
Abstract
Sleep is a behavioral phenomenon conserved among mammals and some invertebrates, yet the biological functions of sleep are still being elucidated. In humans, sleep time becomes shorter, more fragmented, and of poorer quality with advancing age. Epidemiologically, the development of age-related neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease is associated with pronounced sleep disruption, whereas emerging mechanistic studies suggest that sleep disruption may be causally linked to neurodegenerative pathology, suggesting that sleep may represent a key therapeutic target in the prevention of these conditions. In this review, we discuss the physiology of sleep, the pathophysiology of neurodegenerative disease, and the current literature supporting the relationship between sleep, aging, and neurodegenerative disease.
Similar content being viewed by others

Introduction
Sleep changes dramatically throughout the developmental and aging process, and sleep disruption is a common feature of many neurodegenerative diseases, such as Alzheimer’s and Parkinson’s. Interestingly, emerging epidemiological data report that sleep disruption during mid-life may influence risk of dementia in the decades that follow, suggesting that sleep may be causally linked to the development of pathology underlying neurodegenerative conditions. In parallel, recent studies carried out in animal models and human subjects suggest novel mechanistic links between sleep, sleep disruption, and the age-related neuropathology. These studies raise the intriguing possibility that sleep may represent a pivotal therapeutic target for the prevention of age-related neurodegeneration. In this review, we discuss the normal physiology of sleep, the pathophysiology of sleep disruption, and evidence supporting a bidirectional relationship between neurodegeneration and sleep disruption. We also review the evidence for pharmacological and nonpharmacological therapies for modulating sleep that are either in development or currently available for subjects experiencing sleep disturbances.
Overview of Physiological Functions of Sleep
Sleeping and waking reflect 2 distinct behavioral states observed in nearly all living organisms, including humans, birds, fish, and reptiles; sleep-like states are similarly observed in invertebrate organisms such as flies and bees [1]. Although differences between waking and sleep appear obvious to the observer, less obvious are the distinct neurophysiological states that occur cyclically through the course of a single nights’ sleep. Several specific anatomically and chemically defined forebrain and brainstem structures trigger and maintain episodes of sleep and wakefulness [2]. Sleep is assessed most directly via polysomnography, a technique including electroencephalography (EEG), electromyography (EMG), and electrooculography (EOG), routinely used in human and animals to define different sleep stages. In the following paragraphs, we will briefly discuss the sleep/wake cycles which alternate in nearly all living organisms.
Overview of Sleep/Wake Stages
Wakefulness
Wakefulness is a behavioral state defined by voluntary motor activation and responsiveness to internal and external stimuli [3]. During wakefulness, the EEG is characterized by low-amplitude high-frequency beta (14-30 Hz) activity, whereas the EMG shows variable amounts of muscle activity [3, 4]. Wakefulness in maintained through the activity of wake-promoting brain regions, including the cholinergic nuclei of the pontine tegmentum, the adrenergic locus coeruleus, and the serotonergic raphe nuclei. Other regions, including the brainstem, the posterior hypothalamus, and the basal telencephalon, also play a role in the generation of wakefulness. The wake-promoting pathways ascend through the paramedian region of the midbrain, splitting into a dorsal pathway into the thalamus and a large ventral pathway that innervates the hypothalamus, basal forebrain, and cortex [3].
Sleep
Based on EEG recordings, sleep is readily distinguishable from other states of altered consciousness, such as coma and anesthesia, in that it is easily reversible and self-regulating [4]. The American Academy of Sleep Medicine categorizes sleep/wake states into wakefulness (stage W, defined above), 3 nonrapid eye movement (NREM) sleep stages (N1, N2, N3), and REM sleep (R stage).
Stage N1 sleep, termed transitional or light sleep, occurs following wakefulness and is characterized by low-voltage and fast EEG activity, including theta (4-8 Hz) activity and low-amplitude beta (> 13 Hz) activity, coupled with slow eye movements and variable EMG amplitude. Stage N1 is very short and lasts about 1 to 7 min [5, 6]. Stage N2 sleep, also termed intermediate sleep, typically follows stage N1 and is characterized predominantly by theta (4-8 Hz) activity and occasional bursts of faster activity, coupled with no eye movement and a tonically low EMG activity. In addition, stage N1 may show minimal alpha (8-13 Hz) and delta (0-4 Hz) activity, as well as the presence of K complexes (large amplitude waveforms) and sleep spindles (11-16 Hz bursts). N2 sleep is accompanied by progressive diminution of peripheral physiological and metabolic function, including blood pressure, gastrointestinal secretions, and cardiac activity. Stage N2 lasts about 20 min [5, 6]. Stage N3 sleep, termed deep or slow wave sleep, is characterized by high-amplitude slow waves dominated by delta (0-4 Hz) activity as well as further reductions in muscle activity. N3 has the highest threshold for arousal, far greater than the threshold during N1 and N2 sleep [5, 6].
Stage REM (R) sleep, known as paradoxical or active sleep, is characterized by low-amplitude, mixed-frequency EEG theta (4-8 Hz) intermixed with alpha (8-13 Hz) waves, coupled with pronounced rapid eye movements and muscle atonia. Compared with the suppression of physiological and metabolic activity observed in NREM sleep, activity during stage R sleep is greater. For example, blood pressure and heart rate increase and show intermittent fluctuations; respiration becomes irregular, and brain oxygen consumption increases. Stage R is associated with dreaming, penile erections in men, and clitoral engorgement in women [5, 6].
Sleep Cycles
In healthy adults, sleep progresses sequentially from N1, N2, to N3 sleep, then back to REM sleep to complete a sleep cycle over the course of 60 to 120 min, with several sleep cycles occurring throughout the night. Early in the night, the proportion of N3 to REM sleep is greater, while later in the night, the contribution of REM sleep to the cycle increases. N3 sleep accounts for between 10 and 25% of total sleep time, whereas REM sleep typically comprises 20 to 25% of the sleep period [7].
Sleep–Wake Regulation
Wakefulness and sleep are regulated by 2 overlaid processes which interact to determine the time of sleep onset and cessation, as well as the stability of waking neurocognitive function [8,9,10]. The homeostatic sleep drive (termed process S) is cued by the duration of prior wakefulness, whereas the circadian sleep drive (termed process C) is cued by the light/dark cycle. The homeostatic process reflects the drive for sleep that increases during wakefulness and dissipates during sleep, a process that is proposed to correspond to the accumulation and dissipation of a putative “sleep substance” [9, 10]. The circadian process reflects daily rhythms in physiological function and behavior which are entrained by the environmental light–dark cycle. Just as discrete brain regions promote wakefulness, sleep-promoting brain regions, including the anterior hypothalamus and the ventrolateral preoptic nucleus, regulate sleep onset [11,12,13]. When homeostatic sleep drive increases above a certain threshold, sleep is triggered; when it decreases below another threshold, wakefulness is invoked. The circadian process represents daily oscillatory modulation of these thresholds. The rapid switching of the brain between 2 generally stable arousal states is thought to be accomplished through the opposing inhibitory actions of sleep-promoting and wake-promoting regions on one another, termed the “Flip-Flop Switch” model of sleep regulation [14] (for more details on this model, see Saper et al. [15]).
Anesthesia and Sedation
Like sleep, general anesthesia and sedation are associated with altered levels of arousal. Although sleep differs from these states in its reversibility and self-regulation [16], sleep and anesthesia can share electroencephalographic features and in the brain regions activated or inhibited in either state [17]. Slow delta (0.5-4 Hz) activity is a feature both of deep N3 sleep as well as general anesthesia [18]. However, different anesthetics can produce distinct EEG signatures. For example, slow delta and alpha activities are the predominant EEG signatures of the general anesthetic propofol, as well as intravenous GABAA receptor-targeting anesthetics such as benzodiazepines and etomidate. Delta, theta, and alpha activities also characterize EEGs during sevoflurane, desflurane, and isoflurane anesthesia. Dexmedetomidine, an α2 adrenergic agonist sedative, is associated with delta and spindle activity on EEG, which closely resembles that of N2 sleep. Isoelectricity (suppression) is observed when high doses of anesthetics such as sevoflurane and propofol are administered [18].
Physiological Functions of Sleep
The physiological function of sleep has remained the subject of avid scientific interest for centuries, yet, until recently, these functions remained speculative and have historically been best appreciated when the physiological, cognitive, and behavioral consequences of sleep disruption or deprivation were observed. A central role for sleep in the process of memory consolidation has been established through studies carried out both in animals and human subjects. More recently, a role for sleep in the clearance of interstitial metabolic wastes through the activity of the glymphatic system has been described primarily in rodent studies. For the purpose of this review, in addition to the well-known physiological functions of sleep including modulation of pain and immune, metabolic, and cardiovascular function, we will focus primarily on its roles in memory consolidation and on the clearance of interstitial metabolic waste products, including amyloid-β and tau, that accumulate during wakefulness.
Learning and Memory Consolidation
Sleep plays a critical role in memory consolidation, whereas sleep disruption before or after a learning task significantly impairs learning and memory. Chronic sleep deprivation involving complete loss of sleep and chronic sleep restriction involving partial sleep loss over several days to years have been used to define the effects of sleep loss on cognitive function. A consistent finding in these studies is impairment of attention evidenced by slow reaction time and increased lapses during simple vigilance tests. Meta-analysis demonstrates that acute sleep deprivation impairs a wide array of cognitive functions, including substantial impacts on sustained attention and working memory tasks [19]. In one representative study, a single night of total sleep deprivation impaired objective, but not self-estimated working memory performance in young adult women compared with 8 h of sleep opportunity [20]. Ongoing sleep restriction can have similar effects to acute sleep deprivation: restricting sleep to 6 h per night can lead to significant impairment in psychomotor vigilance performance. If prolonged for up 2 weeks, it can reach impairment levels that are comparable to about 2 nights of total sleep deprivation [21]. Sleep restriction to 5 h per night causes increasing impairment in psychomotor vigilance performance, whereas sleep restriction to less than 4 h per night can lead to continued degradation in vigilance performance [21, 22]. Following 5 nights of sleep restricted to 4 h in healthy adults between 22 and 45 years of age, behavioral alertness and subjective sleepiness did not fully recover even with an extended night of sleep recovery [23]. In addition to effects on cognitive function, chronic sleep disruption, particularly when associated with obstructive sleep apnea (OSA) and insomnia, is also associated with several chronic disease states, which will be discussed later in this review.
The role of sleep in the process of memory consolidation is perhaps the most well-established physiological role of sleep, with memory encoding occurring during wakefulness and memory consolidation taking place during subsequent episodes of sleep, enabling memory retrieval during following periods of wakefulness [24]. However, the ability to consolidate memories during sleep depends on several factors, including the type of memory being integrated and the duration and architecture of ensuing sleep [25]. A number of studies have best established roles for both slow wave sleep [26,27,28,29,30] and REM sleep [31,32,33,34,35] in these processes. In animals, in sleep periods following learning tasks, REM sleep is consistently increased [36,37,38,39,40]. Among students, similar changes in REM sleep patterns were observed during intensive study periods [41]. When REM sleep is pharmacologically increased by carbachol injection into the pontine reticular formation or by sleep deprivation [42], memory is improved. Although REM sleep is historically associated with the processes of memory consolidation, recent findings suggest the importance of NREM sleep in these processes [43,44,45]. For example, following periods of motor task learning, both the duration of NREM sleep [43] and the number of sleep spindles [46] increase. Improved performance in a finger-tapping task correlates with the amount of NREM sleep [45], spindle density [47], and delta power [48]. In human subjects, enhancing slow wave sleep by transcranial electrical stimulation enhances memory consolidation [49], whereas selective deprivation of stage N2 sleep reduced memory improvement for a rotor pursuit task [50].
The cellular substrate of sleep’s role in memory consolidation is reflected in the process of scaling synaptic strength and weakness throughout the processes of waking and sleep, which involve processes of long-term potentiation and long-term depression [51,52,53]. During waking, synaptic strength is enhanced (termed synaptic upscaling), whereas sleep counterbalances this process by weakening synaptic connections (synaptic downscaling) [54,55,56,57]. At the molecular level, gene transcripts and proteins implicated in long-term potentiation are upregulated following extended periods of wakefulness, whereas those implicated in long-term depression are increased following periods of sleep [58, 59].
Glymphatic Pathway Function and Interstitial Waste Clearance
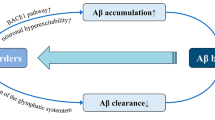
More recently, a role for sleep in the regulation of metabolic waste clearance from the brain has been described by Nedergaard, Iliff, and colleagues. Although the brain reflects only about 2% of the body’s mass, it accounts for about 20% of the body’s overall metabolic output [60]. Compared with wakefulness, metabolic demand during sleep tends to be reduced [61, 62], although this reduction is not uniformly distributed across different sleep stages. This reduction is attributable in part to quiescent “down states” during slow wave activity; brain metabolic activity during REM sleep is not substantively different from that observed during waking. Sleep–wake differences in metabolic activity are believed to underlie diurnal fluctuations in levels of metabolic by-products within the brain interstitium (measured in animals by in vivo microdialysis) or in the cerebrospinal fluid compartment (measured in human subjects by serial CSF sampling). These include lactate and amyloid beta (Aβ) and the microtubule-associated protein tau, which are released in response to synaptic activity [63,64,65,66]. Thus, one proposed role for sleep may be the provision of periods of relative metabolic suppression when metabolic by-products, including pathogenic peptides such as Aβ, may be removed from the brain.
Recent studies conducted in rodents suggest that the clearance of metabolic waste from the brain interstitium during sleep may be more rapid and anatomically organized than previously recognized. These studies demonstrate that during sleep, and under specific anesthetic conditions, CSF moves rapidly into and through the brain parenchyma along perivascular spaces surrounding penetrating arteries to exchange with brain interstitial fluid [67,68,69]. Interstitial solutes, in turn, are cleared along white matter tracts and the deep venous drainage to the subarachnoid CSF compartment where they can then be cleared along CSF reabsorption pathways, including arachnoid villi, the cribriform plate, meningeal lymphatic vessels, or cranial and spinal nerve sheathes [70]. Because it was dependent upon the perivascular astroglial water channel aquaporin-4 (AQP4), this perivascular network that supports CSF–interstitial fluid exchange was termed the “glymphatic” system [67, 71].
Interestingly, both glymphatic exchange and lymphatic drainage appear to be regulated by the sleep–wake cycle. Movement of CSF tracers through brain tissue is more rapid in the sleeping and anesthetized compared with the waking mouse brain; similarly, the clearance of interstitial solutes including Aβ is more rapid from the sleeping and anesthetized compared with the waking mouse brain [72]. Increased CSF–interstitial fluid exchange coincided with a significant expansion of the extracellular space, suggesting that during sleep the physical properties of brain tissue change to support rapid clearance of interstitial solutes and waste. Reduced solute clearance was sensitive to noradrenergic receptor blockade, demonstrating that central noradrenergic tone is one key regulator of glymphatic function. In a second study conducted in mice, lymphatic drainage was more rapid in waking compared with anesthetized animals [73]. These findings suggest that during sleep, glymphatic exchange supports the clearance of solutes and wastes from the brain interstitium to the CSF compartment, while during waking, lymphatic drainage supports the clearance of solutes from the CSF compartment via the deep cervical lymphatic vasculature.
Age-Related Changes in Sleep
Continuous changes in sleep macro- and microarchitecture occur throughout normal human aging. Among these changes are reductions in total sleep time and other measures of sleep quality, including increased sleep latency (the time it takes to fall asleep), reduced sleep efficiency (the amount of time spent asleep vs the amount of time spent in bed), and greater sleep fragmentation [7, 74,75,76]. Interestingly, when good health is maintained throughout the aging process, the trend for declining total sleep time with age tends to cease after age 60, at which point total sleep time plateaus [7]. However, in the presence of comorbidities, age-related sleep changes may be exacerbated. The composition of sleep also changes throughout the aging process, with the proportion of sleep time spent in N1 and N2 sleep increasing and the time spent in N3 (slow wave sleep) declining between early adulthood and old age. A corresponding decline in EEG spectral delta power, sleep spindles and K-complexes, and increasing high-frequency beta power, an indicator of cortical arousal, is commonly observed in older individuals [76,77,78,79]. REM sleep increases between childhood and adolescence, then declines between young adulthood and middle age [7]. Changes in circadian rhythms have also been reported with advancing age, with a decline in the cortisol and melatonin rhythms that entrain day/night activity patterns [80].
Older adults are more likely than other age groups to experience sleep disruption [81], which is attributable to both age-dependent changes in sleep architecture and circadian rhythm, and also to higher rates of sleep disorders such as insomnia and OSA [81]. Although sleep disruption among the elderly is strongly associated with poorer cognitive performance and an increased risk of cognitive decline [82], more recently, results from the Framingham Heart Study and the Atherosclerosis Risk in Communities studies have demonstrated that mid-life sleep disruption predicted the development of dementia in later life [83, 84]. These studies suggest that sleep disruption may not simply be a feature of the aging brain, but that sleep disruption may be a factor rendering the aging brain vulnerable to neurodegenerative processes underlying diseases such as Alzheimer’s disease (AD).
Sleep Disruption in Age-Related Comorbidities
Acute and chronic sleep disruption is a pervasive feature of modern societies due to factors including work demands, family responsibilities, social pressure, and lifestyle choices [85], and more recently, electronic devices such as television, video games, computers, and smartphones. The consequences of sleep disruption span temporal scales between immediate impairments in cognitive function to the long-term changes in the pathogenic processes that may underlie vulnerability to neurodegenerative diseases like AD and Parkinson’s disease (PD).
Insomnia
Insomnia, the most common primary sleep disorder in the general population, is defined as difficulty falling asleep (sleep onset), staying asleep (sleep maintenance), or nonrestorative sleep despite adequate opportunity to sleep; it is associated with impairment in daytime functioning that can be a symptom or cause of several different medical conditions [86,87,88]. Risk factors for insomnia include a large number of genetic predispositions, social (lower socioeconomic status, divorce/separation) and environmental factors, gender (female sex), race (greater risk in African Americans compared with white race), older age, and comorbid medical or mental disorders such as depression, post-traumatic stress disorder (PTSD), and heart disease [89]. Persistent insomnia is associated with an increased risk of development of a psychiatric disorder [90]. Although insomnia and depression are frequently comorbid [91], insomnia confers a greater risk for the development of depression [92,93,94,95,96,97].
Obstructive Sleep Apnea
Sleep apnea is a common sleep disorder in which respiration transiently ceases or decreases considerably in volume during sleep. It can be due to either obstruction of the upper airway (OSA), dysfunction in the neurological drive to breathe (central sleep apnea, Cheyne–Stokes breathing, or secondary to medication or drug use), or their combination (mixed apnea, complex apnea, or obesity-hypoventilation syndrome) [98]. OSA is highly prevalent in the middle-aged population and worsens with age. It is characterized by repetitive transient upper airway obstructions during sleep that result in either complete (apnea) or partial (hypopnea) pauses in breathing [99,100,101,102]. Periods of apnea are accompanied by periods of hypoxemia and by brief electroencephalographic and autonomic arousals that lead to a disruption of normal sleep architecture [101]. OSA is associated with several chronic conditions, including depression, hypertension, risk of stroke and myocardial infarction, and cardiac arrhythmia [103,104,105,106,107]. OSA is also associated with vascular dementia and with an earlier onset of mild cognitive impairment and dementia compared with subjects without OSA [108].
Cardiovascular Effects
Epidemiological evidence indicates that habitually short sleep duration is a risk factor for cardiovascular diseases [109,110,111]. Acute total sleep deprivation causes impaired cardiovascular functions with increased sympathetic activity; for example, restriction of sleep in healthy young adults to 5 h for 1 night is associated with an increase in heart rate of 5 beats/min and in systolic/diastolic blood pressure of 10 mmHg compared with nonsleep-deprived conditions [112]. In a study in which total sleep disruption lasted for 88 h, similar changes in heart rate and systolic/diastolic blood pressure exhibited virtually no signs of recovery on the first day of recovery after sleep deprivation [113]. Consistent with these results, late night sleep deprivation from 4 to 8 a.m. resulted in an increased plasma norepinephrine and epinephrine levels in healthy male participants [114]. Similar to acute total sleep deprivation, continuous sleep restriction over prolonged periods result in changes in heart rate, blood pressure, and measures of cardiac function measures [115, 116]. For example, restriction of sleep 3.6 to 4.5 h per night for 10 days in healthy subjects was associated with increases in heart rate and blood pressure; on the last (10th) day, heart rate was elevated, compared with prerestriction baseline levels by 5 beats/min during sleep periods and by 22 beats/min during sleep deprivation periods, with similar results for blood pressure [113]. Restriction of sleep to < 80% of the habitual duration for 4 weeks in young men resulted in decreased heart rate variability and elevated plasma catecholamine levels [115].
Intensive Care Unit Delirium
Although not considered strictly a cause or consequence of sleep disruption, a frequent and relevant condition encountered in the aging population is postoperative and intensive care unit (ICU) delirium. Delirium, a disturbance of consciousness that develops acutely, tends to fluctuate and is associated with inattention, impaired cognition, and perceptual disturbances which is a prevalent occurrence among patients in the ICU environment [117], an environment that is notable for promoting sleep fragmentation due in part to isolation from natural light/dark cycles, noisy environment, and staff interventions that lead to frequent arousals and awakening [118,119,120]. Among subjects in the ICU, sleep architecture is altered with increased in light sleep and reductions in slow wave and REM sleep. Total sleep time averages between 2.1 and 8.8 h per night and is typically neither continuous nor strictly restricted to night times [118,119,120,121,122]. Most importantly, delirium is frequent among aged populations; a study of 614 patients monitored for delirium over 1 year showed that a majority of patients older than 65 years of age exhibited hypoactive delirium (characterized by decreased psychomotor activity and inattention) at some point during their ICU stay [123]. ICU delirium is associated with functional and cognitive decline [124, 125]; however, whether these outcomes are the result of sleep and circadian disruption in the ICU environment remains unknown and an important avenue for future research. The potential role that age-related changes in sleep play in the development of neurodegenerative diseases and chronic diseases such as insomnia, obstructive sleep apnea, and cardiovascular diseases will be discussed at length in sections to follow.
Sleep–Wake Disruption in the Pathogenesis of Neurodegenerative Disease
In addition to the changes in sleep architecture and circadian rhythm that occur across the lifespan of nondemented individuals [126], pathological changes in sleep often occur in the setting of dementia. Indeed, sleep–wake disturbances are frequently reported as symptomatic features of many neurodegenerative diseases, including AD [127, 128], PD [129,130,131], and chronic traumatic encephalopathy. Sleep disruption is also commonly reported in atypical parkinsonian disorders like progressive supranuclear palsy (PSP), Lewy body dementia, multiple system atrophy (MSA), and corticobasal degeneration [132]. The mechanisms and brain regions involved in these neurodegenerative diseases are diverse, yet the appearance of sleep disturbances across dementias suggests an important relationship between cognitive impairment and sleep.
Alzheimer’s Disease
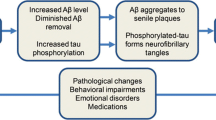
Clinical AD is characterized by progressive memory loss, difficulty completing familiar tasks, trouble understanding spatial relationships, and mood changes. The diagnosis of AD requires the presence of neuritic plaques and neurofibrillary tangles in the brain, composed mostly of Aβ and tau proteins, respectively [133]. Although the severity of cognitive impairment correlates with the burden of neurofibrillary tangles and neuritic plaques [133], these hallmarks of AD can be found in the aging human brain without clinical detection of cognitive impairment, suggesting that individual responses to Aβ and tau aggregation may vary substantially [134, 135]. In patients with AD, complaints of sleep and circadian disruption are common and include insomnia and fragmentation of sleep architecture [136]. These changes result in a reduction in sleep quantity and quality that, along with polypharmacy, can also result in excessive daytime sleepiness. The relationship between the pathophysiology of AD and sleep disruption is complicated by the association of AD with aging and brain atrophy, both of which have also been linked to sleep disruption [137,138,139]. However, recent research has begun to define a relationship between Aβ trafficking in the CNS, aging, and sleep disturbance.
In cognitively intact human subjects, Aβ40 and Aβ42 turnover rates were slowed in the CSF of aged subjects (73.3 ± 6.6 years) in comparison with younger controls (48.0 ± 14.6 years) [140]. A striking 2.5-fold reduction in Aβ half-life was reported between 30- and 80-year-old participants, suggesting that Aβ may have a markedly increased residence time in the aged human CSF. Several studies also report an association between parenchymal Aβ burden and metrics of sleep disruption, including longer sleep latency [141, 142], lower sleep efficiency [143], and worsening sleep quality [144, 145] in subjects without clinical AD.
In a series of studies utilizing serial CSF sampling in human subjects, diurnal fluctuations in CSF Aβ, tau, and alpha-synuclein concentrations have been reported [63]. For example, CSF Aβ levels increase during waking and decline during sleep, a pattern that parallels sleep–wake fluctuation in interstitial Aβ levels measured by microdialysis in rodents [65]. Interestingly, these diurnal fluctuations are blunted in subjects with AD or in rodents in the presence of Aβ pathology [146, 147]. The overnight decline in Aβ levels can be inhibited by sleep deprivation or through specific ablation of slow wave sleep [65, 148, 149]. More recently, an imaging study using an Aβ-binding PET ligand demonstrated that 1 night of sleep deprivation increases parenchymal Aβ burden by 5% in 20 healthy control subjects [150]. These results suggest a clear causal relationship between sleep–wake regulation and CNS Aβ, tau, and alpha-synuclein dynamics independent of the effects of aging. Conversely, other studies report that cortical Aβ disrupts slow wave activity in human subjects, suggesting that sleep disruption and Aβ burden may have bidirectional effects [151].
Parkinson’s Disease
PD is a neurodegenerative disease characterized by a symptom constellation that includes tremor, muscular rigidity, shuffling gait, bradykinesia, and postural instability [152]. Pathological examination of the PD brain typically reveals atrophy of multiple brain regions [153], particularly in the substantia nigra [154]. Histological features of PD include cytoplasmic inclusions called Lewy bodies, which are composed of the protein alpha-synuclein [155, 156] and hyperphosphorylated tau species, both of which are thought to propagate through the CNS in a stereotyped anatomical pattern as the disease progresses [157]. Sleep disruption is also common in PD, with 40 to 60% of patients reporting sleep disturbance [130, 131]. Similar to patients with AD, PD patients report insomnia and psychiatric symptoms that interfere with the quality and quantity of sleep. Furthermore, PD patients often experience disorders related to motor function, such as rapid eye movement behavior disorder (RBD), restless leg syndrome, akinesia, and sleep-disordered breathing, all of which cause significant disruption to normal sleep behavior [158]. Interestingly, patients diagnosed with RBD are at significantly increased risk of developing dementia [159]. Whether RBD is simply a symptom of prodromal PD or the disruption of sleep accelerates the progression of dementia remains unclear.
Although these motor dysfunctions result from the primary pathophysiology of PD, they likely play a secondary role in sleep disruption by either fragmenting sleep or increasing sleep latency. Interestingly, evidence of disrupted circadian rhythms has also been reported in patients with PD, suggesting that the pathophysiology of PD may directly impair sleep rhythms in patients. This includes progressive worsening of motor functions throughout the daytime [160], nighttime dysfunction of cardiovascular regulation [161], and reversal of the circadian blood pressure rhythm [162]. The pattern and magnitude of melatonin secretion is also disrupted in PD patients which may contribute to circadian disturbances [163,164,165]. These findings are further supported by evidence of Lewy bodies and phosphorylated tau deposition in the suprachiasmatic nucleus, suggesting that the central circadian rhythm may be altered in PD [166]. To date, studies focused on the relationship between the pathophysiology of PD and sleep disruption in humans have remained largely associative, but a multivariate relationship between the severity of motor dysfunction, iatrogenic dysregulation of the circadian rhythm through dopaminergic therapy, and possible direct pathophysiological effects on the circadian rhythm is beginning to emerge.
Atypical Parkinsonism
Atypical parkinsonism describes a group of neurodegenerative diseases featuring a wide phenotypic spectrum within and among subgroups. Broadly, this includes DLB, MSA, PSP, and corticobasal degeneration. DLB and MSA are characterized histopathologically by aggregation of alpha-synuclein [167, 168], whereas PSP and corticobasal degeneration feature aggregation of hyperphosphorylated tau species [169, 170]. Unlike AD and PD, the relationship between the pathophysiology of the neurodegenerative disease and sleep disturbances reported by patients with atypical parkinsonism has received relatively little attention, likely owing to the rarity of these diseases. Still, there exists a high incidence of sleep disorders in these diseases, including insomnia, excessive daytime sleepiness, RBD, sleep-disordered breathing, and nighttime stridor [132]. Although there is an overlap between symptoms in disease subtypes, some sleep disturbances appear to be more specific to subtypes than others. For instance, a strong link to RBD has been demonstrated in dementia with Lewy bodies and multiple system atrophy [171].
Glymphatic Dysfunction in the Aging Brain and Neurodegenerative Diseases
Neurodegenerative diseases are characterized by the misaggregation of extracellular and intracellular proteins, now widely believed to propagate in a prion-like manner between neighboring or synaptically connected cells via the extracellular space [172]. Thus, the role of glymphatic pathway function, which supports the clearance of interstitial proteins during sleep in the pathogenesis of neurodegenerative disease, is a subject of substantial ongoing research interest [69, 72, 173].
Age-Related Impairment of Glymphatic Function
Glymphatic function supports the perivascular clearance of interstitial solutes including Aβ, tau, and alpha-synuclein. Aβ clearance is dependent upon the perivascular astroglial water channel AQP4 [173] and is more rapid in the sleeping compared with the waking brain [72]. Aqp4 gene disruption in a mouse model of amyloidosis accelerates Aβ plaque deposition and the development of neurocognitive decline [174]. Since the initial characterization of the glymphatic system, several studies have demonstrated impairment in CSF–interstitial fluid exchange in aging, or in the presence of injury. In aging mice [175], glymphatic function was slowed, including a reduction in the clearance of Aβ. In a mouse model of traumatic brain injury, glymphatic function was slowed a month after injury, while impairment of glymphatic function through Aqp4 gene deletion exacerbated phosphorylated tau accumulation [176]. In a mouse model of microinfarcts, diffuse ischemic lesions impaired glymphatic function [177]. In each case, age- or injury-related impairment of glymphatic pathway function was associated with the loss of perivascular AQP4 localization in reactive astrocytes. Importantly, in human frontal cortical tissue, such a loss in perivascular AQP4 localization is observed in AD subjects and is associated with increasing amyloid and tau pathology [178]. These findings suggest that impairment of glymphatic function may be one feature of the aging or injured brain that render it vulnerable to neurodegeneration.
In considering the relationship between sleep disruption, glymphatic function, and neurodegenerative processes, it is important to distinguish between glymphatic capacity and glymphatic activity. In the preclinical studies outlined above, glymphatic function was evaluated in anesthetized animals [175,176,177], in which sleep–wake differences in glymphatic function are masked. However, both aging and brain injury are also associated with sleep disruption, as detailed above. Thus, overlapping contributions of reduced glymphatic capacity (glymphatic function when sleep can be achieved) and opportunity (due to sleep disruption) likely result in declines in overall glymphatic activity (the composite of capacity and opportunity).
Detecting Vulnerability to Neurodegenerative Processes by Evaluating Glymphatic Function
Although the glymphatic system remains an active subject of research and debate, with our understanding of its underlying mechanisms still being refined [179,180,181], there is much interest in developing methods for noninvasive imaging of glymphatic function in rodents and humans. Because glympahtic function contributes to the clearance of interstitial solutes such as A[beta] and tau, age- and injury-associated impairment of glymphatic function may lay mechanistically upstream of pathological changes such as Aβ or tau deposition. If so, then detection of glymphatic pathway dysfunction might provide a measure of vulnerability to protein misaggregation or widespread aggregate propagation. Thus, the development of biomarkers that reflect glymphatic function may have substantial clinical utility.
After the initial characterization of the glymphatic system using fluorescent tracers and multiphoton microscopy, steps were quickly taken to develop clinically relevant MRI-based approaches to assess glymphatic pathway function in the rodent by dynamic contrast-enhanced-MRI following intrathecal gadolinium-based contrast agent injection [68, 182]. Shortly thereafter, an elegant case study was published by Eide and Ringstad [183] recapitulating these preclinical studies in a human subject with suspected CSF leakage. Through serial imaging of the brain after intrathecal infusion of gadobutrol in this subject, the authors concluded that glymphatic ISF–CSF exchange exists in the human as well as the rodent brain. Further work by Eide and Ringstad has begun to define CSF–ISF exchange in human subjects [183,184,185]. Interestingly, these studies have identified reduced contrast clearance in subjects with normal pressure hydrocephalus, which provides proof of principle for measuring glymphatic function in human disease [184]. Because this technique relies on intrathecal contrast, however, it may face barriers to widespread clinical adoption in detecting vulnerability to neurodegenerative processes.
To date, several noninvasive MRI-based approaches have been developed to measure CSF flow dynamics noninvasively. Real-time phase contrast MRI has been deployed to investigate major effectors of CSF flow and may provide additional insight into abnormalities in CSF flow that precede neurodegenerative disease [184, 186]. More recently, 2 studies in rodent have used diffusion tensor MRI and arterial spin labeling with multiple echo times to measure physiological parameters that are directly relevant to the glymphatic system, including water exchange at the blood–brain interface [187] and CSF flow in the perivascular space [188]. MRI can also be used to measure the diffusivity of water in the human brain to compare apparent diffusion coefficients between waking and sleeping states to assess the magnitude of ISF–CSF exchange in human subjects [189]. This study provides an important noninvasive tool for measuring glymphatic function in cognitively normal and impaired subjects. Once these studies have been recapitulated in human subjects and validated against gold-standard contrast-based measures of CSF–ISF exchange, these MRI techniques may aid investigators in identifying changes in the glymphatic system that reflect vulnerability to neurodegeneration.
To date, AD has received the most attention in the development of diagnostic biomarkers that are detectable in the CSF. Through multiple multicenter and prospective studies in AD patients over the last 2 decades, a highly sensitive and specific signature of elevated total and phospho-tau protein and lowered Aβ42 in the CSF has been identified [190,191,192,193]. The reduction in CSF Aβ42 has been widely interpreted as an increase in Aβ42 deposition in the brain and reduced clearance into the CSF compartment. Several other similar trends also exist in the literature that may provide biomarkers upstream of widespread parenchymal protein aggregation. This includes the study by Roh et al. [147] that described a reduction in diurnal ISF [Aβ] and [lactate] fluctuation with advancing stages in a mouse model of AD. In light of this observation, it is possible that diurnal fluctuation of CNS-derived solutes such as Aβ, tau, or alpha-synuclein within the CSF, or perhaps within the plasma, may serve as a biomarker for the dynamics of ISF–CSF exchange that reflects vulnerability to aggregation of solutes in the brain parenchyma that drive neurodegeneration. Indeed, this hypothesis is corroborated by a study that found reduced CSF levels of Aβ40, Aβ42, and neurogranin in subjects with OSA [148], suggesting that changes in sleep-related exchange kinetics may be detectible in peripheral compartments. Clearly, more studies are needed to determine if changes in the magnitude of diurnal fluctuation of parenchymally derived solutes in the CSF predict cognitive decline across multiple neurodegenerative diseases.
Addressing Sleep–Wake Disruption in the Prevention of Neurodegenerative Disease
Sleep disruption is prevalent within the general population and is linked to a myriad of chronic health problems [194]. Although their impact is spread across all age groups, ethnicities, and degrees of health status, they are more prevalent in elderly populations in which emerging evidence suggests they may contribute to pathogenic processes underlying neurodegenerative conditions such as AD and PD. Age-related sleep disturbances are often independent of any medical condition or primary sleep disorder but are part of the aging process itself. This includes the decline in sleep quantity and quality generally, and particularly in slow wave activity among the elderly [195]. The epidemiological association of mid-life sleep parameters with the development of dementia in the decades that follow [83, 84] suggests that sleep may be a modifiable risk factor in the primary prevention of neurodegenerative disease. Thus, approaches to improve or maintain sleep, and in particular to maintain slow wave sleep in throughout the aging process, may be of substantial future clinical interest.
Pharmacological Approaches to Enhance Slow Wave Sleep
Sedative-hypnotic agents, particularly benzodiazepines (i.e., triazolam and temazepam) and nonbenzodiazapines (such as zolpidem, zaleplon, and eszopiclone), are the most widely used medications for the short-term treatment of sleep disturbances. However, they are associated with increased incidence of sedation, confusion, anterograde amnesia, daytime sleepiness, and rebound insomnia and, thus, may not be appropriate for use in elderly populations [196]. However, several pharmacological agents that enhance slow wave sleep and slow wave activity in both healthy individuals and patients are already in clinical use. For example, GAT-1 inhibitors, such as tiagabine [197]; GABAA agonists which bind to the extrasynaptic GABAA receptor [198] such as gaboxadol; GABAB modulators such as GHB [199]; and 5HT2A antagonists such as seganserin and eplivanserin [200, 201] increase slow wave sleep. Moreover, seganserin, a 5HT2A antagonist, increased slow wave sleep during NREM sleep and significantly enhanced low frequency and theta activity [200]. Another selective 5HT2A antagonist, eplivanserin, was shown to enhance slow wave sleep and reduce stage 2 sleep, without affecting subjective sleep quality in healthy volunteers [201, 202]. Several of these drugs have been demonstrated to be effective in treating insomnia [203,212,213,206].
As previously mentioned, melatonin rhythms that entrain the sleep–wake cycle decline with age [80]. In the USA, exogenous melatonin can be purchased without a prescription for promoting sleep quality; however, many studies on the efficacy of melatonin have reported conflicting findings [207, 208]. These conflicting reports likely reflect the variability in the dosing of melatonin, the severity of insomnia, and age of study subjects. In aging subjects, the use of sustained-release exogenous melatonin increases sleep efficiency and fast-acting melatonin improves sleep initiation [209,218,211]. Importantly, the beneficial effects of melatonin on these sleep parameters do not appear to translate to patients with AD and PD [212, 213]. In all cases, avoidance of supraphysiological melatonin dosing is imperative to avoid further disruption of the circadian rhythm [214].
Modulation of central adrenergic tone to enhancing sleep quality has been studied in populations with PTSD [215, 216] and traumatic brain injury [217]. Although most clinical trials have focused on the effect of prazosin (an alpha 1 adrenergic receptor antagonist) in reducing PTSD-related nightmares, it is possible that prazosin may help reduce sleep fractionation in the general population, thereby increasing opportunity for ISF–CSF exchange, in aging patients without PTSD. Given the widespread use of prazosin to treat hypertension in aging populations, a prospective study considering sleep parameters and nocturnal use of prazosin would provide valuable data in evaluating this possibility. Furthermore, adrenergic inhibition has been shown to increase the magnitude of ISF–CSF exchange in rodents, which may provide further rationale for targeting adrenergic inhibition in aging humans [72] (unpublished data).
Nonpharmacological Approaches to Improving Sleep
Because of the risk for sedating medication side effects, behavioral strategies are employed clinically as the first-line treatment for sleep disturbances in elderly population. This approach includes maintaining regular bedtimes and rising times, limiting daytime napping, and restricting time in bed. In addition, dietary recommendations which include establishing consistent meal times; avoiding alcohol, nicotine, and caffeine; and emptying the bladder before bedtime are strongly recommended. Maintenance of a sleep-promoting sleep environment, avoiding overly hot or cold, bright or noisy environments, also promotes healthy sleep [218]. Cognitive behavioral therapy is as effective as prescription medications for the treatment of chronic insomnia [219]. In OSA patients, positive airway pressure treatment is associated with increased slow wave activity, whereas greater improvement in OSA was associated with greater decreases in CSF Aβ and tau levels [220]. A recent study [221] showed rocking stimulation shortens sleep latency to N2 stage sleep, whereas sleep latencies to N1, N3, and REM sleep were not affected compared with the stationary position. They also found that the rocking stimulation decreases the proportion of sleep time spent in N1 and N2 sleep due to a significant increase in N3 sleep. Most importantly, when the authors investigated the effects of rocking stimulation on waking behavior and memory, they found participants in the rocking stimulation had better memory performance after the rocking night evidenced by the significant decrease in the number of errors and an increase of correct responses. Interestingly, another new study applied rocking stimulation for the first time in a transgenic mouse model lacking functional otoliths making them unable to encode linear acceleration to investigate the involvement of the vestibular system in mediating the effects of rocking stimulation. This study replicated findings in humans and showed that rocking stimulation at a frequency of 1.0 Hz in mice reduced sleep-onset latency, increases NREM sleep time, and reduces active wakefulness when compared with the stationary condition [222].
In conclusion, a combination of nonpharmacologic and pharmacologic interventions should also be considered when treating older adults with sleep disorders in order to help them sleep better at night and function better in their daily activities.
References
Cirelli, C. & Tononi, G. Is sleep essential? PLoS Biol 6, e216, https://doi.org/10.1371/journal.pbio.0060216 (2008).
Rosenwasser, A. M. Functional neuroanatomy of sleep and circadian rhythms. Brain Res Rev 61, 281–306, https://doi.org/10.1016/j.brainresrev.2009.08.001 (2009).
Scammell, T. E., Arrigoni, E. & Lipton, J. O. Neural circuitry of wakefulness and sleep. Neuron 93, 747–765, https://doi.org/10.1016/j.neuron.2017.01.014 (2017).
Fuller, P. M., Gooley, J. J. & Saper, C. B. Neurobiology of the sleep-wake cycle: sleep architecture, circadian regulation, and regulatory feedback. J Biol Rhythm 21, 482–493, https://doi.org/10.1177/0748730406294627 (2006).
Berry, R. B., Brooks, R., Gamaldo, C., et al. AASM scoring manual updates for 2017 (Version 2.4). J Clin Sleep Med 13, 665–666, https://doi.org/10.5664/jcsm.6576 (2017).
Malhotra RK, Avidan AY. in Atlas of sleep medicine (ed Saunders) Ch. 3, 77–99. Elsevier Inc., 2014.
Ohayon, M. M., Carskadon, M. A., Guilleminault, C. & Vitiello, M. V. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: developing normative sleep values across the human lifespan. Sleep 27, 1255–1273 (2004).
Khalsa, S. B. S., Jewett, M. E., Duffy, J. F. & Czeisler, C. A. The timing of the human circadian clock is accurately represented by the core body temperature rhythm following phase shifts to a three-cycle light stimulus near the critical zone. J Biol Rhythm 15, 524–530, https://doi.org/10.1177/074873040001500609 (2000).
Van Dongen, H. P. & Dinges, D. F. Investigating the interaction between the homeostatic and circadian processes of sleep-wake regulation for the prediction of waking neurobehavioural performance. J Sleep Res 12, 181–187 (2003).
Borbely, A. A. A two process model of sleep regulation. Hum Neurobiol 1, 195–204 (1982).
von Economo, C. Sleep as a problem of localization. J Nerv Ment Dis 71, 249–259 (1930).
Nauta, W. J. Hypothalamic regulation of sleep in rats; an experimental study. J Neurophysiol 9, 285–316, https://doi.org/10.1152/jn.1946.9.4.285 (1946).
Saper, C. B., Lu, J., Chou, T. C. & Gooley, J. The hypothalamic integrator for circadian rhythms. Trends Neurosci 28, 152–157, https://doi.org/10.1016/j.tins.2004.12.009 (2005).
Brown, R. E., Basheer, R., McKenna, J. T., Strecker, R. E. & McCarley, R. W. Control of sleep and wakefulness. Physiol Rev 92, 1087–1187, https://doi.org/10.1152/physrev.00032.2011 (2012).
Saper, C. B., Scammell, T. E. & Lu, J. Hypothalamic regulation of sleep and circadian rhythms. Nature 437, 1257–1263, https://doi.org/10.1038/nature04284 (2005).
Marano, G., Traversi, G., Catalano, V., et al. Sleep regulation: a bidirectional interaction between brain and the endocrine system. Clin Neuropsychiatry 8, 192–203 (2011).
Song, J., Um, Y.H., Kim, T.W., Kim, S.M., Kwon, S.Y., & Hong, S.C. Sleep and anesthesia. Sleep Med Res 9, 11–19 (2018).
Akeju, O. & Brown, E. N. Neural oscillations demonstrate that general anesthesia and sedative states are neurophysiologically distinct from sleep. Curr Opin Neurobiol 44, 178–185, https://doi.org/10.1016/j.conb.2017.04.011 (2017).
Lim, J. & Dinges, D. F. A meta-analysis of the impact of short-term sleep deprivation on cognitive variables. Psychol Bull 136, 375–389, https://doi.org/10.1037/a0018883 (2010).
Rangtell, F. H. et al. A single night of sleep loss impairs objective but not subjective working memory performance in a sex-dependent manner. J Sleep Res, https://doi.org/10.1111/jsr.12651 (2018).
Van Dongen, H. P., Maislin, G., Mullington, J. M. & Dinges, D. F. The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep 26, 117–126 (2003).
Belenky, G. et al. Patterns of performance degradation and restoration during sleep restriction and subsequent recovery: a sleep dose-response study. J Sleep Res 12, 1–12 (2003).
Banks, S., Van Dongen, H. P., Maislin, G. & Dinges, D. F. Neurobehavioral dynamics following chronic sleep restriction: dose-response effects of one night for recovery. Sleep 33, 1013–1026 (2010).
McGaugh, J. L. Memory—a century of consolidation. Science 287, 248–251 (2000).
Stickgold, R. & Walker, M. P. Sleep-dependent memory triage: evolving generalization through selective processing. Nat Neurosci 16, 139–145, https://doi.org/10.1038/nn.3303 (2013).
Ji, D. & Wilson, M. A. Coordinated memory replay in the visual cortex and hippocampus during sleep. Nat Neurosci 10, 100–107, https://doi.org/10.1038/nn1825 (2007).
Massimini, M. et al. Triggering sleep slow waves by transcranial magnetic stimulation. Proc Natl Acad Sci U S A 104, 8496–8501, https://doi.org/10.1073/pnas.0702495104 (2007).
Peigneux, P. et al. Are spatial memories strengthened in the human hippocampus during slow wave sleep? Neuron 44, 535–545, https://doi.org/10.1016/j.neuron.2004.10.007 (2004).
Rasch, B., Buchel, C., Gais, S. & Born, J. Odor cues during slow-wave sleep prompt declarative memory consolidation. Science 315, 1426–1429, https://doi.org/10.1126/science.1138581 (2007).
Wilson, M. A. & McNaughton, B. L. Reactivation of hippocampal ensemble memories during sleep. Science 265, 676–679, https://doi.org/10.1126/science.8036517 (1994).
Poe, G. R., Walsh, C. M. & Bjorness, T. E. Cognitive neuroscience of sleep. Prog Brain Res 185, 1–19, https://doi.org/10.1016/B978-0-444-53702-7.00001-4 (2010).
Portell Cortes, I. & Morgado Bernal, I. [Learning and subsequent paradoxical sleep]. Arch Neurobiol (Madr) 51, 305–315 (1988).
Smith, C. Sleep states and learning: a review of the animal literature. Neurosci Biobehav Rev 9, 157–168 (1985).
Smith, C. Sleep states, memory processes and synaptic plasticity. Behav Brain Res 78, 49–56 (1996).
Smith, C. Sleep states and memory processing in rodents: a review. Sleep Med Clinics Sleep Med Clin 6, 59–70 (2011).
Portell-Cortes, I., Marti-Nicolovius, M., Segura-Torres, P. & Morgado-Bernal, I. Correlations between paradoxical sleep and shuttle-box conditioning in rats. Behav Neurosci 103, 984–990 (1989).
Smith, C. Sleep states and memory processes in humans: procedural versus declarative memory systems. Sleep Med Rev 5, 491–506, https://doi.org/10.1053/smrv.2001.0164 (2001).
Smith, C. & Peters, K. R. Sleep, memory, and molecular neurobiology. Handb Clin Neurol 98, 259–272, https://doi.org/10.1016/B978-0-444-52006-7.00017-4 (2011).
Smith, C. & Wong, P. T. Paradoxical sleep increases predict successful learning in a complex operant task. Behav Neurosci 105, 282–288 (1991).
Smith, C., Young, J. & Young, W. Prolonged increases in paradoxical sleep during and after avoidance-task acquisition. Sleep 3, 67–81 (1980).
Smith, C. & Lapp, L. Increases in number of REMS and REM density in humans following an intensive learning period. Sleep 14, 325–330 (1991).
Wetzel, W., Wagner, T. & Balschun, D. REM sleep enhancement induced by different procedures improves memory retention in rats. Eur J Neurosci 18, 2611–2617 (2003).
Fogel, S. M. & Smith, C. T. Learning-dependent changes in sleep spindles and stage 2 sleep. J Sleep Res 15, 250–255, https://doi.org/10.1111/j.1365-2869.2006.00522.x (2006).
Laventure, S. et al. NREM2 and sleep spindles are instrumental to the consolidation of motor sequence memories. PLoS Biol 14, e1002429, https://doi.org/10.1371/journal.pbio.1002429 (2016).
Walker, M. P., Brakefield, T., Morgan, A., Hobson, J. A. & Stickgold, R. Practice with sleep makes perfect: sleep-dependent motor skill learning. Neuron 35, 205–211 (2002).
Morin, A. et al. Motor sequence learning increases sleep spindles and fast frequencies in post-training sleep. Sleep 31, 1149–1156 (2008).
Nishida, M. & Walker, M. P. Daytime naps, motor memory consolidation and regionally specific sleep spindles. PLoS One 2, e341, https://doi.org/10.1371/journal.pone.0000341 (2007).
Tamaki, M. et al. Enhanced spontaneous oscillations in the supplementary motor area are associated with sleep-dependent offline learning of finger-tapping motor-sequence task. J Neurosci 33, 13894–13902, https://doi.org/10.1523/JNEUROSCI.1198-13.2013 (2013).
Marshall, L., Helgadottir, H., Molle, M. & Born, J. Boosting slow oscillations during sleep potentiates memory. Nature 444, 610–613, https://doi.org/10.1038/nature05278 (2006).
Smith, C. & MacNeill, C. Impaired motor memory for a pursuit rotor task following stage 2 sleep loss in college students. J Sleep Res 3, 206–213 (1994).
Collingridge, G. L., Peineau, S., Howland, J. G. & Wang, Y. T. Long-term depression in the CNS. Nat Rev Neurosci 11, 459–473, https://doi.org/10.1038/nrn2867 (2010).
Kandel, E. R. The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038, https://doi.org/10.1126/science.1067020 (2001).
Tsanov, M. & Manahan-Vaughan, D. Synaptic plasticity from visual cortex to hippocampus: systems integration in spatial information processing. Neuroscientist 14, 584–597, https://doi.org/10.1177/1073858408315655 (2008).
de Vivo, L. et al. Ultrastructural evidence for synaptic scaling across the wake/sleep cycle. Science 355, 507–510, https://doi.org/10.1126/science.aah5982 (2017).
Gilestro, G. F., Tononi, G. & Cirelli, C. Widespread changes in synaptic markers as a function of sleep and wakefulness in Drosophila. Science 324, 109–112, https://doi.org/10.1126/science.1166673 (2009).
Tononi, G. & Cirelli, C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron 81, 12–34, https://doi.org/10.1016/j.neuron.2013.12.025 (2014).
Vyazovskiy, V. V., Cirelli, C., Pfister-Genskow, M., Faraguna, U. & Tononi, G. Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat Neurosci 11, 200–208, https://doi.org/10.1038/nn2035 (2008).
Cirelli, C. & Tononi, G. Differential expression of plasticity-related genes in waking and sleep and their regulation by the noradrenergic system. J Neurosci 20, 9187–9194 (2000).
Cirelli, C. & Tononi, G. Gene expression in the brain across the sleep-waking cycle. Brain Res 885, 303–321 (2000).
Magistretti, P. J. & Pellerin, L. Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos Trans R Soc Lond Ser B Biol Sci 354, 1155–1163, https://doi.org/10.1098/rstb.1999.0471 (1999).
Maquet, P. Sleep function(s) and cerebral metabolism. Behav Brain Res 69, 75–83 (1995).
Madsen, P. L. & Vorstrup, S. Cerebral blood flow and metabolism during sleep. Cerebrovasc Brain Metab Rev 3, 281–296 (1991).
Holth, J. K. et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans Science (2019) https://doi.org/10.1126/science.aav2546.
Roh, J. H. et al. Disruption of the sleep-wake cycle and diurnal fluctuation of beta-amyloid in mice with Alzheimer’s disease pathology. Sci Transl Med 4, 150ra122 (2012) https://doi.org/10.1126/scitranslmed.3004291.
Kang, J. E. et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007 (2009) https://doi.org/10.1126/science.1180962.
Lundgaard, I. et al. Glymphatic clearance controls state-dependent changes in brain lactate concentration. J Cereb Blood Flow Metab 37, 2112–2124 (2017) https://doi.org/10.1177/0271678X16661202.
Iliff, J. J. et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 4, 147ra111 (2012) https://doi.org/10.1126/scitranslmed.3003748.
Iliff, J. J. et al. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest 123, 1299–1309, https://doi.org/10.1172/JCI67677 (2013).
Simon, M. J. & Iliff, J. J. Regulation of cerebrospinal fluid (CSF) flow in neurodegenerative, neurovascular and neuroinflammatory disease. Biochim Biophys Acta 1862, 442–451, https://doi.org/10.1016/j.bbadis.2015.10.014 (2016).
Louveau, A. et al. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J Clin Invest 127, 3210–3219, https://doi.org/10.1172/JCI90603 (2017).
Mestre, H. et al. Aquaporin-4-dependent glymphatic solute transport in the rodent brain. Elife 7 (2018) https://doi.org/10.7554/eLife.40070.
Xie, L. et al. Sleep drives metabolite clearance from the adult brain. Science 342, 373–377, https://doi.org/10.1126/science.1241224 (2013).
Ma, Q. et al. Rapid lymphatic efflux limits cerebrospinal fluid flow to the brain. Acta Neuropathol 137, 151–165, https://doi.org/10.1007/s00401-018-1916-x (2019).
Moraes, W. et al. Effects of aging on sleep structure throughout adulthood: a population-based study. Sleep Med 15, 401–409, https://doi.org/10.1016/j.sleep.2013.11.791 (2014).
Redline, S. et al. The effects of age, sex, ethnicity, and sleep-disordered breathing on sleep architecture. Arch Intern Med 164, 406–418, https://doi.org/10.1001/archinte.164.4.406 (2004).
Schwarz, J. F. A. et al. Age affects sleep microstructure more than sleep macrostructure. J Sleep Res 26, 277–287, https://doi.org/10.1111/jsr.12478 (2017).
Carrier, J., Land, S., Buysse, D. J., Kupfer, D. J. & Monk, T. H. The effects of age and gender on sleep EEG power spectral density in the middle years of life (ages 20–60 years old). Psychophysiology 38, 232–242 (2001).
Crowley, K., Trinder, J., Kim, Y., Carrington, M. & Colrain, I. M. The effects of normal aging on sleep spindle and K-complex production. Clin Neurophysiol 113, 1615–1622 (2002).
Martin, N. et al. Topography of age-related changes in sleep spindles. Neurobiol Aging 34, 468–476, https://doi.org/10.1016/j.neurobiolaging.2012.05.020 (2013).
Mazzoccoli, G. et al. Circadian variations of cortisol, melatonin and lymphocyte subpopulations in geriatric age. Int J Immunopathol Pharmacol 23, 289–296, https://doi.org/10.1177/039463201002300127 (2010).
Bloom, H. G. et al. Evidence-based recommendations for the assessment and management of sleep disorders in older persons. J Am Geriatr Soc 57, 761–789 (2009).
Yaffe, K., Falvey, C. M. & Hoang, T. Connections between sleep and cognition in older adults. Lancet Neurol 13, 1017–1028, https://doi.org/10.1016/S1474-4422(14)70172-3 (2014).
Li, J., Ogrodnik, M., Kolachalama, V. B., Lin, H. & Au, R. Assessment of the mid-life demographic and lifestyle risk factors of dementia using data from the Framingham Heart Study Offspring Cohort. J Alzheimers Dis 63, 1119–1127, https://doi.org/10.3233/JAD-170917 (2018).
Lutsey, P. L. et al. Sleep characteristics and risk of dementia and Alzheimer’s disease: the Atherosclerosis Risk in Communities Study. Alzheimers Dement 14, 157–166, https://doi.org/10.1016/j.jalz.2017.06.2269 (2018).
Reynolds, A. C. & Banks, S. Total sleep deprivation, chronic sleep restriction and sleep disruption. Prog Brain Res 185, 91–103, https://doi.org/10.1016/B978-0-444-53702-7.00006-3 (2010).
Anderson, K. N. & Bradley, A. J. Sleep disturbance in mental health problems and neurodegenerative disease. Nat Sci Sleep 5, 61–75, https://doi.org/10.2147/NSS.S34842 (2013).
Riemann, D. et al. The hyperarousal model of insomnia: a review of the concept and its evidence. Sleep Med Rev 14, 19–31, https://doi.org/10.1016/j.smrv.2009.04.002 (2010).
McCrae, C. S. & Lichstein, K. L. Secondary insomnia: diagnostic challenges and intervention opportunities. Sleep Med Rev 5, 47–61, https://doi.org/10.1053/smrv.2000.0146 (2001).
Ohayon, M. M. Epidemiology of insomnia: what we know and what we still need to learn. Sleep Med Rev 6, 97–111 (2002).
Ford, D. E. & Kamerow, D. B. Epidemiologic study of sleep disturbances and psychiatric disorders. An opportunity for prevention? JAMA 262, 1479–1484 (1989).
NIH State-of-the-Science Conference Statement on manifestations and management of chronic insomnia in adults. NIH Consens State Sci Statements 22, 1–30 (2005).
Cole, M. G. & Dendukuri, N. Risk factors for depression among elderly community subjects: a systematic review and meta-analysis. Am J Psychiatry 160, 1147–1156, https://doi.org/10.1176/appi.ajp.160.6.1147 (2003).
Dryman, A. & Eaton, W. W. Affective symptoms associated with the onset of major depression in the community: findings from the US National Institute of Mental Health Epidemiologic Catchment Area Program. Acta Psychiatr Scand 84, 1–5 (1991).
Fava, M. Daytime sleepiness and insomnia as correlates of depression. J Clin Psychiatry 65 Suppl 16, 27–32 (2004).
Livingston, G., Blizard, B. & Mann, A. Does sleep disturbance predict depression in elderly people? A study in inner London Br J Gen Pract 43, 445–448 (1993).
Nowell, P. D. & Buysse, D. J. Treatment of insomnia in patients with mood disorders. Depress Anxiety 14, 7–18 (2001).
Roberts, R. E., Shema, S. J., Kaplan, G. A. & Strawbridge, W. J. Sleep complaints and depression in an aging cohort: a prospective perspective. Am J Psychiatry 157, 81–88, https://doi.org/10.1176/ajp.157.1.81 (2000).
Gooneratne, N. S. & Vitiello, M. V. Sleep in older adults: normative changes, sleep disorders, and treatment options. Clin Geriatr Med 30, 591–627, https://doi.org/10.1016/j.cger.2014.04.007 (2014).
Ancoli-Israel, S., Klauber, M. R., Butters, N., Parker, L. & Kripke, D. F. Dementia in institutionalized elderly: relation to sleep apnea. J Am Geriatr Soc 39, 258–263 (1991).
Young, T. et al. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med 328, 1230–1235, https://doi.org/10.1056/NEJM199304293281704 (1993).
El-Ad, B. & Lavie, P. Effect of sleep apnea on cognition and mood. Int Rev Psychiatry 17, 277–282, https://doi.org/10.1080/09540260500104508 (2005).
Kerner, N. A. et al. Association of obstructive sleep apnea with episodic memory and cerebral microvascular pathology: a preliminary study. Am J Geriatr Psychiatry 25, 316–325, https://doi.org/10.1016/j.jagp.2016.11.009 (2017).
Ohayon, M. M. The effects of breathing-related sleep disorders on mood disturbances in the general population. J Clin Psychiatry 64, 1195–1200; quiz, 1274–1196 (2003).
Peppard, P. E., Szklo-Coxe, M., Hla, K. M. & Young, T. Longitudinal association of sleep-related breathing disorder and depression. Arch Intern Med 166, 1709–1715, https://doi.org/10.1001/archinte.166.16.1709 (2006).
Wheaton, A. G., Perry, G. S., Chapman, D. P. & Croft, J. B. Sleep disordered breathing and depression among U.S. adults: National Health and Nutrition Examination Survey, 2005–2008. Sleep 35, 461–467, https://doi.org/10.5665/sleep.1724 (2012).
Culebras, A. Sleep apnea and stroke. Curr Neurol Neurosci Rep 15, 503, https://doi.org/10.1007/s11910-014-0503-3 (2015).
Culebras, A. & Anwar, S. Sleep apnea is a risk factor for stroke and vascular dementia. Curr Neurol Neurosci Rep 18, 53, https://doi.org/10.1007/s11910-018-0855-1 (2018).
Osorio, R. S. et al. Sleep-disordered breathing advances cognitive decline in the elderly. Neurology 84, 1964–1971 (2015).
Faraut, B., Boudjeltia, K. Z., Vanhamme, L. & Kerkhofs, M. Immune, inflammatory and cardiovascular consequences of sleep restriction and recovery. Sleep Med Rev 16, 137–149, https://doi.org/10.1016/j.smrv.2011.05.001 (2012).
Gangwisch, J. E. et al. Short sleep duration as a risk factor for hypertension: analyses of the first National Health and Nutrition Examination Survey. Hypertension 47, 833–839, https://doi.org/10.1161/01.HYP.0000217362.34748.e0 (2006).
Hoevenaar-Blom, M. P., Spijkerman, A. M., Kromhout, D., van den Berg, J. F. & Verschuren, W. M. Sleep duration and sleep quality in relation to 12-year cardiovascular disease incidence: the MORGEN study. Sleep 34, 1487–1492, https://doi.org/10.5665/sleep.1382 (2011).
Lusardi, P. et al. Effects of a restricted sleep regimen on ambulatory blood pressure monitoring in normotensive subjects. Am J Hypertens 9, 503–505 (1996).
Meier-Ewert, H. K. et al. Effect of sleep loss on C-reactive protein, an inflammatory marker of cardiovascular risk. J Am Coll Cardiol 43, 678–683, https://doi.org/10.1016/j.jacc.2003.07.050 (2004).
Irwin, M., Thompson, J., Miller, C., Gillin, J. C. & Ziegler, M. Effects of sleep and sleep deprivation on catecholamine and interleukin-2 levels in humans: clinical implications. J Clin Endocrinol Metab 84, 1979–1985, https://doi.org/10.1210/jcem.84.6.5788 (1999).
Verrier, R. L. & Josephson, M. E. Impact of sleep on arrhythmogenesis. Circ Arrhythm Electrophysiol 2, 450–459, https://doi.org/10.1161/CIRCEP.109.867028 (2009).
Palagini, L. et al. Sleep loss and hypertension: a systematic review. Curr Pharm Des 19, 2409–2419 (2013).
Miller, R. R., 3rd & Ely, E. W. Delirium and cognitive dysfunction in the intensive care unit. Curr Psychiatry Rep 9, 26–34 (2007).
Freedman, N. S., Gazendam, J., Levan, L., Pack, A. I. & Schwab, R. J. Abnormal sleep/wake cycles and the effect of environmental noise on sleep disruption in the intensive care unit. Am J Respir Crit Care Med 163, 451–457, https://doi.org/10.1164/ajrccm.163.2.9912128 (2001).
Friese, R. S., Diaz-Arrastia, R., McBride, D., Frankel, H. & Gentilello, L. M. Quantity and quality of sleep in the surgical intensive care unit: are our patients sleeping? J Trauma 63, 1210–1214, https://doi.org/10.1097/TA.0b013e31815b83d7 (2007).
Gabor, J. Y. et al. Contribution of the intensive care unit environment to sleep disruption in mechanically ventilated patients and healthy subjects. Am J Respir Crit Care Med 167, 708–715, https://doi.org/10.1164/rccm.2201090 (2003).
Aurell, J. & Elmqvist, D. Sleep in the surgical intensive care unit: continuous polygraphic recording of sleep in nine patients receiving postoperative care. Br Med J (Clin Res Ed) 290, 1029–1032 (1985).
Cooper, A. B. et al. Sleep in critically ill patients requiring mechanical ventilation. Chest 117, 809–818 (2000).
Peterson, J. F. et al. Delirium and its motoric subtypes: a study of 614 critically ill patients. J Am Geriatr Soc 54, 479–484, https://doi.org/10.1111/j.1532-5415.2005.00621.x (2006).
Jackson, J. C., Gordon, S. M., Hart, R. P., Hopkins, R. O. & Ely, E. W. The association between delirium and cognitive decline: a review of the empirical literature. Neuropsychol Rev 14, 87–98 (2004).
McCusker, J., Cole, M., Dendukuri, N., Belzile, E. & Primeau, F. Delirium in older medical inpatients and subsequent cognitive and functional status: a prospective study. CMAJ 165, 575–583 (2001).
Rodriguez, J. C., Dzierzewski, J. M. & Alessi, C. A. Sleep problems in the elderly. Med Clin North Am 99, 431–439, https://doi.org/10.1016/j.mcna.2014.11.013 (2015).
Carpenter, B. D., Strauss, M. & Patterson, M. B. Sleep disturbances in community-dwelling patients with Alzheimer’s disease. Clin Gerontol 16, 35–49, https://doi.org/10.1300/J018v16n02_04 (1996).
Moran, M. et al. Sleep disturbance in mild to moderate Alzheimer’s disease. Sleep Med 6, 347–352, https://doi.org/10.1016/j.sleep.2004.12.005 (2005).
Askenasy, J. J. M. Sleep disturbances in Parkinsonism. J Neural Transm 110, 125–150, https://doi.org/10.1007/s007020300001 (2003).
Kumar, S., Bhatia, M. & Behari, M. Sleep disorders in Parkinson’s disease. Mov Disord 17, 775–781, https://doi.org/10.1002/mds.10167 (2002).
Tandberg, E., Larsen, J. P. & Karlsen, K. A community-based study of sleep disorders in patients with Parkinson’s disease. Mov Disord 13, 895–899, https://doi.org/10.1002/mds.870130606 (1998).
Abbott, S. M. & Videnovic, A. Sleep disorders in atypical parkinsonism. Mov Disord Clin Pract 1, 89–96, https://doi.org/10.1002/mdc3.12025 (2014).
Hyman, B. T. et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8, 1–13, https://doi.org/10.1016/J.JALZ.2011.10.007 (2012).
Bennett, D. A. et al. in Neurology. 1837–1844.
Rodrigue, K. M. et al. β-Amyloid burden in healthy aging: regional distribution and cognitive consequences. Neurology 78, 387–395, https://doi.org/10.1212/WNL.0b013e318245d295 (2012).
Weldemichael, D. A. & Grossberg, G. T. Circadian rhythm disturbances in patients with Alzheimer’s disease: a review. Int J Alzheimers Dis 2010, 1–9, https://doi.org/10.4061/2010/716453 (2010).
Landry, G. J., Best, J. R. & Liu-Ambrose, T. Measuring sleep quality in older adults: a comparison using subjective and objective methods. Front Aging Neurosci 7, 166, https://doi.org/10.3389/fnagi.2015.00166 (2015).
Myers, B. L. & Badia, P. Changes in circadian rhythms and sleep quality with aging: mechanisms and interventions. Neurosci Biobehav Rev 19, 553–571 (1995).
Sexton, C. E., Storsve, A. B., Walhovd, K. B., Johansen-Berg, H. & Fjell, A. M. Poor sleep quality is associated with increased cortical atrophy in community-dwelling adults. Neurology 83, 967–973 (2014) https://doi.org/10.1212/WNL.0000000000000774.
Patterson, B. W. et al. Age and amyloid effects on human central nervous system amyloid-beta kinetics. Ann Neurol 78, 439–453, https://doi.org/10.1002/ana.24454 (2015).
Brown, B. M., Rainey-Smith, S. R., Bucks, R. S., Weinborn, M. & Martins, R. N. Exploring the bi-directional relationship between sleep and beta-amyloid. Curr Opin Psychiatry 29, 397–401 (2016) https://doi.org/10.1097/YCO.0000000000000285.
Brown, B. M. et al. The relationship between sleep quality and brain amyloid burden. Sleep 39, 1063–1068, https://doi.org/10.5665/sleep.5756 (2016).
Ju, Y.-E. S. et al. Sleep quality and preclinical Alzheimer disease. JAMA Neurology 70, 587, https://doi.org/10.1001/jamaneurol.2013.2334 (2013).
Spira, A. P. et al. Self-reported sleep and β-amyloid deposition in community-dwelling older adults. JAMA Neurology 70, 1537–1543, https://doi.org/10.1001/jamaneurol.2013.4258 (2013).
Sprecher, K. E. et al. Poor sleep is associated with CSF biomarkers of amyloid pathology in cognitively normal adults. Neurology 89, 445–453 (2017) https://doi.org/10.1212/WNL.0000000000004171.
Huang, Y. et al. Effects of age and amyloid deposition on Abeta dynamics in the human central nervous system. Arch Neurol 69, 51–58, https://doi.org/10.1001/archneurol.2011.235 (2012).
Roh, J. H. et al. Disruption of the sleep-wake cycle and diurnal fluctuation of β-amyloid in mice with Alzheimer’s disease pathology. Sci Transl Med 4, 150ra122, https://doi.org/10.1126/scitranslmed.3004291 (2012).
Ju, Y. S. et al. Slow wave sleep disruption increases cerebrospinal fluid amyloid-beta levels. Brain 140, 2104–2111, https://doi.org/10.1093/brain/awx148 (2017).
Ooms, S. et al. Effect of 1 night of total sleep deprivation on cerebrospinal fluid beta-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol 71, 971–977, https://doi.org/10.1001/jamaneurol.2014.1173 (2014).
Shokri-Kojori, E. et al. β-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad Sci 115, 4483–4488, https://doi.org/10.1073/pnas.1721694115 (2018).
Mander, B. A. et al. β-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat Neurosci 18, 1051–1057, https://doi.org/10.1038/nn.4035 (2015).
Parkinson, J. in J Neuropsychiatry Clin Neurosci.
Burton, E. J., McKeith, I. G., Burn, D. J., Williams, E. D. & O’Brien, J. T. Cerebral atrophy in Parkinson’s disease with and without dementia: a comparison with Alzheimer’s disease, dementia with Lewy bodies and controls. Brain 127, 791–800, https://doi.org/10.1093/brain/awh088 (2004).
Hughes, A. J., Daniel, S. E., Kilford, L. & Lees, A. J. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55, 181–184, https://doi.org/10.1136/JNNP.55.3.181 (1992).
Baba, M. et al. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol 152, 879–884 (1998).
Spillantini, M. G. et al. α-Synuclein in Lewy bodies. Nature 388, 839–840, https://doi.org/10.1038/42166 (1997).
Braak, H. et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24, 197–211, https://doi.org/10.1016/S0197-4580(02)00065-9 (2003).
Dhawan, V., Healy, D. G., Pal, S. & Chaudhuri, K. R. Sleep-related problems of Parkinson’s disease. Age Ageing 35, 220–228, https://doi.org/10.1093/ageing/afj087 (2006).
Iranzo, A. et al. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. Lancet Neurol 12, 443–453, https://doi.org/10.1016/S1474-4422(13)70056-5 (2013).
Bonuccelli, U. et al. Diurnal motor variations to repeated doses of Levodopa in Parkinson’s disease. Clin Neuropharmacol 23, 28–33, https://doi.org/10.1097/00002826-200001000-00006 (2000).
Pursiainen, V. et al. Circadian heart rate variability in Parkinson’s disease. J Neurol 249, 1535–1540, https://doi.org/10.1007/s00415-002-0884-0 (2002).
Ahsan Ejaz, A., Sekhon, I. S. & Munjal, S. Characteristic findings on 24-h ambulatory blood pressure monitoring in a series of patients with Parkinson’s disease. Eur J Intern Med 17, 417–420, https://doi.org/10.1016/J.EJIM.2006.02.020 (2006).
Bordet, R. et al. Study of circadian melatonin secretion pattern at different stages of Parkinson’s disease. Clin Neuropharmacol 26, 65–72, https://doi.org/10.1097/00002826-200303000-00005 (2003).
Breen, D. P. et al. Sleep and circadian rhythm regulation in early Parkinson disease. JAMA Neurology 71, 589, https://doi.org/10.1001/jamaneurol.2014.65 (2014).
Videnovic, A. et al. Circadian melatonin rhythm and excessive daytime sleepiness in Parkinson disease. JAMA Neurology 71, 463, https://doi.org/10.1001/jamaneurol.2013.6239 (2014).
De Pablo-Fernández, E., Courtney, R., Warner, T. T. & Holton, J. L. A histologic study of the circadian system in Parkinson disease, multiple system atrophy, and progressive supranuclear palsy. JAMA Neurology 75, 1008, https://doi.org/10.1001/jamaneurol.2018.0640 (2018).
Gilman, S. et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 71, 670–676, https://doi.org/10.1212/01.wnl.0000324625.00404.15 (2008).
McKeith, I. G. et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 47, 1113–1124, https://doi.org/10.1212/WNL.47.5.1113 (1996).
Litvan, I. et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 47, 1–9 (1996).
Armstrong, M. J. et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 80, 496–503, https://doi.org/10.1212/WNL.0b013e31827f0fd1 (2013).
Boeve, B. F. et al. Clinicopathologic correlations in 172 cases of rapid eye movement sleep behavior disorder with or without a coexisting neurologic disorder. Sleep Med 14, 754–762, https://doi.org/10.1016/j.sleep.2012.10.015 (2013).
Polymenidou, M. & Cleveland, D. W. Prion-like spread of protein aggregates in neurodegeneration. J Exp Med 209, 889–893, https://doi.org/10.1084/jem.20120741 (2012).
Iliff, J. J. et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid. Sci Transl Med 4, 147ra111, https://doi.org/10.1126/scitranslmed.3003748 (2012).
Xu, Z. et al. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Abeta accumulation and memory deficits. Mol Neurodegener 10, 58, :https://doi.org/10.1186/s13024-015-0056-1 (2015).
Kress, B. T. et al. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol 76, 845–861, :https://doi.org/10.1002/ana.24271 (2014).
Iliff, J. J. et al. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci 34, 16180–16193, :https://doi.org/10.1523/JNEUROSCI.3020-14.2014 (2014).
Wang, M. et al. Focal solute trapping and global glymphatic pathway impairment in a murine model of multiple microinfarcts. J Neurosci 37, 2870–2877, :https://doi.org/10.1523/JNEUROSCI.2112-16.2017 (2017).
Zeppenfeld, D. M. et al. Association of perivascular localization of aquaporin-4 with cognition and Alzheimer disease in aging brains. JAMA Neurol 74, 91–99, :https://doi.org/10.1001/jamaneurol.2016.4370 (2017).
Abbott, N. J., Pizzo, M. E., Preston, J. E., Janigro, D. & Thorne, R. G. The role of brain barriers in fluid movement in the CNS: is there a ‘glymphatic’ system? Acta Neuropathol 135, 387–407, :https://doi.org/10.1007/s00401-018-1812-4 (2018).
Holter, K. E. et al. Interstitial solute transport in 3D reconstructed neuropil occurs by diffusion rather than bulk flow. Proc Natl Acad Sci 114, 9894–9899, https://doi.org/10.1073/pnas.1706942114 (2017).
Lee, H. et al. The effect of body posture on brain glymphatic transport. J Neurosci 35, 11034–11044, https://doi.org/10.1523/JNEUROSCI.1625-15.2015 (2015).
Yang, L. et al. Evaluating glymphatic pathway function utilizing clinically relevant intrathecal infusion of CSF tracer. J Transl Med 11, 107, https://doi.org/10.1186/1479-5876-11-107 (2013).
Ringstad, G., Vatnehol, S. A. S. & Eide, P. K. Glymphatic MRI in idiopathic normal pressure hydrocephalus. Brain J Neurol 140, 2691–2705, https://doi.org/10.1093/brain/awx191 (2017).
Eide, P. K. & Ringstad, G. Delayed clearance of cerebrospinal fluid tracer from entorhinal cortex in idiopathic normal pressure hydrocephalus: a glymphatic magnetic resonance imaging study. J Cereb Blood Flow Metab. 271678X18760974, https://doi.org/10.1177/0271678X18760974 (2018).
Ringstad, G. et al. Brain-wide glymphatic enhancement and clearance in humans assessed with MRI. JCI Insight 3, https://doi.org/10.1172/jci.insight.121537 (2018).
Joseph, A. A. et al. Real-time phase-contrast MRI of cardiovascular blood flow using undersampled radial fast low-angle shot and nonlinear inverse reconstruction. NMR Biomed 25, 917–924, https://doi.org/10.1002/nbm.1812 (2012).
Ohene, Y. et al. Non-invasive MRI of brain clearance pathways using multiple echo time arterial spin labelling: an aquaporin-4 study. Neuroimage 188, 515–523, https://doi.org/10.1016/j.neuroimage.2018.12.026 (2018).
Harrison, I. F. et al. Non-invasive imaging of CSF-mediated brain clearance pathways via assessment of perivascular fluid movement with diffusion tensor MRI. eLife 7, https://doi.org/10.7554/eLife.34028 (2018).
Demiral, Ş. B. et al. Apparent diffusion coefficient changes in human brain during sleep—does it inform on the existence of a glymphatic system? NeuroImage 185, 263–273, https://doi.org/10.1016/j.neuroimage.2018.10.043 (2018).
Mattsson, N. et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 302, 385, https://doi.org/10.1001/jama.2009.1064 (2009).
Paterson, R. W. et al. CSF in the differential diagnosis of Alzheimer’s disease: clinical utility of an extended panel of biomarkers in a specialist cognitive clinic. Alzheimers Dement 12, P203-P204, https://doi.org/10.1016/J.JALZ.2016.06.356 (2016).
Riemenschneider, M. et al. Cerebrospinal fluid tau and beta-amyloid 42 proteins identify Alzheimer disease in subjects with mild cognitive impairment. Arch Neurol 59, 1729–1734 (2002).
Visser, P. J. et al. Prevalence and prognostic value of CSF markers of Alzheimer’s disease pathology in patients with subjective cognitive impairment or mild cognitive impairment in the DESCRIPA study: a prospective cohort study. Lancet Neurol 8, 619–627, https://doi.org/10.1016/S1474-4422(09)70139-5 (2009).
Ohayon, M.M. [Prevalence and comorbidity of sleep disorders in general population]. Rev Prat 57, 1521–1528 (2007).
Dijk, D. J. Slow-wave sleep deficiency and enhancement: implications for insomnia and its management. World J Biol Psychiatry 11 Suppl 1, 22–28, https://doi.org/10.3109/15622971003637645 (2010).
Deschenes, C. L. & McCurry, S. M. Current treatments for sleep disturbances in individuals with dementia. Curr Psychiatry Rep 11, 20–26 (2009).
Mathias, S., Steiger, A. & Lancel, M. The GABA(A) agonist gaboxadol improves the quality of post-nap sleep. Psychopharmacology 157, 299–304, https://doi.org/10.1007/s002130100819 (2001).
Dijk, D. J., James, L. M., Peters, S., Walsh, J. K. & Deacon, S. Sex differences and the effect of gaboxadol and zolpidem on EEG power spectra in NREM and REM sleep. J Psychopharmacol 24, 1613–1618, https://doi.org/10.1177/0269881109105788 (2010).
Pardi, D. & Black, J. gamma-Hydroxybutyrate/sodium oxybate: neurobiology, and impact on sleep and wakefulness. CNS Drugs 20, 993–1018, https://doi.org/10.2165/00023210-200620120-00004 (2006).
Dijk, D. J., Beersma, D. G., Daan, S. & van den Hoofdakker, R. H. Effects of seganserin, a 5-HT2 antagonist, and temazepam on human sleep stages and EEG power spectra. Eur J Pharmacol 171, 207–218 (1989).
Landolt, H. P. et al. Serotonin-2 receptors and human sleep: effect of a selective antagonist on EEG power spectra. Neuropsychopharmacology 21, 455–466, https://doi.org/10.1016/S0893-133X(99)00052-4 (1999).
Landolt, H. P. & Wehrle, R. Antagonism of serotonergic 5-HT2A/2C receptors: mutual improvement of sleep, cognition and mood? Eur J Neurosci 29, 1795–1809, https://doi.org/10.1111/j.1460-9568.2009.06718.x (2009).
Deacon, S. et al. Effect of short-term treatment with gaboxadol on sleep maintenance and initiation in patients with primary insomnia. Sleep 30, 281–287 (2007).
Lankford, D. A. et al. Effect of gaboxadol on sleep in adult and elderly patients with primary insomnia: results from two randomized, placebo-controlled, 30-night polysomnography studies. Sleep 31, 1359–1370 (2008).
Roth, T., Wright, K. P., Jr. & Walsh, J. Effect of tiagabine on sleep in elderly subjects with primary insomnia: a randomized, double-blind, placebo-controlled study. Sleep 29, 335–341 (2006).
Walsh, J. K. et al. Tiagabine increases slow-wave sleep in a dose-dependent fashion without affecting traditional efficacy measures in adults with primary insomnia. J Clin Sleep Med 2, 35–41 (2006).
Brzezinski, A. et al. Effects of exogenous melatonin on sleep: a meta-analysis. Sleep Med Rev 9, 41–50, https://doi.org/10.1016/j.smrv.2004.06.004 (2005).
Buscemi, N. et al. The efficacy and safety of exogenous melatonin for primary sleep disorders. A meta-analysis. J Gen Intern Med 20, 1151–1158, https://doi.org/10.1111/j.1525-1497.2005.0243.x (2005).
Haimov, I. et al. Melatonin replacement therapy of elderly insomniacs. Sleep 18, 598–603 (1995).
Pawlikowski, M., Kolomecka, M., Wojtczak, A. & Karasek, M. Effects of six months melatonin treatment on sleep quality and serum concentrations of estradiol, cortisol, dehydroepiandrosterone sulfate, and somatomedin C in elderly women. Neuro Endocrinol Lett 23 Suppl 1, 17–19 (2002).
Zhdanova, I. V. et al. Melatonin treatment for age-related insomnia. J Clin Endocrinol Metab 86, 4727–4730, https://doi.org/10.1210/jcem.86.10.7901 (2001).
Dowling, G. A. et al. Melatonin for sleep disturbances in Parkinson’s disease. Sleep Med 6, 459–466, https://doi.org/10.1016/j.sleep.2005.04.004 (2005).
Singer, C. et al. A multicenter, placebo-controlled trial of melatonin for sleep disturbance in Alzheimer’s disease. Sleep 26, 893–901 (2003).
Vural, E. M., van Munster, B. C. & de Rooij, S. E. Optimal dosages for melatonin supplementation therapy in older adults: a systematic review of current literature. Drugs Aging 31, 441–451, https://doi.org/10.1007/s40266-014-0178-0 (2014).
Raskind, M. A. et al. A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry 61, 928–934, https://doi.org/10.1016/j.biopsych.2006.06.032 (2007).
Taylor, F. B. et al. Prazosin effects on objective sleep measures and clinical symptoms in civilian trauma posttraumatic stress disorder: a placebo-controlled study. Biol Psychiatry 63, 629–632, https://doi.org/10.1016/j.biopsych.2007.07.001 (2008).
Ruff, R. L., Ruff, S. S. & Wang, X. F. Improving sleep: initial headache treatment in OIF/OEF veterans with blast-induced mild traumatic brain injury. J Rehabil Res Dev 46, 1071–1084 (2009).
McCurry, S. M. & Ancoli-Israel, S. Sleep dysfunction in Alzheimer’s disease and other dementias. Curr Treat Options Neurol 5, 261–272 (2003).
Ancoli-Israel, S. Sleep and aging: prevalence of disturbed sleep and treatment considerations in older adults. J Clin Psychiatry 66 Suppl 9, 24–30; quiz 42–23 (2005).
Ju, Y. S., Zangrilli, M. A., Finn, M. B., Fagan, A. M. & Holtzman, D. M. Obstructive sleep apnea treatment, slow wave activity, and amyloid-beta. Ann Neurol 85, 291–295, https://doi.org/10.1002/ana.25408 (2019).
Perrault, A. A. et al. Whole-night continuous rocking entrains spontaneous neural oscillations with benefits for sleep and memory. Curr Biol 29, 402–411 e403, https://doi.org/10.1016/j.cub.2018.12.028 (2019).
Kompotis, K. et al. Rocking promotes sleep in mice through rhythmic stimulation of the vestibular system. Curr Biol 29, 392–401 e394, https://doi.org/10.1016/j.cub.2018.12.007 (2019).
Acknowledgments
This work was supported by grant from the National Institutes of Health to Jeffrey J. Iliff (AG054456, NS089709) and to James Goodman (AG060681).
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure Statement
This study was not industry-supported and the authors declare that they have no financial conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 506 kb)
Rights and permissions
About this article

Cite this article
Bah, T.M., Goodman, J. & Iliff, J.J. Sleep as a Therapeutic Target in the Aging Brain. Neurotherapeutics 16, 554–568 (2019). https://doi.org/10.1007/s13311-019-00769-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-019-00769-6