
Abstract
Most of the glycogen metabolism disorders that affect skeletal muscle involve enzymes in glycogenolysis (myophosphorylase (PYGM), glycogen debranching enzyme (AGL), phosphorylase b kinase (PHKB)) and glycolysis (phosphofructokinase (PFK), phosphoglycerate mutase (PGAM2), aldolase A (ALDOA), β-enolase (ENO3)); however, 3 involve glycogen synthesis (glycogenin-1 (GYG1), glycogen synthase (GSE), and branching enzyme (GBE1)). Many present with exercise-induced cramps and rhabdomyolysis with higher-intensity exercise (i.e., PYGM, PFK, PGAM2), yet others present with muscle atrophy and weakness (GYG1, AGL, GBE1). A failure of serum lactate to rise with exercise with an exaggerated ammonia response is a common, but not invariant, finding. The serum creatine kinase (CK) is often elevated in the myopathic forms and in PYGM deficiency, but can be normal and increase only with rhabdomyolysis (PGAM2, PFK, ENO3). Therapy for glycogen storage diseases that result in exercise-induced symptoms includes lifestyle adaptation and carefully titrated exercise. Immediate pre-exercise carbohydrate improves symptoms in the glycogenolytic defects (i.e., PYGM), but can exacerbate symptoms in glycolytic defects (i.e., PFK). Creatine monohydrate in low dose may provide a mild benefit in PYGM mutations.
Similar content being viewed by others


Introduction
The myopathies associated with glucose metabolism are generally categorized into disorders of glycolysis, glycogenolysis, and glycogen synthesis. These disorders usually present during periods of increased metabolic energy demand (i.e., exercise) and are considered metabolic myopathies; however, some can present with a fixed myopathy and resemble structural myopathies such as muscular dystrophy or congenital myopathies. In this chapter, I shall review the metabolism of glucose within skeletal muscle and the relationship to exercise, present a summary of the main disorders of glucose metabolism, and then summarize therapies. Although Pompe disease is sometimes considered to be a metabolic glycogen storage disease (GSD II), it is better categorized as a lysosomal storage disease and is considered separately in the article by Raben et al. (this issue). Furthermore, the enzyme defect responsible for Pompe disease, acid-alpha glucosidase (GAA), is not involved in metabolic energy provision during exercise in skeletal muscle [1].
Pathways of Glucose Metabolism in Skeletal Muscle
Glucose is taken up from the plasma by 2 glucose transporters called GLUT4 and GLUT1. GLUT4 is found partially in the sarcolemma and partially within intracellular vesicles that can migrate to the sarcolemma in response to insulin and muscle contraction to enhance glucose uptake. In contrast, GLUT1 is considered a constitutive glucose transporter and does not respond to either insulin or contraction-induced migration. In the postprandial state, when insulin is high, the insulin receptor binding activates IRS1 > Akt that leads to a migration of GLUT4 to the sarcolemma to enhance glucose uptake for glycogen synthesis [2]. In response to skeletal muscle contraction-induced signals (calcium and reactive oxygen species) [3], GLUT4 also migrates to the sarcolemma to enhance glucose entry for glycolytic flux during prolonged exercise and the GLUT4 remains at the sarcolemma for some time after exercise cessation to facilitate the resynthesis of glycogen [3].
Once glucose enters the muscle, it is phosphorylated by hexokinase II and ATP to form glucose-6-phosphate. Glucose-6-phosphate is then committed to either glycogen synthesis or glycolysis (depending on the hormonal environment) and cannot be used for replenishing blood glucose since skeletal muscle does not have glucose-6-phosphatase, unlike the gluconeogenic organs, the liver and kidney. If epinephrine is high (fasting and exercise), the glucose-6-phosphate will enter the glycolytic pathway to form pyruvate (see below), while if insulin is high, the glucose-6-phosphate will enter the glycogen synthesis pathway for storage for future use.
In the postprandial state, when insulin is high, the glycogen synthesis pathway starts with the conversion of glucose-6-phosphate to glucose-1-phosphate by phosphoglucomutase-1. Glucose-1-phosphate is converted to UDP-glucose through UDP-glucose pyrophosphorylase, and the UDP-glucose moieties are joined end to end in a 1,4-configuration upon the glycogenin-1 protein backbone as a form of priming step. Glycogen synthase is activated by dephosphorylation in the presence of insulin and catalyzes the further linear extension of glycogen through alpha 1,4 linkages. Branching enzyme is also active and catalyzes the branching of the growing glycogen molecules through formation of an alpha 1,6 linkage. Glycogen is found as a granule-like structure both under the sarcolemma (subsarcolemmal) and between the actin–myosin contractile elements (intermyofibrillar) (Fig. 1). Skeletal muscle glycogen concentration is 200 to 400 mmol/kg dry muscle weight (~ 400 g total in a 70-kg human) but can double (up to 800 mmol/kg dry muscle mass) in endurance athletes [4] and patients with glycogen storage disease (see Fig. 1). Skeletal muscle glycogen concentration in healthy humans can be manipulated by diet with a very high carbohydrate percentage for 3 to 4 days, leading to a near doubling of glycogen concentration (glycogen loading) [5].

Electron microscopy of human skeletal muscle. IMCL = intramyocellular lipid; IMF = intermyofibrillar; SS = subsarcolemmal
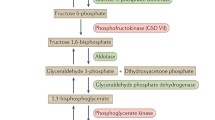
During periods of metabolic demand, the increase in epinephrine leads to a phosphorylation of phosphorylase b kinase that in turn phosphorylates and activates myophosphorylase and hydrolyzes the alpha 1,4-glycosidic links of glycogen to initiate glycogenolysis. Debranching enzyme hydrolyzes the alpha 1,6-glycosidic glycogen bonds to yield glucose-1-phosphate which is then converted to glucose-6-phosphate by phosphoglucomutase-1. Glucose-6-phosphate is then converted to fructose-1-phosphate by G-6-P-isomerase, and the process of glycolysis is initiated. Fructose-6-phosphate is then converted to fructose 1,6-bisphosphate by phosphofructokinase (PFK), the rate-limiting enzyme for glycolysis. PFK is activated by inorganic phosphate liberated during energy demand by ATP hydrolysis and possibly creatine [6]. Fructose 1,6-bisphosphate (6 carbons) is then hydrolyzed to glyceraldehyde 3-phosphate (3 carbons) and dihydroxyacetone phosphate (3 carbons) by aldolase. During high-energy demand, dihydroxyacetone phosphate is converted back to glyceraldehyde 3-phosphate by triosephosphate isomerase. Glyceraldehyde 3-phosphate is converted to 1,3-bisphosphoglycerate by glyceraldehyde phosphate dehydrogenase. 1,3-Bisphosphoglycerate is converted to 3-phosphoglycerate by phosphoglycerate kinase (PGK) and that is then converted to 2-phosphoglycerate by phosphoglycerate mutase 2 (PGAM-2). The 2-phosphoglycerate is then converted to phosphoenolpyruvate (PEP) by β-enolase. Finally, PEP is converted to pyruvate by pyruvate kinase (PK).
In the rest-to-exercise transition and/or under anaerobic conditions, pyruvate is converted to lactate by lactate dehydrogenase in order to regenerate NAD+ to keep glycolysis going at the glyceraldehyde phosphate dehydrogenase step. The lactate is then exported from muscle to plasma through monocarboxylate transporter-4 (MCT4) [7]. If the energy demand is sustained and there is oxygen availability, the pyruvate can then enter the tricarboxylic acid cycle after dehydrogenation and decarboxylation by the pyruvate dehydrogenase (PDH) complex. A summary of these pathways can be found in Fig. 2.

Glycogen metabolism in skeletal muscle. GLUT = glucose transporter; DHAP = dihydroxyacetone phosphate; GYS1 = glycogen synthase; BE = branching enzyme; MCT = monocarboxylate transporter; TCA = tricarboxylic acid cycle.
Exercise Metabolism
At the onset of exercise, there is an immediate need for ATP regeneration due to ATPase-mediated hydrolysis of ATP. ATPases are required for many processes associated with muscle contraction including sodium/potassium ATPase (membrane potential), calcium ATPase (reuptake of calcium into the sarcoplasmic reticulum), and myosin ATPase (actin–myosin contraction). The increase in ADP immediately fluxes through adenylate kinase to form ATP (ADP + ADP > ATP + AMP). To keep this equilibration reaction moving in the forward direction, the AMP is removed by myoadenylate deaminase (AMPD1) to form inosine monophosphate (IMP) and ammonia. The IMP is converted to inosine which is converted to hypoxanthine, and this is in turn converted to xanthine and, finally, uric acid by xanthine oxidase. At the onset of exercise, there is also an immediate rephosphorylation of ADP by phosphocreatine (PCr) and by cytosolic creatine kinase (in the presence of a proton (H+) from glycolysis) to generate ATP and free creatine (Cr). Glycolysis is activated rapidly (within 4-6 s), to provide the bulk of ADP rephosphorylation during the rest > exercise transition and with ischemia [8].
About half of the ATP generation within the first 8 to 10 s of exercise is from PCr hydrolysis [8], with glycolysis providing the majority of the ATP from 8 to 10 s to about 60 s (~ 400 m runner). Although the energy derivation is rapid from anaerobic glycolysis, the resultant proton from the lactic acid (lactate + H+) will inhibit enzymatic pathways and limit exercise performance. At 2 min of running (~ 800 m runner), most of the energy is coming from aerobic glycogenolysis/glycolysis through pyruvate dehydrogenase (PDH) to the TCA cycle and the reducing equivalents are then used by the mitochondria to generate ATP in the presence of molecular oxygen. Top athletes have nearly 2 h of glycogen stores in active skeletal muscle to support exercise, and top marathon runners working at 80% of maximal oxygen consumption (VO2peak) will use glycogen stores for nearly 100% of a marathon (just over 2 h) [9]. As exercise duration increases beyond 2 h, or if the intensity of the exercise is lower (< 65% VO2peak), there will be a proportionate increase in the oxidation of free fatty acids through β-oxidation to supply reducing equivalents to the electron transport chain of the mitochondria [10, 11]. The source of the free fatty acids during exercise is both plasma and intramyocellular lipids (IMCL). Given that FFAs are more reduced than glucose, the amount of oxygen required for oxidation is higher and, thus, when carbohydrate stores are reduced, a runner must slow down and this is perceived during a long run such as a marathon as “hitting the wall.”
Energy Crisis in Glycolytic and Glycogenolytic Defects and Rhabdomyolysis
Most patients with glycogenolytic and glycolytic defects will experience an energy crisis during a sustained isometric contraction (ischemic) or repetitive higher-intensity activity for the reasons outlined above. Given that carbohydrate (pyruvate) availability is required for maximal efficiency of mitochondrial electron transport, patients with glycogenolytic and glycolytic defects will have a lower VO2peak and experience exercise limitations even at lower exercise intensities [12]. Some glycogenolytic and glycolytic defects will experience a “second wind” phenomenon where the rating of perceived exertion and physiological stress metrics (i.e., heart rate and minute ventilation) will increase rapidly during the first few minutes of exercise and then will be rapidly alleviated if the intensity is lowered and time is given for capillary dilatation to provide exogenous substrate and oxygen to the working muscle and for substrate mobilization from liver (glycogenolysis) and adipocytes (lipolysis) [12].
If a patient with a glycolytic or glycogenolytic defect performs unaccustomed exercise or increases the intensity too rapidly and does not heed the perception of cramps and pain within the contracting muscle, they can experience rhabdomyolysis. Rhabdomyolysis is often defined as an increase in the plasma creatine kinase (CK) of > 10 times the upper limit of normal (ULN = ~ 200 iU/L). The increase in CK is reflective of a breach of the sarcolemma allowing the release of cytosolic enzymes into the plasma. The damage to muscle during rhabdomyolysis from a metabolic myopathy is due to energy deficiency-mediated processes including reactive oxygen species, calcium overload, and ionic imbalance. Although CK is conveniently used as the marker for rhabdomyolysis (and damage from fixed myopathies), it is important to note that many cytoplasmic enzymes are also released including aspartate amino transferase (AST), alanine amino transferase (ALT), lactate dehydrogenase (LDH), aldolase, myoglobin, fatty acid-binding protein, etc. and in the context of rhabdomyolysis, proteins such as ALT, AST, and LDH are reflective of skeletal muscle damage and not liver damage.
The rhabdomyolysis-induced increase in some of these proteins (i.e., myoglobin) and likely cellular debris (apoptotic bodies) can damage the nephron and lead to acute tubular necrosis and acute renal failure. Consequently, many patients with glycogenolytic and glycolytic defects will often report episodes of dark urine (pigmenturia) cotemporal with exercise that they often describe as “tea,” “red,” “cola,” “brown,” “blood-tinged,” or “very dark and frothy.” A full description of the assessment and treatment of rhabdomyolysis is beyond the scope of the current chapter; however, the general approach will be discussed. The first aspect of treatment for a patient is preventative recognition and avoidance of the precipitating factors such as unaccustomed exercise, forced exercise, and/or repetitive exercise. With acute rhabdomyolysis, it is essential to take baseline bloodwork (CK activity, creatinine, urea, uric acid, electrolytes (sodium, potassium, chloride, calcium, and magnesium)) and a urinalysis (routine and microscopic, and myoglobin if available). Intravenous fluids with normal saline to maintain a high urine output is the mainstay of therapy [13], with specific electrolytes added as needed if specific deficiencies are identified. The use of bicarbonate to alkalinize the urine remains unclear. With severe rhabdomyolysis, there can be an acute compartment syndrome that requires wick pressure monitoring and plastic surgery assessment for fasciotomy. If the creatinine is elevated in spite of fluid therapy (often with high urea and potassium), a nephrology consultation is recommended to consider hemodialysis.
Defects in Glycogenolysis (Table 1)
Myophosphorylase
Myophosphorylase is encoded for by the PYGM gene. Mutations in the PYGM gene represent the most common inborn error of metabolism affecting glucose/glycogen metabolism, with an incidence of ~ 1/50,000 to 1/150,000 [14,15,16]. Heterozygous or homozygous mutations in the PYGM gene are responsible for GSD V (McArdle disease [17]), with the p.R50X mutation being the most common in Caucasians [16]. Patients with GSD V usually present in young adulthood (~ 60% < 10 years of age) with recurrent muscle cramps and pigmenturia with higher-intensity exercise, and most report a second-wind phenomenon [16]. Some patients will also have several bouts of gout due to myogenic hyperuricemia due to compensatory flux through the AK/AMPD1 pathway [18]. Most patients will have a normal neurological examination; however, older patients can sometimes show proximal weakness (including paraspinal muscles) [19], possibly from multiple bouts of rhabdomyolysis and incomplete interevent skeletal muscle repair.
The diagnosis is suspected from the history and blood testing showing high CK activity and often high urate [16]. The classical forearm exercise test consists of the measurement of lactate and ammonia from the antecubital vein before and after repetitive exercise with sphygmomanometer-induced ischemia and showing a failure of the normal lactate rise and an exaggerated ammonia rise [17, 20]. Due to concerns about the potential for inducing compartment syndrome in the forearm, we, and others [21], have adopted a nonischemic forearm exercise test and have shown that the changes in lactate (or lack thereof) and ammonia are similar to those we found using cuff-induced ischemia, likely due to the fact that repetitive isometric contractions powerfully activates glycogenolysis and glycolysis. Our protocol involves placement of a 22-Ga catheter into the antecubital vein, with blood samples taken in tubes with glycolytic inhibitors for lactate (gray = fluoride oxalate) and ammonia (green = sodium heparin) on ice. We then have the subject squeeze a handgrip dynamometer (squeezing a rolled-up towel or tennis ball will suffice) for 9 s with 1 s relaxation, repetitively for 60 s (6 bouts). We then measure lactate and ammonia at 1 min post exercise. As with our earlier ischemic results [20], we find very high sensitivity and specificity for GSD V (and other glycolytic defects—see below) with absence of a lactate rise (< 2.0-fold but most are < 50% increase) and an exaggerated ammonia rise (> 2.5-fold). The forearm exercise or aerobic bike test is an adjunct to the history and physical examination and can assist in the interpretation of genetic test results, especially if the history is not classical for a GSD. For example, a normal lactate and ammonia rise with exercise testing would help to eliminate a variant of uncertain significance in the PYGM gene found with genetic testing such as a “myopathy panel” or whole exome sequencing. In fact, a normal lactate and ammonia response to exercise has high sensitivity to eliminate nearly all metabolic GSDs from further consideration.
The muscle biopsy in GSD V, and other GSDs, can be misleading if taken after an acute bout of rhabdomyolysis for it can show nonspecific necrosis, neutrophils, and macrophages that can mask classical features. Usually, patients will show absence of myophosphorylase histochemical activity, although this can be falsely positive due to transient upregulation of the embryonic isoform following acute rhabdomyolysis. Other features include an increase in glycogen seen with periodic acid-Schiff staining and vacuoles (glycogen filled) with routine hematoxylin and eosin staining. Electron microscopy shows a nonspecific increase in non–membrane bound glycogen in the subsarcolemmal and intermyofibrillar regions (Fig. 1, inset).
The standard for molecular diagnosis of GSD V has traditionally been targeted mutation panel analysis for common pathogenic gene mutations (i.e., p.R50X, p.W798R, p.G205S in Caucasians and p.Phe710del in Japanese). Given that there have been ~ 175 pathogenic mutations reported (https://databases.lovd.nl/shared/variants/PYGM), many groups have resorted to Sanger sequencing of all of the coding regions (and intron/exon boundaries) of the PYGM gene. Given that the clinical features of GSD V are similar to several of the other metabolic glycogenolytic and glycolytic defects (see below), and that patients with structural myopathies can present with exercise-induced cramps and pigmenturia (pseudometabolic), several companies now offer next-generation sequencing-based “myopathy panels” that include many of the structural genes that can lead to exercise-induced symptoms (i.e., DYS, SCGA, SCGB, SCGD, TTN, etc.) and all of the GSD-associated genes.
Phosphorylase b Kinase
The phosphorylase b kinase enzyme is a hexadecameric enzyme that has a number of subunits, and I will only consider the 2 subunits that are expressed in muscle and associated with metabolic myopathy: phosphorylase b kinase α skeletal muscle isoform (PHKA1, PHKA1 gene, X-linked recessive, GSD IXd) and phosphorylase b kinase β isoform (PHKB, PHKB gene, X-linked recessive). PKHA1 deficiency (GSD IXd) usually presents with exercise intolerance/cramps and occasionally rhabdomyolysis and muscle weakness [22,23,24,25]. Some have questioned the pathogenicity of mutations in the PHKA1 gene since patients usually have mild symptoms and show a normal rise in lactate in response to forearm exercise [26]. The same group also reported mild hyperCKemia, mild myalgias, a normal lactate response to forearm exercise, but a blunted lactate rise in response to aerobic exercise in 2 men and concluded that GSD IXd is a mild disorder [27]. Although the PHKB gene is expressed in skeletal muscle, mutations typically result in a mild liver phenotype [28, 29].
Debranching Enzyme
Debranching enzyme is a monomeric enzyme but has 2 catalytic activities: amylo-1,6-glucosidase and 4-α-glucanotransferase. It is encoded for by the AGL gene, and homozygous or compound heterozygous autosomal recessive mutations result in 2 main subcategories: GSD IIIa and GSD IIIb [30,31,32,33]. Subcategory IIIb is less common (15%) and exclusively hepatic, IIIa being the most common (85%), and presents with hepatopathy, cardiomyopathy, and myopathy with weakness and hyperCKemia [31, 33, 34]. Some patients can present only with muscle weakness and hyperCKemia and not other overt clinical features [35,36,37,38].
Defects in Glycogen Synthesis (Table 1)
Glycogenin-1
Glycogenin-1 is a predominantly cardiac and skeletal muscle-specific enzyme that functions as a catalytic scaffold-like protein to initiate the end-to-end 1,4 glycosidic links and autoglycosylation of UDP-glucose-forming linear glycogen molecules of ~ 8 glucose molecules in length before glycogen synthase and branching enzyme further elongate the growing glycogen molecule. Glycogenin-1 is also involved in glycogen breakdown [39]. Glycogenin-1 is encoded for by the GYG1 gene, and mutations have been described in GSD XV. The first report of a mutation in the GYG1 was in a young man with postexertional arrhythmia, mild ventricular thickening, and mild muscle weakness [40]. Muscle biopsy showed reduced glycogen, mitochondrial proliferation, and type I fiber predominance [40]. The next report of GYG1 mutations was a case series with 7 adults (onset 17-65 years) with slowly progressive proximal weakness, no cardiac issues, a myopathic EMG, normal CK, and polyglucosan bodies in the muscle biopsy [41]. Another report also found late-onset (50-60 years) proximal weakness with polyglucosan bodies and no cardiomyopathy in 5 patients with homozygous GYG1 mutations [42]. In contrast, severe cardiomyopathy (2 needing heart transplant) without proximal muscle weakness was reported in 3 patients (34-52 years) with homozygous GYG1 mutations [43, 44]. Finally, 9 patients from 5 GSD XV families were reported with a later-onset limb–girdle and/or scapuloperoneal weakness pattern with no cardiomyopathy [45]. Patients with GYG1 mutations do show improvements in exercise capacity with glucose infusion [39], suggesting that pre-exercise glucose ingestion will be therapeutically useful.
Glycogen Synthase
The glycogen synthase-1 enzyme is encoded for by the GYS1 gene, and mutations in this gene result in GSD 0 [46]. Patients with GSD 0 can present with exercise-induced arrhythmia, cardiomyopathy, and/or exercise intolerance and possibly epilepsy [46, 47]. Interestingly, mutations in GYS1 are a common cause of exertional rhabdomyolysis and polysaccharide storage myopathy (PSSM) in racehorses [48, 49]. A characteristic feature on muscle biopsy in humans is the absence of glycogen and polyglucosan bodies with mitochondrial proliferation that can mimic mitochondrial myopathies [46].
Branching Enzyme
Branching enzyme is encoded for by the GBE1 gene, and mutations can result in GSD IV with severe hepatopathy with cirrhosis and varices, severe neuromuscular hypotonia and even arthrogryposis in infancy with hepatomegaly [50,51,52,53,54], or adult-onset polyglucosan body disease [55]. Adult-onset polyglucosan body disease presents usually after age 50 years with progressive spastic paraparesis, neurogenic bladder, and axonal neuropathy with severe vibration loss [55, 56]. Polyglucosan body disease can mimic ALS, MS, and CMT neuropathy [57, 58]. Polyglucosan bodies can be seen in peripheral nerve and muscle biopsies [56, 57].
Defects in Glycolysis (Table 2)
Phosphofructokinase
Phosphofructokinase (PFK) is the rate-limiting enzyme of glycolysis and catalyzes the conversion of fructose-6-phosphate to fructose 1,6-bisphosphate. The PFK enzyme is encoded for by the PFKM gene, and most mutations have been seen in the Japanese and Ashkenazi-Jewish populations [59,60,61,62,63]. The clinical description was first described by Tarui and colleagues in 3 Japanese patients with exercise-induced myalgia/stiffness, exercise intolerance, muscle weakness, and a positive forearm exercise test [64]. Clinically, these patients do not show a second-wind phenomenon [65]. Others have confirmed the muscle symptoms [66], but also reported hemolytic anemia, rhabdomyolysis, and abdominal pain with activity [67,68,69]. Myogenic hyperuricemia and gout have been seen due to a compensatory increased flux through the AK > AMPD1 pathway to form uric acid [18]. There have been a few reports of a severe infantile form with weakness, epilepsy, cortical blindness, corneal clouding, and cardiomyopathy [70,71,72,73]. The muscle biopsy shows increased non–membrane bound glycogen, and there is a histochemical enzyme stain available for PFK activity. Given the rarity of GSD VII and the even more rare severe infantile-onset form, it is possible that the children with the severe symptoms had a second genetic disease accounting for the CNS phenotype (molecular double trouble).
Phosphoglycerate Kinase-1
Phosphoglycerate kinase-1 (PGK-1) catalyzes the conversion of 1,3-bisphosphoglycerate to 3-phosphoglycerate. It is an X-linked recessive GSD with the protein encoded for by the PGK1 gene. The first patient with exercise intolerance and rhabdomyolysis symptoms was reported by DiMauro and colleagues in 1983 [74], and this phenotype has been reported by others [75]. Mutations in this gene often result in a chronic hemolytic anemia, intellectual disability, and occasionally late-onset Parkinson disease features [74, 76,77,78,79,80].
Phosphoglycerate Mutase
The phosphoglycerate mutase enzyme (PGAM2) is a distal glycolytic enzyme that converts 3-phosphoglycerate to 2-phosphoglycerate. The PGAM2 protein is encoded for by the PGAM gene, and the first autosomal recessive pathogenic mutations were reported in African–Americans with exercise intolerance, exercise-induced muscle cramps, and rhabdomyolysis [81,82,83,84]. Others have been reported from Italian, Japanese, and Pakistani ethnicity [84,85,86,87,88]. In addition to the accumulation of non–membrane bound glycogen, there are often tubular aggregates on muscle biopsy [81]. Although most patients show an attenuated lactate rise with forearm exercise testing, some show a normal rise [81]. In contrast to GSD V, a second-wind phenomenon is often absent [89].
β-Enolase
The β-enolase protein is a distal glycolytic enzyme yielding phosphoenolpyruvate from 2-phosphoglycerate and is composed of 3 subunits (α, β, γ) with the β-subunit being the muscle predominant subunit. The β-subunit is encoded for by the ENO3 gene. The first patient described with what was called GSD XIII experienced exercise intolerance with muscle myalgias, no rhabdomyolysis, and no lactate rise with forearm ischemic testing [90]. The mutation was a compound heterozygous for an in trans ENO3 mutation resulting in 5% of residual enzyme activity in skeletal muscle [90]. With only 1 patient diagnosed with GSD XIII, this was considered extremely rare and rather mild; however, 2 other unrelated patients were reported with homozygous mutations resulting in somewhat higher residual activities (10 and 20% residual), yet more severe muscle cramps, postexertional myalgias, rhabdomyolysis, and no significant lactate rise with forearm ischemic testing [91]. Although rare, it should be considered in the differential of a patient with typical myopathic GSD symptoms and/or an attenuated lactate rise on forearm testing.
Lactate Dehydrogenase A
The muscle-specific isoform of lactate dehydrogenase (A) (LDHA) converts pyruvate to lactate in skeletal muscle. The LDHA protein is encoded for by the LDHA gene with compound heterozygous or homozygous mutations leading to symptoms of exercise intolerance, often with myalgia and rhabdomyolysis [92,93,94]. Uterine stiffness during pregnancy was also reported, and some patients have typical erythematous skin lesions [92, 95].
Treatment of Myopathic Metabolic Glycogen Metabolism Defects
Given the rarity of most of the metabolic GSDs, most of the research into therapies has been done with GSD V (McArdle disease) patients [96,97,98,99,100,101,102,103,104]; however, many of the general recommendations can be applied to other myopathic GSDs with some caveats. Prevention is the mainstay of therapy, with patients being educated on the signs and symptoms of a muscle energy crisis and taking steps to avoid triggering situations such as heavy snow shoveling, prolonged isometric contractions (i.e., moving furniture or carrying heavy items), stair climbing, etc. Unfortunately, some patients restrict activity to the point of major inactivity that ultimately leads to obesity, dysglycemia, cardiovascular disease, and other sedentary lifestyle-associated disorders. In fact, many GSD patients have low work capacity and low VO2peak [12], and a low VO2peak and physical inactivity are associated with a higher risk of all-cause mortality [105, 106]. Fortunately, many studies are showing that properly conducted endurance exercise can increase exercise capacity/fitness and raise the threshold for triggering myalgia > cramps > rhabdomyolysis in GSD V patients [98, 101, 107]; however, these suggestions are likely applicable to the other metabolic GSDs. Studies have shown increases in VO2peak, cardiac output, work capacity, and increases in enzyme markers of mitochondrial mass and β-oxidation following 14 weeks of gradually progressive endurance exercise 4 times a week for 30 to 40 min @ ~ 65% of heart rate peak in GSD V patients [98].
We and others [108, 109] have GSD V and GSD VII patients who have gradually and carefully performed resistance exercise, but this must be done with caution and carefully following all of the suggestions below including dietary state. To minimize the risk of rhabdomyolysis for a metabolic GSD patient who wishes to engage in resistance training, it is important for them to start with low repetitions and low weight and only very gradually increase the weight but to keep the repetitions generally < 10 (as opposed to the usual 12-15 repetitions) in order not to deplete phosphocreatine stores. Given that it takes ~ 2 min for aerobic phosphocreatine replenishment between bouts of contractions, we would recommend not re-exercising the same muscle group for a repeat set for ≥ 3 min. The latter is best accomplished using a circuit-set routine where one does an arm exercise, a leg exercise, and a core exercise and back to the initial exercise. It is essential for the patient to “listen to their body” and wait for a longer period of time if a muscle remains sore or to stop immediately if the muscle goes into a contracture. The provision of milk-based protein in the early postexercise period can help to maximize muscle protein synthesis and attenuate protein degradation [110]. I would also suggest 48 h (or longer if any myalgia exists) between exercise sessions.
In general, patients should start exercise at a low intensity in a well-hydrated condition (urine light yellow to clear) and avoid exercise with superimposed metabolic stressors such as fever or infection. In disorders with a second wind, it is important to back off on exercise intensity early on if shortness of breath and/or tachycardia is significant and drop the intensity to mitigate myalgias and cramps. It is also important to cease activity and stretch and hydrate if the myalgia and cramps do not subside and to monitor for pigmenturia. If a muscle group is stiff and sore, it is important to wait until that subsides before another exercise bout. For GSD V and presumably phosphorylase b kinase deficiency, the consumption of glucose or sucrose (glucose–fructose) 15 to 40 min before exercise will allow for some pyruvate availability by bypassing the defect and allowing for easier access to the second wind and improved exercise capacity [12, 65, 97, 104]. It is likely that the consumption of sucrose/glucose too early before exercise (≥ 1 h) can lead to an insulin peak and uptake of glucose and glycogen synthesis, rendering the glucose unavailable for the exercise bout. Such a practice can also lead to conversion of glucose to triglyceride storage in adipocytes and obesity, especially if the subsequent exercise does not occur or is of short duration. From a practical perspective, most fruit juices contain varying proportions of glucose and fructose and provide a convenient pre-exercise drink for patients with glycogenolytic defects.
In contrast, the consumption of carbohydrate before exercise in disorders of glycolysis (i.e., GSD VII) has the complete opposite effect and seriously inhibits exercise capacity [111], for the glucose remains unavailable for pyruvate production and the resultant increase in insulin inhibits the availability of substrates such as free fatty acids (from peripheral and IMCL lipolysis). We have a patient with GSD VII who exercises 6 days per week for the past 10 years, but he must fast overnight and exercise in the morning before eating and early on noted a dramatic attenuation of exercise capacity if he exercised after eating.
From a habitual diet perspective, it is attractive to hypothesize that increasing the proportion of dietary fat and/or protein could provide alternative substrates for exercise metabolism and/or lead to a form of substrate reduction therapy. It is known that an increase in dietary protein (or branched-chain amino acids) can increase the oxidation of branched-chain amino acids [112], and these can be oxidized by skeletal muscle during endurance exercise [113]. Consequently, it has been suggested that a high dietary protein intake plus alanine to provide pyruvate through the alanine amino transferase reaction improved symptoms in GSD V patients; however, this was an open trial [114]. Furthermore, oral branched-chain amino acids did not improve exercise capacity in GSD V patients [96]. A high dietary protein intake (~ 25% of energy) has been recommended for patients with GSD III, IV, and IX [115], with dramatic case report evidence in GSD III [116].
Collectively, the data would suggest that GSD myopathy patients consume at least 1.2 g protein/kg/day to maintain muscle mass, especially with aging [117]; however, high intakes of greater than 1.6 g/kg/day cannot currently be recommended given that this level is the maximal one for stimulation of protein synthesis even for athletes performing heavy training [112]. Given that the oxidation of free fatty acids is an important compensatory mechanism in patients with GSD V [118, 119](and presumably other myopathic GSDs), there has been interest in the use of a ketogenic diet by some patients who have anecdotally reported a significant reduction in GSD-associated symptoms and increased exercise capacity [120]. One report found exercise capacity improvements with a combination of a ketogenic diet and creatine monohydrate [121]. Given the practical challenges in maintaining ketosis and the unknown long-term cardiometabolic effects from a diet containing > 80% dietary fat, it would seem prudent to not routinely recommend such a diet at this time to patients with muscle metabolic GSDs.
Since myophosphorylase binds to vitamin B6 (pyridoxine) and that pyridoxine is involved in various amino acid transamination reactions that can facilitate non–glucose substrate oxidation, it is possible that pyridoxine could benefit GSD V patients. A case report found improved exercise tolerance and better glycolytic flux after pyridoxine supplementation in a GSD V patient [99]. Another case study reported improved phosphorylase activity, improved exercise capacity, and glycolytic flux after 60 to 90 mg of pyridoxine in a GSD patient [122]. The overall conclusion from a meta-analysis was that pyridoxine was not likely of benefit in GSD V patients [123]; however, the research in the area is very limited. Given the consequences of pyridoxine deficiency, it would be reasonable for GSD V patients with stop codon mutations (that lead to nonsense mediated protein decay) to ingest a multivitamin that contains pyridoxine to avoid a deficiency state.
Because of the compensatory increase in flux through the phosphocreatine > creatine pathway with impaired glycogenolysis/glycolysis, and the fact that dietary creatine ingestion can lead to increases in phosphocreatine [124], it was logical to consider that creatine ingestion could improve exercise capacity in metabolic GSD patients [102, 103]. One study found mild evidence for improvement in exercise capacity in GSD V patients with low-dose creatine ingestion [102], but an impairment in function with high doses (20 g) [103]. It is likely that the lack of dramatic improvement in exercise capacity with creatine loading was due to the fact that the forward reaction requires a proton and that is lacking in disorders of glycogenolysis/glycolysis. Furthermore, high amounts of intracellular free creatine likely inhibit the PFK step in glycolysis [6]. Given the increase in muscle mass and strength seen in a meta-analysis of muscular dystrophies with creatine supplementation [125], it is plausible that the GSDs that result in fixed proximal weakness may also benefit.
One possible metabolic bypass strategy for the treatment of the glycolytic and glycogenolytic defects could be the anaplerotic TCA cycle precursor triheptanoin. Triheptanoin is a triglyceride with 3 × 7 carbon (7C—odd chain) free fatty acids. The 7 carbon FFAs are metabolized to β-ketopentanoate (5C) and β-hydroxypentanoate (5C) that can enter the TCA cycle as propionyl-CoA > succinyl-CoA and provide an anaplerotic function [126]. In addition to the metabolic bypass potential for triheptanoin [127], there also may be an enzyme-membrane-stabilizing effect that enhances the activity of some GBE1 variants that could have a role in the treatment of polyglucosan body disease [128], and likely GSD IV patients. We have demonstrated an increase in oxidative stress in skeletal muscle from GSD V patients [129, 130]; consequently, it would be of interest to see if antioxidants could be of some clinical benefit. Finally, it is likely that future research efforts in the area of myopathic GSDs will focus on enzyme replacement therapy (ERT) and genetic manipulation/editing interventions including viral-mediated gene delivery, liposome-mediated gene delivery, and CRISPR-Cas9 therapy.
References
Preisler N, Laforet P, Madsen KL, et al. Fat and carbohydrate metabolism during exercise in late-onset Pompe disease. Mol Genet Metab 2012;107(3):462–8.
Thorell A, Hirshman MF, Nygren J et al. Exercise and insulin cause GLUT-4 translocation in human skeletal muscle. Am J Physiol 1999;277(4 Pt 1):E733–41.
Hayashi T, Wojtaszewski JF, Goodyear LJ. Exercise regulation of glucose transport in skeletal muscle. Am J Physiol 1997;273(6 Pt 1):E1039–51.
Costill DL, Sherman WM, Fink WJ, Maresh C, Witten M, Miller JM. The role of dietary carbohydrates in muscle glycogen resynthesis after strenuous running. Am J Clin Nutr 1981;34(9):1831–6.
Tarnopolsky MA, Atkinson SA, Phillips SM, MacDougall JD. Carbohydrate loading and metabolism during exercise in men and women. J Appl Physiol (1985) 1995;78(4):1360–8.
Storey KB, Hochachka PW. Activation of muscle glycolysis: a role for creatine phosphate in phosphofructokinase regulation. FEBS Lett 1974;46(1):337–9.
Benton CR, Yoshida Y, Lally J, Han XX, Hatta H, Bonen A. PGC-1alpha increases skeletal muscle lactate uptake by increasing the expression of MCT1 but not MCT2 or MCT4. Physiol Genomics 2008;35(1):45–54.
Gaitanos GC, Williams C, Boobis LH, Brooks S. Human muscle metabolism during intermittent maximal exercise. J Appl Physiol (1985) 1993;75(2):712–9.
O'Brien MJ, Viguie CA, Mazzeo RS, Brooks GA. Carbohydrate dependence during marathon running. Med Sci Sports Exerc 1993;25(9):1009–17.
Romijn JA, Coyle EF, Sidossis LS, et al. Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am J Physiol 1993;265(3 Pt 1):E380–91.
Romijn JA, Coyle EF, Sidossis LS, Rosenblatt J, Wolfe RR. Substrate metabolism during different exercise intensities in endurance-trained women. J Appl Physiol (1985) 2000;88(5):1707–14.
Haller RG, Vissing J. Spontaneous “second wind” and glucose-induced second “second wind” in McArdle disease: oxidative mechanisms. Arch Neurol 2002;59(9):1395–402.
Better OS, Abassi ZA. Early fluid resuscitation in patients with rhabdomyolysis. Nat Rev Nephrol 2011;7(7):416–22.
De Castro M, Johnston J, Biesecker L. Determining the prevalence of McArdle disease from gene frequency by analysis of next-generation sequencing data. Genet Med 2015;17(12):1002–6.
Santalla A, Nogales-Gadea G, Encinar AB, et al. Genotypic and phenotypic features of all Spanish patients with McArdle disease: a 2016 update. BMC Genomics 2017;18(Suppl 8):819.
Lucia A, Ruiz JR, Santalla A, et al. Genotypic and phenotypic features of McArdle disease: insights from the Spanish national registry. J Neurol Neurosurg Psychiatry 2012;83(3):322–8.
Mc AB. Myopathy due to a defect in muscle glycogen breakdown. Clin Sci 1951;10(1):13–35.
Mineo I, Tarui S. Myogenic hyperuricemia: what can we learn from metabolic myopathies? Muscle Nerve Suppl 1995;3:S75–81.
Cheraud C, Froissart R, Lannes B, Echaniz-Laguna A. Novel variant in the PYGM gene causing late-onset limb-girdle myopathy, ptosis, and camptocormia. Muscle Nerve 2018;57(1):157–60.
Tarnopolsky M, Stevens L, MacDonald JR, et al. Diagnostic utility of a modified forearm ischemic exercise test and technical issues relevant to exercise testing. Muscle Nerve 2003;27(3):359–66.
Kazemi-Esfarjani P, Skomorowska E, Jensen TD, Haller RG, Vissing J. A nonischemic forearm exercise test for McArdle disease. Ann Neurol 2002;52(2):153–9.
Clemens PR, Yamamoto M, Engel AG. Adult phosphorylase b kinase deficiency. Ann Neurol 1990;28(4):529–38.
Wilkinson DA, Tonin P, Shanske S, Lombes A, Carlson GM, DiMauro S. Clinical and biochemical features of 10 adult patients with muscle phosphorylase kinase deficiency. Neurology 1994;44(3 Pt 1):461–6.
Echaniz-Laguna A, Akman HO, Mohr M, et al. Muscle phosphorylase b kinase deficiency revisited. Neuromuscul Disord 2010;20(2):125–7.
Wuyts W, Reyniers E, Ceuterick C, Storm K, de Barsy T, Martin JJ. Myopathy and phosphorylase kinase deficiency caused by a mutation in the PHKA1 gene. Am J Med Genet A 2005;133A(1):82–4.
Orngreen MC, Schelhaas HJ, Jeppesen TD, et al. Is muscle glycogenolysis impaired in X-linked phosphorylase b kinase deficiency? Neurology 2008;70(20):1876–82.
Preisler N, Orngreen MC, Echaniz-Laguna A, et al. Muscle phosphorylase kinase deficiency: a neutral metabolic variant or a disease? Neurology 2012;78(4):265–8.
Burwinkel B, Hu B, Schroers A, et al. Muscle glycogenosis with low phosphorylase kinase activity: mutations in PHKA1, PHKG1 or six other candidate genes explain only a minority of cases. Eur J Hum Genet 2003;11(7):516–26.
Beauchamp NJ, Dalton A, Ramaswami U, et al. Glycogen storage disease type IX: high variability in clinical phenotype. Mol Genet Metab 2007;92(1–2):88–99.
Aoyama Y, Ozer I, Demirkol M, et al. Molecular features of 23 patients with glycogen storage disease type III in Turkey: a novel mutation p.R1147G associated with isolated glucosidase deficiency, along with 9 AGL mutations. J Hum Genet 2009;54(11):681–6.
Endo Y, Horinishi A, Vorgerd M, et al. Molecular analysis of the AGL gene: heterogeneity of mutations in patients with glycogen storage disease type III from Germany, Canada, Afghanistan, Iran, and Turkey. J Hum Genet 2006;51(11):958–63.
Lucchiari S, Fogh I, Prelle A, et al. Clinical and genetic variability of glycogen storage disease type IIIa: seven novel AGL gene mutations in the Mediterranean area. Am J Med Genet 2002;109(3):183–90.
Okubo M, Horinishi A, Takeuchi M, et al. Heterogeneous mutations in the glycogen-debranching enzyme gene are responsible for glycogen storage disease type IIIa in Japan. Hum Genet 2000;106(1):108–15.
Coleman RA, Winter HS, Wolf B, Gilchrist JM, Chen YT. Glycogen storage disease type III (glycogen debranching enzyme deficiency): correlation of biochemical defects with myopathy and cardiomyopathy. Ann Intern Med 1992;116(11):896–900.
Cornelio F, Bresolin N, Singer PA, DiMauro S, Rowland LP. Clinical varieties of neuromuscular disease in debrancher deficiency. Arch Neurol 1984;41(10):1027–32.
Kiechl S, Kohlendorfer U, Thaler C, et al. Different clinical aspects of debrancher deficiency myopathy. J Neurol Neurosurg Psychiatry 1999;67(3):364–8.
DiMauro S, Hartwig GB, Hays A, et al. Debrancher deficiency: neuromuscular disorder in 5 adults. Ann Neurol 1979;5(5):422–36.
Marbini A, Gemignani F, Saccardi F, Rimoldi M. Debrancher deficiency neuromuscular disorder with pseudohypertrophy in two brothers. J Neurol 1989;236(7):418–20.
Stemmerik MG, Madsen KL, Laforet P, Buch AE, Vissing J. Muscle glycogen synthesis and breakdown are both impaired in glycogenin-1 deficiency. Neurology 2017;89(24):2491–4.
Moslemi AR, Lindberg C, Nilsson J, Tajsharghi H, Andersson B, Oldfors A. Glycogenin-1 deficiency and inactivated priming of glycogen synthesis. N Engl J Med 2010;362(13):1203–10.
Malfatti E, Nilsson J, Hedberg-Oldfors C, et al. A new muscle glycogen storage disease associated with glycogenin-1 deficiency. Ann Neurol 2014;76(6):891–8.
Akman HO, Aykit Y, Amuk OC, et al. Late-onset polyglucosan body myopathy in five patients with a homozygous mutation in GYG1. Neuromuscul Disord 2016;26(1):16–20.
High KA. The Jeremiah Metzger Lecture: gene therapy for inherited disorders: from Christmas disease to Leber’s amaurosis. Trans Am Clin Climatol Assoc 2009;120:331–59.
Holloszy JO, Oscai LB, Don IJ, Mole PA. Mitochondrial citric acid cycle and related enzymes: adaptive response to exercise. Biochem Biophys Res Commun 1970;40(6):1368–73.
Ben Yaou R, Hubert A, Nelson I, et al. Clinical heterogeneity and phenotype/genotype findings in 5 families with GYG1 deficiency. Neurol Genet 2017;3(6):e208.
Kollberg G, Tulinius M, Gilljam T, et al. Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. N Engl J Med 2007;357(15):1507–14.
Cameron JM, Levandovskiy V, MacKay N, et al. Identification of a novel mutation in GYS1 (muscle-specific glycogen synthase) resulting in sudden cardiac death, that is diagnosable from skin fibroblasts. Mol Genet Metab 2009;98(4):378–82.
McCue ME, Valberg SJ, Miller MB, et al. Glycogen synthase (GYS1) mutation causes a novel skeletal muscle glycogenosis. Genomics 2008;91(5):458–66.
Maile CA, Hingst JR, Mahalingan KK, et al. A highly prevalent equine glycogen storage disease is explained by constitutive activation of a mutant glycogen synthase. Biochim Biophys Acta 2017;1861(1 Pt A):3388–98.
Bao Y, Kishnani P, Wu JY, Chen YT. Hepatic and neuromuscular forms of glycogen storage disease type IV caused by mutations in the same glycogen-branching enzyme gene. J Clin Invest 1996;97(4):941–8.
Bruno C, van Diggelen OP, Cassandrini D, et al. Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV). Neurology 2004;63(6):1053–8.
Bruno C, DiRocco M, Lamba LD, et al. A novel missense mutation in the glycogen branching enzyme gene in a child with myopathy and hepatopathy. Neuromuscul Disord 1999;9(6–7):403–7.
Servidei S, Riepe RE, Langston C, et al. Severe cardiopathy in branching enzyme deficiency. J Pediatr 1987;111(1):51–6.
Malfatti E, Barnerias C, Hedberg-Oldfors C, et al. A novel neuromuscular form of glycogen storage disease type IV with arthrogryposis, spinal stiffness and rare polyglucosan bodies in muscle. Neuromuscul Disord 2016;26(10):681–7.
Bruno C, Servidei S, Shanske S, et al. Glycogen branching enzyme deficiency in adult polyglucosan body disease. Ann Neurol 1993;33(1):88–93.
Hellmann MA, Kakhlon O, Landau EH, et al. Frequent misdiagnosis of adult polyglucosan body disease. J Neurol 2015;262(10):2346–51.
McDonald TD, Faust PL, Bruno C, DiMauro S, Goldman JE. Polyglucosan body disease simulating amyotrophic lateral sclerosis. Neurology 1993;43(4):785–90.
Paradas C, Akman HO, Ionete C, et al. Branching enzyme deficiency: expanding the clinical spectrum. JAMA Neurol 2014;71(1):41–7.
Toscano A, Musumeci O. Tarui disease and distal glycogenoses: clinical and genetic update. Acta Myol 2007;26(2):105–7.
Sherman JB, Raben N, Nicastri C, et al. Common mutations in the phosphofructokinase-M gene in Ashkenazi Jewish patients with glycogenesis VII—and their population frequency. Am J Hum Genet 1994;55(2):305–13.
Tani K, Fujii H, Takegawa S, et al. Two cases of phosphofructokinase deficiency associated with congenital hemolytic anemia found in Japan. Am J Hematol 1983;14(2):165–74.
Hamaguchi T, Nakajima H, Noguchi T, et al. A new variant of muscle phosphofructokinase deficiency in a Japanese case with abnormal RNA splicing. Biochem Biophys Res Commun 1994;202(1):444–9.
Shimizu T, Kono N, Kiyokawa H, et al. Erythrocyte glycolysis and its marked alterations by muscular exercise in type VII glycogenosis. Blood 1988;71(4):1130–4.
Tarui S, Okuno G, Ikura Y, Tanaka T, Suda M, Nishikawa M. Phosphofructokinase deficiency in skeletal muscle. a new type of glycogenosis. Biochem Biophys Res Commun 1965;19:517–23.
Haller RG, Vissing J. No spontaneous second wind in muscle phosphofructokinase deficiency. Neurology 2004;62(1):82–6.
Musumeci O, Bruno C, Mongini T, et al. Clinical features and new molecular findings in muscle phosphofructokinase deficiency (GSD type VII). Neuromuscul Disord 2012;22(4):325–30.
Etiemble J, Kahn A, Boivin P, Bernard JF, Goudemand M. Hereditary hemolytic anemia with erythrocyte phosphofructokinase deficiency: studies of some properties of erythrocyte and muscle enzyme. Hum Genet 1976;31(1):83–91.
Vives-Corrons JL, Koralkova P, Grau JM, Manu Pereira Mdel M, Van Wijk R. First description of phosphofructokinase deficiency in spain: identification of a novel homozygous missense mutation in the PFKM gene. Front Physiol 2013;4:393.
Lin HC, Young C, Wang PJ, Shen YZ. Muscle phosphofructokinase deficiency (Tarui’s disease): report of a case. J Formos Med Assoc 1999;98(3):205–8.
Wu PL, Yang YN, Tey SL, Yang CH, Yang SN, Lin CS. Infantile form of muscle phosphofructokinase deficiency in a premature neonate. Pediatr Int 2015;57(4):746–9.
Amit R, Bashan N, Abarbanel JM, Shapira Y, Sofer S, Moses S. Fatal familial infantile glycogen storage disease: multisystem phosphofructokinase deficiency. Muscle Nerve 1992;15(4):455–8.
Garcia M, Pujol A, Ruzo A, et al. Phosphofructo-1-kinase deficiency leads to a severe cardiac and hematological disorder in addition to skeletal muscle glycogenosis. PLoS Genet 2009;5(8):e1000615.
Servidei S, Bonilla E, Diedrich RG, et al. Fatal infantile form of muscle phosphofructokinase deficiency. Neurology 1986;36(11):1465–70.
DiMauro S, Dalakas M, Miranda AF. Phosphoglycerate kinase deficiency: another cause of recurrent myoglobinuria. Ann Neurol 1983;13(1):11–9.
Aasly J, van Diggelen OP, Boer AM, Bronstad G. Phosphoglycerate kinase deficiency in two brothers with McArdle-like clinical symptoms. Eur J Neurol 2000;7(1):111–3.
Beutler E. PGK deficiency. Br J Haematol 2007;136(1):3–11.
Cohen-Solal M, Valentin C, Plassa F, et al. Identification of new mutations in two phosphoglycerate kinase (PGK) variants expressing different clinical syndromes: PGK Creteil and PGK Amiens. Blood 1994;84(3):898–903.
Fermo E, Bianchi P, Chiarelli LR, et al. A new variant of phosphoglycerate kinase deficiency (p.I371K) with multiple tissue involvement: molecular and functional characterization. Mol Genet Metab 2012;106(4):455–61.
Sugie H, Sugie Y, Nishida M, et al. Recurrent myoglobinuria in a child with mental retardation: phosphoglycerate kinase deficiency. J Child Neurol 1989;4(2):95–9.
Svaasand EK, Aasly J, Landsem VM, Klungland H. Altered expression of PGK1 in a family with phosphoglycerate kinase deficiency. Muscle Nerve 2007;36(5):679–84.
Salameh J, Goyal N, Choudry R, Camelo-Piragua S, Chong PS. Phosphoglycerate mutase deficiency with tubular aggregates in a patient from Panama. Muscle Nerve 2013;47(1):138–40.
Koo B, Oskarsson B. Phosphoglycerate mutase deficiency (glycogen storage disease X) caused by a novel variant in PGAM-M. Neuromuscul Disord 2016;26(10):688–90.
DiMauro S, Miranda AF, Khan S, Gitlin K, Friedman R. Human muscle phosphoglycerate mutase deficiency: newly discovered metabolic myopathy. Science 1981;212(4500):1277–9.
Tsujino S, Shanske S, Sakoda S, Toscano A, DiMauro S. Molecular genetic studies in muscle phosphoglycerate mutase (PGAM-M) deficiency. Muscle Nerve Suppl 1995;3:S50–3.
Tonin P, Bruno C, Cassandrini D, et al. Unusual presentation of phosphoglycerate mutase deficiency due to two different mutations in PGAM-M gene. Neuromuscul Disord 2009;19(11):776–8.
Toscano A, Tsujino S, Vita G, Shanske S, Messina C, Dimauro S. Molecular basis of muscle phosphoglycerate mutase (PGAM-M) deficiency in the Italian kindred. Muscle Nerve 1996;19(9):1134–7.
Naini A, Toscano A, Musumeci O, Vissing J, Akman HO, DiMauro S. Muscle phosphoglycerate mutase deficiency revisited. Arch Neurol 2009;66(3):394–8.
Kawashima N, Mishima M, Shindo R, et al. Partial deficiency of phosphoglycerate mutase with diabetic polyneuropathy: the first Japanese patient. Intern Med 1996;35(10):799–802.
Vissing J, Quistorff B, Haller RG. Effect of fuels on exercise capacity in muscle phosphoglycerate mutase deficiency. Arch Neurol 2005;62(9):1440–3.
Comi GP, Fortunato F, Lucchiari S, et al. Beta-enolase deficiency, a new metabolic myopathy of distal glycolysis. Ann Neurol 2001;50(2):202–7.
Musumeci O, Brady S, Rodolico C, et al. Recurrent rhabdomyolysis due to muscle beta-enolase deficiency: very rare or underestimated? J Neurol 2014;261(12):2424–8.
Kanno T, Sudo K, Maekawa M, Nishimura Y, Ukita M, Fukutake K. Lactate dehydrogenase M-subunit deficiency: a new type of hereditary exertional myopathy. Clin Chim Acta 1988;173(1):89–98.
Kanno T, Sudo K, Takeuchi I, et al. Hereditary deficiency of lactate dehydrogenase M-subunit. Clin Chim Acta 1980;108(2):267–76.
Maekawa M, Sudo K, Li SS, Kanno T. Genotypic analysis of families with lactate dehydrogenase A (M) deficiency by selective DNA amplification. Hum Genet 1991;88(1):34–8.
Kanno T, Maekawa M. Lactate dehydrogenase M-subunit deficiencies: clinical features, metabolic background, and genetic heterogeneities, Muscle Nerve Suppl 1995;3:S54–60.
MacLean D, Vissing J, Vissing SF, Haller RG. Oral branched-chain amino acids do not improve exercise capacity in McArdle disease. Neurology 1998;51(5):1456–9.
Andersen ST, Haller RG, Vissing J. Effect of oral sucrose shortly before exercise on work capacity in McArdle disease. Arch Neurol 2008;65(6):786–9.
Haller RG, Wyrick P, Taivassalo T, Vissing J. Aerobic conditioning: an effective therapy in McArdle’s disease. Ann Neurol 2006;59(6):922–8.
Izumi R, Suzuki N, Kato K, et al. A case of McArdle disease: efficacy of vitamin B6 on fatigability and impaired glycogenolysis. Intern Med 2010;49(15):1623–5.
Martinuzzi A, Liava A, Trevisi E, Antoniazzi L, Frare M. Chronic therapy for McArdle disease: the randomized trial with ACE inhibitor. Acta Myol 2007;26(1):64–6.
Quinlivan R, Vissing J, Hilton-Jones D, Buckley J. Physical training for McArdle disease. Cochrane Database Syst Rev 2011(12):CD007931.
Vorgerd M, Grehl T, Jager M, Muller K, Freitag G, Patzold T, et al. Creatine therapy in myophosphorylase deficiency (McArdle disease): a placebo-controlled crossover trial. Arch Neurol 2000;57(7):956–63.
Vorgerd M, Zange J, Kley R, Grehl T, Husing A, Jager M, et al. Effect of high-dose creatine therapy on symptoms of exercise intolerance in McArdle disease: double-blind, placebo-controlled crossover study. Arch Neurol 2002;59(1):97–101.
Vissing J, Haller RG. The effect of oral sucrose on exercise tolerance in patients with McArdle’s disease. N Engl J Med 2003;349(26):2503–9.
Blair SN, Kohl HW, 3rd, Paffenbarger RS, Jr., Clark DG, Cooper KH, Gibbons LW. Physical fitness and all-cause mortality. A prospective study of healthy men and women. JAMA 1989;262(17):2395–401.
Wei M, Kampert JB, Barlow CE, Nichaman MZ, Gibbons LW, Paffenbarger RS, Jr., et al. Relationship between low cardiorespiratory fitness and mortality in normal-weight, overweight, and obese men. JAMA 1999;282(16):1547–53.
Mate-Munoz JL, Moran M, Perez M, Chamorro-Vina C, Gomez-Gallego F, Santiago C, et al. Favorable responses to acute and chronic exercise in McArdle patients. Clin J Sport Med 2007;17(4):297–303.
Garcia-Benitez S, Fleck SJ, Naclerio F, Martin MA, Lucia A. Resistance (weight lifting) training in an adolescent with McArdle disease. J Child Neurol 2013;28(6):805–8.
Santalla A, Munguia-Izquierdo D, Brea-Alejo L, Pagola-Aldazabal I, Diez-Bermejo J, Fleck SJ, et al. Feasibility of resistance training in adult McArdle patients: clinical outcomes and muscle strength and mass benefits. Front Aging Neurosci 2014;6:334.
Morton RW, McGlory C, Phillips SM. Nutritional interventions to augment resistance training-induced skeletal muscle hypertrophy. Front Physiol 2015;6:245.
Haller RG, Lewis SF. Glucose-induced exertional fatigue in muscle phosphofructokinase deficiency. N Engl J Med 1991;324(6):364–9.
Tarnopolsky MA, Atkinson SA, MacDougall JD, Chesley A, Phillips S, Schwarcz HP. Evaluation of protein requirements for trained strength athletes. J Appl Physiol (1985) 1992;73(5):1986–95.
McKenzie S, Phillips SM, Carter SL, Lowther S, Gibala MJ, Tarnopolsky MA. Endurance exercise training attenuates leucine oxidation and BCOAD activation during exercise in humans. Am J Physiol Endocrinol Metab 2000;278(4):E580–7.
Slonim AE, Goans PJ. Myopathy in McArdle’s syndrome. Improvement with a high-protein diet. N Engl J Med 1985;312(6):355–9.
Goldberg T, Slonim AE. Nutrition therapy for hepatic glycogen storage diseases. J Am Diet Assoc 1993;93(12):1423–30.
Slonim AE, Weisberg C, Benke P, Evans OB, Burr IM. Reversal of debrancher deficiency myopathy by the use of high-protein nutrition. Ann Neurol 1982;11(4):420–2.
Traylor DA, Gorissen SHM, Phillips SM. Perspective: Protein requirements and optimal intakes in aging: are we ready to recommend more than the recommended daily allowance? Adv Nutr 2018.
Andersen ST, Jeppesen TD, Taivassalo T, Sveen ML, Heinicke K, Haller RG, et al. Effect of changes in fat availability on exercise capacity in McArdle disease. Arch Neurol 2009;66(6):762–6.
Orngreen MC, Jeppesen TD, Andersen ST, Taivassalo T, Hauerslev S, Preisler N, et al. Fat metabolism during exercise in patients with McArdle disease. Neurology 2009;72(8):718–24.
Baranano KW, Hartman AL. The ketogenic diet: uses in epilepsy and other neurologic illnesses. Curr Treat Options Neurol 2008;10(6):410–9.
Vorgerd M, Zange J. Treatment of glycogenosys type V (McArdle disease) with creatine and ketogenic diet with clinical scores and with 31P-MRS on working leg muscle. Acta Myol 2007;26(1):61–3.
Sato S, Ohi T, Nishino I, Sugie H. Confirmation of the efficacy of vitamin B6 supplementation for McArdle disease by follow-up muscle biopsy. Muscle Nerve 2012;45(3):436–40.
Quinlivan R, Martinuzzi A, Schoser B. Pharmacological and nutritional treatment for McArdle disease (glycogen storage disease type V). Cochrane Database Syst Rev 2014;11:CD003458.
Hultman E, Soderlund K, Timmons JA, Cederblad G, Greenhaff PL. Muscle creatine loading in men. J Appl Physiol (1985) 1996;81(1):232–7.
Kley RA, Tarnopolsky MA, Vorgerd M. Creatine for treating muscle disorders. Cochrane Database Syst Rev 2013;6:CD004760.
Roe CR, Mochel F. Anaplerotic diet therapy in inherited metabolic disease: therapeutic potential. J Inherit Metab Dis 2006;29(2–3):332–40.
Orngreen MC, Vissing J. Treatment opportunities in patients with metabolic myopathies. Curr Treat Options Neurol 2017;19(11):37.
Alvarez R, Casas J, Lopez DJ, Ibarguren M, Suari-Rivera A, Teres S, et al. Triacylglycerol mimetics regulate membrane interactions of glycogen branching enzyme: implications for therapy. J Lipid Res 2017;58(8):1598–612.
Kaczor JJ, Robertshaw HA, Tarnopolsky MA. Higher oxidative stress in skeletal muscle of McArdle disease patients. Mol Genet Metab Rep 2017;12:69–75.
Kitaoka Y, Ogborn DI, Nilsson MI, Mocellin NJ, MacNeil LG, Tarnopolsky MA. Oxidative stress and Nrf2 signaling in McArdle disease. Mol Genet Metab 2013;110(3):297–302.
Acknowledgments
I would like to thank Mr. Gordon Reid and Giant Tiger stores for the kind donations for the Giant Tiger Metabolic Testing facility and research into McArdle disease. The Canadian Institute for Health Research (CIHR) has supported Dr. Tarnopolsky’s work evaluating therapies for metabolic muscle disorders.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(PDF 1225 kb)
Rights and permissions
About this article

Cite this article
Tarnopolsky, M.A. Myopathies Related to Glycogen Metabolism Disorders. Neurotherapeutics 15, 915–927 (2018). https://doi.org/10.1007/s13311-018-00684-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-018-00684-2