;6(3):258–68.
Fukuda S, Fini CA, Mabuchi T, Koziol JA, Eggleston LL Jr, del Zoppo GJ. Focal cerebral ischemia induces active proteases that degrade microvascular matrix. Stroke 2004;35(4):998–1004.
Duan X, Wen Z, Shen H, Shen M, Chen G. Intracerebral hemorrhage, oxidative stress, and antioxidant therapy. Oxidative Med Cell Longev 2016;2016:1203285.
Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke 2011;42(6):1781–6.
Urday S, Kimberly WT, Beslow LA, et al. Targeting secondary injury in intracerebral haemorrhage—perihaematomal oedema. Nat Rev Neurol 2015;11(2):111–22.
Shima K. Hydrostatic brain edema: basic mechanisms and clinical aspect. Presented at: Brain Edema XII.
Halperin JL, Levine GN, Al-Khatib SM, et al. Further evolution of the ACC/AHA Clinical Practice Guideline Recommendation Classification System: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2016;133(14):1426–8.
Qureshi AI, Suarez JI. Use of hypertonic saline solutions in treatment of cerebral edema and intracranial hypertension. Crit Care Med 2000;28(9):3301–13.
Scallan J, Huxley VH, Korthuis RJ. Chapter 1, Fluid movement across the endothelial barrier. In: Capillary fluid exchange: regulation, functions, and pathology. Anonymous Morgan & Claypool Life Sciences, San Rafael (CA); 2010.
Diringer MN. New trends in hyperosmolar therapy? Curr Opin Crit Care 2013;19(2):77–82.
Javid M, Settlage P. Effect of urea on cerebrospinal fluid pressure in human subjects: preliminary report. JAMA 1956;160(11):943–9.
Raslan A, Bhardwaj A. Medical management of cerebral edema. Neurosurg Focus 2007;22(5):E12.
Otvos B, Kshettry VR, Benzel EC. The history of urea as a hyperosmolar agent to decrease brain swelling. Neurosurg Focus FOC 2014;36(4):E3.
Javid M, Settlage P, Monfore T. Urea in the management of increased intracranial pressure. Surg Forum 1957;7:528–32.
Clasen RA, Cooke PM, Pandolfi S, Carnecki G, Bryar G. Hypertonic urea in experimental cerebral edema. NEUR 1965;12(4):424–34.
Wise BL, Chater N. Use of hypertonic mannitol solutions to lower cerebrospinal fluid pressure and decrease brain bulk in man. Surg Forum 1961;12:398–9.
Palma L, Bruni G, Fiaschi AI, Mariottini A. Passage of mannitol into the brain around gliomas: a potential cause of rebound phenomenon. A study on 21 patients. J Neurosurg Sci 2006;50(3):63–6.
Sorani MD, Manley GT. Dose-response relationship of mannitol and intracranial pressure: a metaanalysis. J Neurosurg 2008;108(1):80–7.
Muizelaar JP, Wei EP, Kontos HA, Becker DP. Mannitol causes compensatory cerebral vasoconstriction and vasodilation in response to blood viscosity changes. J Neurosurg 1983;59(5):822–8.
Fandino W. Understanding the physiological changes induced by mannitol: from the theory to the clinical practice in neuroanaesthesia. J Neuroanaesthesiol Crit Care 2017;4(3):138–46.
Boone MD, Oren-Grinberg A, Robinson TM, Chen CC, Kasper EM. Mannitol or hypertonic saline in the setting of traumatic brain injury: what have we learned? Surg Neurol Int 2015;6:177–7806.170248. eCollection 2015.
Fisher B, Thomas D, Peterson B. Hypertonic saline lowers raised intracranial pressure in children after head trauma. J Neurosurg Anesthesiol 1992;4(1):4–10.
Bhardwaj A, Ulatowski JA. Cerebral edema: hypertonic saline solutions. Curr Treat Options Neurol 1999;1(3):179–88.
Koenig MA, Bryan M, Lewin JL, Mirski MA, Geocadin RG, Stevens RD. Reversal of transtentorial herniation with hypertonic saline. Neurology 2008;70(13):1023–9.
Qureshi AI, Suarez JI, Bhardwaj A, et al. Use of hypertonic (3%) saline/acetate infusion in the treatment of cerebral edema: effect on intracranial pressure and lateral displacement of the brain. Crit Care Med 1998;26(3):440–6.
Stevens RD, Huff JS, Duckworth J, Papangelou A, Weingart SD, Smith WS. Emergency neurological life support: intracranial hypertension and herniation. Neurocrit Care 2012;17(Suppl 1):S60–5.
Pfortmueller CA, Schefold JC. Hypertonic saline in critical illness—a systematic review. J Crit Care 2017;42:168–77.
Todd MM, Cutkomp J, Brian JE. Influence of mannitol and furosemide, alone and in combination, on brain water content after fluid percussion injury. Anesthesiology 2006;105(6):1176–81.
Jha SK. Cerebral edema and its management. Med J Armed Forces India 2003;59(4):326–31.
Bassin SL, Bleck TP. Barbiturates for the treatment of intracranial hypertension after traumatic brain injury. Crit Care 2008;12(5):185–5.
Roberts I, Sydenham E. Barbiturates for acute traumatic brain injury. Cochrane Database Syst Rev 2012;12:CD000033.
Carney N, Totten AM, O'Reilly C, et al. Guidelines for the management of severe traumatic brain injury, fourth edition. Neurosurgery 2017;80(1):6–15.
Zheng YY, Lan YP, Tang HF, Zhu SM. Propofol pretreatment attenuates aquaporin-4 over-expression and alleviates cerebral edema after transient focal brain ischemia reperfusion in rats. Anesth Analg 2008;107(6):2009–16.
Ding Z, Zhang J, Xu J, Sheng G, Huang G. Propofol administration modulates AQP-4 expression and brain edema after traumatic brain injury. Cell Biochem Biophys 2013;67(2):615–22.
Alnemari AM, Krafcik BM, Mansour TR, Gaudin D. A comparison of pharmacologic therapeutic agents used for the reduction of intracranial pressure after traumatic brain injury. World Neurosurg 2017;106:509–28.
Fong JJ, Sylvia L, Ruthazer R, Schumaker G, Kcomt M, Devlin JW. Predictors of mortality in patients with suspected propofol infusion syndrome. Crit Care Med 2008;36(8):2281–7.
Krajcova A, Waldauf P, Andel M, Duska F. Propofol infusion syndrome: a structured review of experimental studies and 153 published case reports. Crit Care 2015;19:398–015–1112-5.
Lazaridis C, Robertson CS. Hypothermia for increased intracranial pressure: is it dead? Curr Neurol Neurosci Rep 2016;16(9):78–016–0681-2.
Baker AJ, Zornow MH, Grafe MR, et al. Hypothermia prevents ischemia-induced increases in hippocampal glycine concentrations in rabbits. Stroke 1991;22(5):666–73.
van der Worp HB, Sena ES, Donnan GA, Howells DW, Macleod MR. Hypothermia in animal models of acute ischaemic stroke: a systematic review and meta-analysis. Brain 2007;130(Pt 12):3063–74.
Karibe H, Zarow GJ, Graham SH, Weinstein PR. Mild intraischemic hypothermia reduces postischemic hyperperfusion, delayed postischemic hypoperfusion, blood-brain barrier disruption, brain edema, and neuronal damage volume after temporary focal cerebral ischemia in rats. J Cereb Blood Flow Metab 1994;14(4):620–7.
Choi HA, Badjatia N, Mayer SA. Hypothermia for acute brain injury—mechanisms and practical aspects. Nat Rev Neurol 2012;8(4):214–22.
Andrews PJ, Sinclair HL, Rodriguez A, et al. Hypothermia for intracranial hypertension after traumatic brain injury. N Engl J Med 2015;373(25):2403–12.
Murayi R, Chittiboina P. Glucocorticoids in the management of peritumoral brain edema: a review of molecular mechanisms. Childs Nerv Syst 2016;32(12):2293–302.
Dietrich J, Rao K, Pastorino S, Kesari S. Corticosteroids in brain cancer patients: benefits and pitfalls. Expert Rev Clin Pharmacol 2011;4(2):233–42.
Witek E, Hickman D, Lahiri DK, Srinivasan M. Glucocorticoid induced leucine zipper in lipopolysaccharide induced neuroinflammation. Front Aging Neurosci 2019;10:432.
Newton R. Anti-inflammatory glucocorticoids: changing concepts. Eur J Pharmacol 2014;724:231–6.
Barnes PJ. Molecular mechanisms and cellular effects of glucocorticosteroids. Immunol Allergy Clin N Am 2005;25(3):451–68.
Poungvarin N, Bhoopat W, Viriyavejakul A, et al. Effects of dexamethasone in primary supratentorial intracerebral hemorrhage. N Engl J Med 1987;316(20):1229–33.
Sandercock PA, Soane T. Corticosteroids for acute ischaemic stroke. Cochrane Database Syst Rev 2011;(9):CD000064.
Feigin VL, Anderson N, Rinkel GJ, Algra A, van Gijn J, Bennett DA. Corticosteroids for aneurysmal subarachnoid haemorrhage and primary intracerebral haemorrhage. Cochrane Database Syst Rev 2005;(3):CD004583.
Walcott BP, Kahle KT, Simard JM. Novel treatment targets for cerebral edema. Neurotherapeutics 2012;9(1):65–72.
Jha RM, Kochanek PM. A precision medicine approach to cerebral edema and intracranial hypertension after severe traumatic brain injury: quo vadis? Curr Neurol Neurosci Rep 2018;18(12):105-018-0912-9.
Maki T, Hayakawa K, Pham LD, Xing C, Lo EH, Arai K. Biphasic mechanisms of neurovascular unit injury and protection in CNS diseases. CNS Neurol Disord Drug Targets 2013;12(3):302–15.
Xing C, Hayakawa K, Lok J, Arai K, Lo EH. Injury and repair in the neurovascular unit. Neurol Res 2012;34(4):325–30.
Lok J, Wang XS, Xing CH, et al. Targeting the neurovascular unit in brain trauma. CNS Neurosci Ther 2015;21(4):304–8.
Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013;62(16):e147–239.
Wilkinson CM, Fedor BA, Aziz JR, et al. Failure of bumetanide to improve outcome after intracerebral hemorrhage in rat. PLoS One 2019;14(1):e0210660.
Zhang J, Pu H, Zhang H, et al. Inhibition of Na(+)-K(+)-2Cl(-) cotransporter attenuates blood-brain-barrier disruption in a mouse model of traumatic brain injury. Neurochem Int 2017;111:23–31.
O'Donnell ME, Tran L, Lam TI, Liu XB, Anderson SE. Bumetanide inhibition of the blood-brain barrier Na-K-Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J Cereb Blood Flow Metab 2004;24(9):1046–56.
Wallace BK, Foroutan S, O’Donnell ME. Ischemia-induced stimulation of Na-K-Cl cotransport in cerebral microvascular endothelial cells involves AMP kinase. Am J Phys Cell Physiol 2011;301(2):C316–26.
Lu KT, Wu CY, Yen HH, Peng JH, Wang CL, Yang YL. Bumetanide administration attenuated traumatic brain injury through IL-1 overexpression. Neurol Res 2007;29(4):404–9.
Lam TI, Anderson SE, Glaser N, O'Donnell ME. Bumetanide reduces cerebral edema formation in rats with diabetic ketoacidosis. Diabetes 2005;54(2):510–6.
Lu KT, Huang TC, Tsai YH, Yang YL. Transient receptor potential vanilloid type 4 channels mediate Na-K-Cl-co-transporter-induced brain edema after traumatic brain injury. J Neurochem 2017;140(5):718–27.
Papadopoulos MC, Verkman AS. Aquaporin water channels in the nervous system. Nat Rev Neurosci 2013;14(4):265–77.
Papadopoulos MC, Manley GT, Krishna S, Verkman AS. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J 2004;18(11):1291–3.
Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci 1997;17(1):171–80.
Mader S, Brimberg L. Aquaporin-4 water channel in the brain and its implication for health and disease. Cells 2019;8(2).
Filippidis AS, Carozza RB, Rekate HL. Aquaporins in brain edema and neuropathological conditions. Int J Mol Sci 2016;18(1). https://doi.org/10.3390/ijms18010055.
Hubbard JA, Szu JI, Binder DK. The role of aquaporin-4 in synaptic plasticity, memory and disease. Brain Res Bull 2018;136:118–29.
Taya K, Marmarou CR, Okuno K, Prieto R, Marmarou A. Effect of secondary insults upon aquaporin-4 water channels following experimental cortical contusion in rats. J Neurotrauma 2010;27(1):229–39.
Kiening KL, van Landeghem FKH, Schreiber S, et al. Decreased hemispheric aquaporin-4 is linked to evolving brain edema following controlled cortical impact injury in rats. Neurosci Lett 2002;324(2):105–8.
Yao X, Uchida K, Papadopoulos MC, Zador Z, Manley GT, Verkman AS. Mildly reduced brain swelling and improved neurological outcome in aquaporin-4 knockout mice following controlled cortical impact brain injury. J Neurotrauma 2015;32(19):1458–64.
Ren Z, Iliff JJ, Yang L, et al. ‘Hit & run’ model of closed-skull traumatic brain injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. J Cereb Blood Flow Metab 2013;33(6):834–45.
Papadopoulos MC, Verkman AS. Aquaporin-4 gene disruption in mice reduces brain swelling and mortality in pneumococcal meningitis. J Biol Chem 2005;280(14):13906–12.
Dardiotis E, Paterakis K, Tsivgoulis G, et al. AQP4 tag single nucleotide polymorphisms in patients with traumatic brain injury. J Neurotrauma 2014;31(23):1920–6.
Saadoun S, Papadopoulos MC, Davies DC, Krishna S, Bell BA. Aquaporin-4 expression is increased in oedematous human brain tumours. J Neurol Neurosurg Psychiatry 2002;72(2):262.
Aoki K, Uchihara T, Tsuchiya K, Nakamura A, Ikeda K, Wakayama Y. Enhanced expression of aquaporin 4 in human brain with infarction. Acta Neuropathol 2003;106(2):121–4.
Badaut J, Brunet JF, Grollimund L et al. Aquaporin 1 and aquaporin 4 expression in human brain after subarachnoid hemorrhage and in peritumoral tissue. Presented at: Brain Edema XII.
Warth A, Kröger S, Wolburg H. Redistribution of aquaporin-4 in human glioblastoma correlates with loss of agrin immunoreactivity from brain capillary basal laminae. Acta Neuropathol 2004;107(4):311–8.
Wallisch JS, Janesko-Feldman K, Alexander H, et al. The aquaporin-4 inhibitor AER-271 blocks acute cerebral edema and improves early outcome in a pediatric model of asphyxial cardiac arrest. Pediatr Res 2019;85(4):511–7.
Farr GW, Hall CH, Farr SM, et al. Functionalized phenylbenzamides inhibit aquaporin-4 reducing cerebral edema and improving outcome in two models of CNS injury. Neuroscience 2019;404:484–98.
Laird MD, Sukumari-Ramesh S, Swift AEB, Meiler SE, Vender JR, Dhandapani KM. Curcumin attenuates cerebral edema following traumatic brain injury in mice: a possible role for aquaporin-4? J Neurochem 2010;113(3):637–48.
Ito H, Yamamoto N, Arima H, et al. Interleukin-1beta induces the expression of aquaporin-4 through a nuclear factor-kappaB pathway in rat astrocytes. J Neurochem 2006;99(1):107–18.
Yu L, Fan Y, Ye G, et al. Curcumin alleviates brain edema by lowering AQP4 expression levels in a rat model of hypoxia-hypercapnia-induced brain damage. Exp Ther Med 2016;11(3):709–16.
Li W, Suwanwela NC, Patumraj S. Curcumin prevents reperfusion injury following ischemic stroke in rats via inhibition of NF‑ΰB, ICAM-1, MMP-9 and caspase-3 expression. Mol Med Rep 2017;16(4):4710–20.
Wang BF, Cui ZW, Zhong ZH, et al. Curcumin attenuates brain edema in mice with intracerebral hemorrhage through inhibition of AQP4 and AQP9 expression. Acta Pharmacol Sin 2015;36(8):939–48.
Klinger NV, Mittal S. Therapeutic potential of curcumin for the treatment of brain tumors. Oxidative Med Cell Longev 2016;2016:14.
Simard JM, Kilbourne M, Tsymbalyuk O, et al. Key role of sulfonylurea receptor 1 in progressive secondary hemorrhage after brain contusion. J Neurotrauma 2009;26(12):2257–67.
Simard JM, Woo SK, Schwartzbauer GT, Gerzanich V. Sulfonylurea receptor 1 in central nervous system injury: a focused review. J Cereb Blood Flow Metab 2012;32(9):1699–717.
Patel AD, Gerzanich V, Geng Z, Simard JM. Glibenclamide reduces hippocampal injury and preserves rapid spatial learning in a model of traumatic brain injury. J Neuropathol Exp Neurol 2010;69(12):1177–90.
Zweckberger K, Hackenberg K, Jung CS, et al. Glibenclamide reduces secondary brain damage after experimental traumatic brain injury. Neuroscience 2014;272:199–206.
Jha RM, Molyneaux BJ, Jackson TC, et al. Glibenclamide produces region-dependent effects on cerebral edema in a combined injury model of traumatic brain injury and hemorrhagic shock in mice. J Neurotrauma 2018;35(17):2125–35.
Khalili H, Derakhshan N, Niakan A, et al. Effects of oral glibenclamide on brain contusion volume and functional outcome of patients with moderate and severe traumatic brain injuries: a randomized double-blind placebo-controlled clinical trial. World Neurosurg 2017;101:130–6.
Zafardoost P, Ghasemi AA, Salehpour F, Piroti C, Ziaeii E. Evaluation of the effect of glibenclamide in patients with diffuse axonal injury due to moderate to severe head trauma. Trauma Mon 2016;21(5):e25113.
Sheth KN, Elm JJ, Beslow LA, Sze GK, Kimberly WT. Glyburide Advantage in Malignant Edema and Stroke (GAMES-RP) Trial: rationale and design. Neurocrit Care 2016;24(1):132–9.
Kimberly WT, Bevers MB, von Kummer R, et al. Effect of IV glyburide on adjudicated edema endpoints in the GAMES-RP Trial. Neurology 2018;91(23):e2163–9.
Turner RJ, Sharp FR. Implications of MMP9 for blood brain barrier disruption and hemorrhagic transformation following ischemic stroke. Front Cell Neurosci 2016;10:56.
ROSENBERG GA. Matrix metalloproteinases in brain injury. J Neurotrauma 1995;12(5):833–42.
Shigemori Y, Katayama Y, Mori T, Maeda T, Kawamata T. Matrix metalloproteinase-9 is associated with blood-brain barrier opening and brain edema formation after cortical contusion in rats. Acta Neurochir Suppl 2006;96:130–3.
Nguyen JH, Yamamoto S, Steers J, et al. Matrix metalloproteinase-9 contributes to brain extravasation and edema in fulminant hepatic failure mice. J Hepatol 2006;44(6):1105–14.
Feiler S, Plesnila N, Thal SC, Zausinger S, Scholler K. Contribution of matrix metalloproteinase-9 to cerebral edema and functional outcome following experimental subarachnoid hemorrhage. Cerebrovasc Dis 2011;32(3):289–95.
Hadass O, Tomlinson BN, Gooyit M, et al. Selective inhibition of matrix metalloproteinase-9 attenuates secondary damage resulting from severe traumatic brain injury. PLoS One 2013;8(10):e76904.
Gu Z, Cui J, Brown S, et al. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci 2005;25(27):6401.
Cui J, Chen S, Zhang C, et al. Inhibition of MMP-9 by a selective gelatinase inhibitor protects neurovasculature from embolic focal cerebral ischemia. Mol Neurodegener 2012;7:21–1.
Jia F, Yin YH, Gao GY, Wang Y, Cen L, Jiang J. MMP-9 inhibitor SB-3CT attenuates behavioral impairments and hippocampal loss after traumatic brain injury in rat. J Neurotrauma 2014;31(13):1225–34.
Chesler M. Regulation and modulation of pH in the brain. Physiol Rev 2003;83(4):1183–221.
Suzuki Y, Matsumoto Y, Ikeda Y, Kondo K, Ohashi N, Umemura K. SM-20220, a Na(+)/H(+) exchanger inhibitor: effects on ischemic brain damage through edema and neutrophil accumulation in a rat middle cerebral artery occlusion model. Brain Res 2002;945(2):242–8.
O'Donnell ME, Chen YJ, Lam TI, Taylor KC, Walton JH, Anderson SE. Intravenous HOE-642 reduces brain edema and Na uptake in the rat permanent middle cerebral artery occlusion model of stroke: evidence for participation of the blood-brain barrier Na/H exchanger. J Cereb Blood Flow Metab 2013;33(2):225–34.
Soltoff SP, Mandel LJ. Amiloride directly inhibits the Na,K-ATPase activity of rabbit kidney proximal tubules. Science 1983;220(4600):957–8.
Vaz R, Sarmento A, Borges N, Cruz C, Azevedo I. Effect of mechanogated membrane ion channel blockers on experimental traumatic brain oedema. Acta Neurochir 1998;140(4):371–5.
Buijs RM. Immunocytochemical demonstration of vasopressin and oxytocin in the rat brain by light and electron microscopy. J Histochem Cytochem 1980;28(4):357–60.
Schrier RW. The sea within us: disorders of body water homeostasis. Curr Opin Investig Drugs 2007;8(4):304–11.
Kleindienst A, Dunbar JG, Glisson R, Marmarou A. The role of vasopressin V1A receptors in cytotoxic brain edema formation following brain injury. Acta Neurochir 2013;155(1):151–64.
Vakili A, Kataoka H, Plesnila N. Role of arginine vasopressin V1 and V2 receptors for brain damage after transient focal cerebral ischemia. J Cereb Blood Flow Metab 2005;25(8):1012–9.
Trabold R, Krieg S, Schöller K, Plesnila N. Role of vasopressin V1a and V2 receptors for the development of secondary brain damage after traumatic brain injury in mice. J Neurotrauma 2008;25(12):1459–65.
Manaenko A, Fathali N, Khatibi NH, et al. Post-treatment with SR49059 improves outcomes following an intracerebral hemorrhagic stroke in mice. Acta Neurochir Suppl 2011;111:191–6.
Kleindienst A, Fazzina G, Dunbar JG, Glisson R, Marmarou A. Protective effect of the V1a receptor antagonist SR49059 on brain edema formation following middle cerebral artery occlusion in the rat. Acta Neurochir Suppl 2006;96:303–6.
Krieg SM, Trabold R, Plesnila N. Time-dependent effects of arginine-vasopressin V1 Receptor inhibition on secondary brain damage after traumatic brain injury. J Neurotrauma 2017;34(7):1329–36.
Ansari S, Krishnan R, Shahripour RB, et al. Combined antagonism of vasopressin receptor subtypes with conivaptan attenuates cerebral edema following ischemic stroke (P5.202). Neurology 2018;90(15):P5.202.
Zeynalov E, Jones SM, Elliott JP. Therapeutic time window for conivaptan treatment against stroke-evoked brain edema and blood-brain barrier disruption in mice. PLoS One 2017;12(8):e0183985.
Allen CJ, Subhawong TK, Hanna MM, et al. Does vasopressin exacerbate cerebral edema in patients with severe traumatic brain injury? Am Surg 2018;84(1):43–50.
Rossi JL, Todd T, Bazan NG, Belayev L. Inhibition of myosin light-chain kinase attenuates cerebral edema after traumatic brain injury in postnatal mice. J Neurotrauma 2013;30(19):1672–9.
Luh C, Kuhlmann CR, Ackermann B, et al. Inhibition of myosin light chain kinase reduces brain edema formation after traumatic brain injury. J Neurochem 2010;112(4):1015–25.
Chavez A, Smith M, Mehta D. Chapter six—new insights into the regulation of vascular permeability. In: International review of cell and molecular biology (volume 290). Jeon KW(Ed.), Academic Press, p. 205–248, 2011.
Licht T, Keshet E. Delineating multiple functions of VEGF-A in the adult brain. Cell Mol Life Sci 2013;70(10):1727–37.
Zhang ZG, Zhang L, Jiang Q, et al. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest 2000;106(7):829–38.
Geiseler SJ, Morland C. The Janus face of VEGF in stroke. Int J Mol Sci 2018;19(5). https://doi.org/10.3390/ijms19051362.
Rosenstein JM, Krum JM, Ruhrberg C. VEGF in the nervous system. Organogenesis 2010;6(2):107–14.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011;473:298.
Stockhammer G, Obwegeser A, Kostron H, et al. Vascular endothelial growth factor (VEGF) is elevated in brain tumor cysts and correlates with tumor progression. Acta Neuropathol 2000;100(1):101–5.
Kim W, Lee H. Brain angiogenesis in developmental and pathological processes: mechanism and therapeutic intervention in brain tumors. FEBS J 2009;276(17):4653–64.
Vredenburgh JJ, Desjardins A, Herndon JE 2nd, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res 2007;13(4):1253–9.
van Bruggen N, Thibodeaux H, Palmer JT, et al. VEGF antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J Clin Invest 1999;104(11):1613–20.
Pignataro G, Ziaco B, Tortiglione A, et al. Neuroprotective effect of VEGF-mimetic peptide QK in experimental brain ischemia induced in rat by middle cerebral artery occlusion. ACS Chem Neurosci. 2015;6(9):1517–25.
Chodobski A, Chung I, KoÅ°niewska E et al. Early neutrophilic expression of vascular endothelial growth factor after traumatic brain injury. Neuroscience 122(4), 853–867 (2003).
Koyama J, Miyake S, Sasayama T, Kondoh T, Kohmura E. Effect of VEGF receptor antagonist (VGA1155) on brain edema in the rat cold injury model. Kobe J Med Sci 2007;53(5):199–207.
Shore PM, Clark RSB, Jackson EK, Wisniewski SR, Adelson PD, Kochanek PM. Vascular endothelial growth factor is increased in cerebrospinal fluid after traumatic brain injury in infants and children. Neurosurgery 2004;54(3):605–12.
Vink R, Gabrielian L, Thornton E. The role of substance P in secondary pathophysiology after traumatic brain injury. Front Neurol 2017;8:304.
Gabrielian L, Helps SC, Thornton E, Turner RJ, Leonard AV, Vink R. Substance P antagonists as a novel intervention for brain edema and raised intracranial pressure. Acta Neurochir Suppl 2013;118:201–4.
Donkin JJ, Nimmo AJ, Cernak I, Blumbergs PC, Vink R. Substance P is associated with the development of brain edema and functional deficits after traumatic brain injury. J Cereb Blood Flow Metab 2009;29(8):1388–98.
Leonard AV, Vink R. The effect of an NK1 receptor antagonist on blood spinal cord barrier permeability following balloon compression-induced spinal cord injury. Acta Neurochir Suppl 2013;118:303–6.
Newbold P, Brain SD. An investigation into the mechanism of capsaicin-induced oedema in rabbit skin. Br J Pharmacol 1995;114(3):570–7.
Donkin JJ, Turner RJ, Hassan I, Vink R. Substance P in traumatic brain injury. Prog Brain Res 2007;161:97–109.
Turner R, Vink R. Inhibition of neurogenic inflammation as a novel treatment for ischemic stroke. Timely Top Med Cardiovasc Dis 2007;11:E24.
Bruno G, Tega F, Bruno A, et al. The role of substance P in cerebral ischemia. Int J Immunopathol Pharmacol 2003;16(1):67–72.
Yu Z, Cheng G, Huang X, Li K, Cao X. Neurokinin-1 receptor antagonist SR140333: a novel type of drug to treat cerebral ischemia. Neuroreport 1997;8(9–10):2117–9.
Lorente L, Martin MM, Almeida T, et al. Serum substance P levels are associated with severity and mortality in patients with severe traumatic brain injury. Crit Care 2015;19:192–015-0911-z.
Mandrekar-Colucci S, Sauerbeck A, Popovich PG, McTigue DM. PPAR agonists as therapeutics for CNS trauma and neurological diseases. ASN Neuro 2013;5(5):e00129.
Moreno S, Farioli-Vecchioli S, Cerù MP. Immunolocalization of peroxisome proliferator-activated receptors and retinoid x receptors in the adult rat CNS. Neuroscience 2004;123(1):131–45.
Thal SC, Neuhaus W. The blood-brain barrier as a target in traumatic brain injury treatment. Arch Med Res 2014;45(8):698–710.
Besson VC, Chen XR, Plotkine M, Marchand-Verrecchia C. Fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, exerts neuroprotective effects in traumatic brain injury. Neurosci Lett 2005;388(1):7–12.
Pilipovic K, Zupan Z, Dolenec P, Mrsic-Pelcic J, Zupan G. A single dose of PPARgamma agonist pioglitazone reduces cortical oxidative damage and microglial reaction following lateral fluid percussion brain injury in rats. Prog Neuro-Psychopharmacol Biol Psychiatry 2015;59:8–20.
Yi J, Park S, Brooks N, Lang B, Vemuganti R. PPARgamma agonist rosiglitazone is neuroprotective after traumatic brain injury via anti-inflammatory and anti-oxidative mechanisms (Volume 1244). 2008.
Sobrado M, Pereira MP, Ballesteros I, et al. Synthesis of lipoxin A4 by 5-lipoxygenase mediates PPARÎ3-dependent neuroprotective effects of rosiglitazone in experimental stroke. J Neurosci 2009;29(12):3875.
Gautier S, Ouk T, Pétrault M, Pétrault O, Bérézowski V, Bordet R. PPAR-alpha agonist used at the acute phase of experimental ischemic stroke reduces occurrence of thrombolysis-induced hemorrhage in rats. PPAR Res 2015;2015(6).
Lee S, Kim H, Hong J, Baek W, Park J. PPARÎ3 agonist pioglitazone reduces matrix metalloproteinase-9 activity and neuronal damage after focal cerebral ischemia. Biochem Biophys Res Commun 2009;380(1):17–21.
Chehaibi K, le Maire L, Bradoni S, Escola JC, Blanco-Vaca F, Slimane MN. Effect of PPAR-beta/delta agonist GW0742 treatment in the acute phase response and blood-brain barrier permeability following brain injury. Transl Res 2017;182:27–48.
Hyong A, Jadhav V, Lee S et al. Rosiglitazone, a PPAR gamma agonist, attenuates inflammation after surgical brain injury in rodents (Volume 1215) (2008).
Zhao XR, Gonzales N, Aronowski J. Pleiotropic role of PPARgamma in intracerebral hemorrhage: an intricate system involving Nrf2, RXR, and NF-kappaB. CNS Neurosci Ther 2015;21(4):357–66.
Zhao X, Sun G, Zhang J, et al. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol 2007;61(4):352–62.
Laird MD, Shields JS, Sukumari-Ramesh S, et al. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of Toll-like receptor 4. Glia 2014;62(1):26–38.
Aucott H, Lundberg J, Salo H, et al. Neuroinflammation in Response to Intracerebral Injections of Different HMGB1 Redox Isoforms. J Innate Immun 2018;10(3):215–27.
Yang L, Wang F, Yang L, et al. HMGB1 a-Box Reverses Brain Edema and Deterioration of Neurological Function in a Traumatic Brain Injury Mouse Model. Cell Physiol Biochem 2018;46(6):2532–42.
Wang D, Liu K, Wake H, Teshigawara K, Mori S, Nishibori M. Anti-high mobility group box-1 (HMGB1) antibody inhibits hemorrhage-induced brain injury and improved neurological deficits in rats. Sci Rep 2017;7:46243.
Haruma J, Teshigawara K, Hishikawa T, et al. Anti-high mobility group box-1 (HMGB1) antibody attenuates delayed cerebral vasospasm and brain injury after subarachnoid hemorrhage in rats. Sci Rep 2016;6:37755.
Musumeci D, Roviello GN, Montesarchio D. An overview on HMGB1 inhibitors as potential therapeutic agents in HMGB1-related pathologies. Pharmacol Ther 2014;141(3):347–57.
Robbins N, Koch SE, Tranter M, Rubinstein J. The history and future of probenecid. Cardiovasc Toxicol 2012;12(1):1–9.
Xiong XX, Gu LJ, Shen J, et al. Probenecid protects against transient focal cerebral ischemic injury by inhibiting HMGB1 release and attenuating AQP4 expression in mice. Neurochem Res 2014;39(1):216–24.
Sun N, Shen Y, Han W, et al. Selective sphingosine-1-phosphate receptor 1 modulation attenuates experimental intracerebral hemorrhage. Stroke 2016;47(7):1899–906.
Lu L, Barfejani AH, Qin T, Dong Q, Ayata C, Waeber C. Fingolimod exerts neuroprotective effects in a mouse model of intracerebral hemorrhage. Brain Res 2014;1555:89–96.
Li H, Zhou X, Li Y et al. The selective sphingosine 1-phosphate receptor 1 modulator RP101075 improves microvascular circulation after cerebrovascular thrombosis. FASEB J 2019;fj201900282R.
Bleck TP. Historical aspects of critical care and the nervous system. Crit Care Clin 2009;25(1):153–64, ix.
Korbakis G, Bleck T. The evolution of neurocritical care. Crit Care Clin 2014;30(4):657–71.
Marcolini EG, Seder DB, Bonomo JB, et al. The present state of neurointensivist training in the united states: a comparison to other critical care training programs. Crit Care Med 2018;46(2):307–15.
Donkin JJ, Vink R. Mechanisms of cerebral edema in traumatic brain injury: therapeutic developments. Curr Opin Neurol 2010;23(3):293–9.
Askenase MH, Sansing LH. Stages of the inflammatory response in pathology and tissue repair after intracerebral hemorrhage. Semin Neurol 2016;36(3):288–97.
Acknowledgments:
Dr. Halstead and Dr. Geocadin do not have any commercial support. Dr. Halstead is supported in part by an educational fellowship from the American Academy of Neurology. Dr. Geocadin is supported in part by a grant from the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Halstead, M.R., Geocadin, R.G. The Medical Management of Cerebral Edema: Past, Present, and Future Therapies. Neurotherapeutics 16, 1133–1148 (2019). https://doi.org/10.1007/s13311-019-00779-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-019-00779-4