
Abstract
A reliable model of a disease pathomechanism is the first step to develop targeted treatment. In facioscapulohumeral muscular dystrophy (FSHD), the third most common muscular dystrophy, recent advances in understanding the complex genetics and epigenetics have led to the identification of a disease mechanism, moving the field towards targeted therapy development. FSHD is caused by expression of DUX4, a retrogene located on the D4Z4 macrosatellite repeat array on chromosome 4q35, a gene expressed in the germline but typically repressed in somatic tissue. DUX4 derepression results from opening of the chromatin structure either by contraction of the number of repeats (FSHD1) or by chromatin hypomethylation of the D4Z4 repeats resulting from mutations in SMCHD1, a gene involved in chromatin methylation (FSHD2). The resulting expression of DUX4, a transcriptional regulator, and its target genes is toxic to skeletal muscle. Efforts for targeted treatment currently focus on disrupting DUX4 expression or blocking 1 or more of several downstream effects of DUX4. This review article focuses on the underlying FSHD genetics, current understanding of the pathomechanism, and potential treatment strategies in FSHD. In addition, recent advances in the development of new clinical outcome measures as well as biomarkers, critical for the success of future clinical trials, are reviewed.
Similar content being viewed by others

Introduction
Facioscapulohumeral muscular dystrophy (FSHD) is the third most common muscular dystrophy after Duchenne muscular dystrophy and myotonic dystrophy with an estimated prevalence of 1:15,000 [1]. However, due to the high degree of clinical variability with up to 20% of genetically affected but asymptomatic individuals [2], the disease frequency is likely underestimated. A recent study in the Netherlands estimated the prevalence rate almost twice as high (1:8300) [3]. In the majority of cases, the disease is inherited in an autosomal dominant pattern with about 10% of de novo mutations with a high frequency of somatic mosaicism [4]. In contrast to Duchenne muscular dystrophy and myotonic dystrophy, bulbar, respiratory, and cardiac involvement is relatively rare in FSHD and most patients have a normal life expectancy. However, physical limitations are significant, resulting in disability or job modifications and a 6-year risk of wheelchair use of 24% [5]. There is currently no disease-modifying treatment available for FSHD, but recent advances in discovering the complex molecular pathophysiology of FSHD have led to a better understanding of the phenotypic variability and allow for development of targeted treatments.
Molecular Genetics
While the genetic mutation causing FSHD was mapped to chromosome 4q35 in 1990 [6, 7] and shortly thereafter a pathogenic loss of D4Z4 macrosatellite repeats was identified [8], the exact molecular pathophysiology of the disease remained uncertain for many years. Recent discoveries of a second pathogenic mechanism and epigenetic factors have moved the field forward towards drug development.
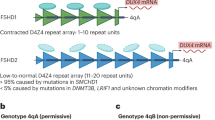
Each D4Z4 unit on chromosome 4q35 contains a copy of DUX4 (double homeobox 4) retrogene, which is a transcription factor expressed in the germline [9]. Healthy individuals carry 11 to 100 D4Z4 repeats (each 3.3 kb size), located within heterochromatin and do not undergo transcription in somatic tissues (Fig. 1). Patients with FSHD carry a reduced number of 1 to 10 repeats, referred to as a “contraction.” The presence of at least 1 repeat, containing a copy of the DUX4 gene, is required to cause disease. This contraction of D4Z4 repeats results in hypomethylation and a decrease in the repressive heterochromatin of the D4Z4 repeats, often referred to as “chromatin relaxation” or “opening of the chromatin structure.” This chromatin relaxation allows DUX4 to be transcribed. However, the transcribed DUX4 full-length mRNA is not stable due to the lack of a polyadenylation signal in the DUX4 sequence. Therefore, repeat contraction and chromatin relaxation are necessary, but not sufficient to cause FSHD. Distal to the last D4Z4 repeat, the chromosome comes in 2 major haplotypes: A or B [10]. The most prevalent 4qA haplotype, but not 4qB, contains a polymorphic polyadenylation signal (PAS), which stabilizes the transcribed DUX4 mRNA and allows for DUX4 protein expression in skeletal muscle [11]. Hence, chromatin relaxation must occur on the specific “permissive” A haplotype to be pathogenic, while contraction on a B variant does not cause the disease [12]. Of all patients with clinical symptoms and signs of FSHD, 95% have a repeat contraction on a chromosome with an A haplotype, termed FSHD type 1 (FSHD1).

This figure displays the spectrum of the genetic mechanisms in FSHD. Normal: in healthy individuals, both copies of 4q35 contain 11 to 100 repeats with normal methylation or, rarely, a contraction with hypomethylation on a nonpermissive B allele. In this figure, we display only 1 copy of 4q35 with a permissive A allele, which is necessary to cause FSHD. In FSHD1, 1 copy of the 4q35 is contracted with hypomethylation of the D4Z4 repeat array. In patients with 1 to 6 repeats, the repeat number is associated with disease severity. In patients with 7 to 10 repeats, nonpenetrance is more common and epigenetic factors (such as mutations in SMCHD1) play a larger role. In FSHD2, 1 copy of the 4q35 contains 11 to 20 repeats. A mutation in SMCHD1 or DNMT3B gene is present and D4Z4 repeat arrays are hypomethylated on both 4q35 copies
FSHD2
The remaining 5% of patients with clinical signs and symptoms of FSHD, phenotypically indistinguishable from FSHD1, typically have a low normal number of repeats on chromosome 4q35, but in addition show a, contraction independent, profound DNA hypomethylation [13] on both copies of D4Z4 [14], with at least 1 4qA variant, and are termed FSHD type 2 (FSHD2). While the D4Z4 repeat number in FSHD2 is normal, most FSHD2 patients have less repeats than the average repeat number in the control population [15], typically ranging from 11 to 20 repeats. Similar to FSHD1, hypomethylation and chromatin relaxation is necessary but not sufficient to result in disease, unless a permissive A allele with a PAS is present, to stabilize the DUX4 mRNA in skeletal muscle. While DNA hypomethylation and chromatin relaxation in FSHD1 is only seen on the contracted allele, both alleles of chromosome 4q35 and similar D4Z4 repeats on chromosome 10 are hypomethylated in FSHD2. This widespread hypomethylation suggests a problem with a gene regulating chromatin methylation. In individuals with FSHD2, 85% have a mutation in the SMCHD1 (structural maintenance of chromosomes flexible hinge domain containing 1) gene on chromosome 18 [15, 16]. SMCHD1 functions as an epigenetic repressor (i.e., it turns genes “off”) and is involved in X chromosome inactivation. SMCHD1 binds to D4Z4 and, if reduced in skeletal muscle, results in DUX4 expression [16]. However, not all FSHD2 patients carry SMCHD1 mutations. In 2 SMCHD1-negative families with FSHD2, a heterozygous mutation in DNA methyltransferase 3B (DNMT3B) gene was identified as another cause of D4Z4 derepression [17]. FSHD2 is consequently a digenic disease requiring the occurrence of 2 genetic variants on separate chromosomes: an SMCHD1 or DNMT3B mutation along with at least 1 4qA variant on chromosome 4q35.
The Concept of Epigenetic Susceptibility
Among patients with FSHD, there is marked clinical variability between and within families with incomplete penetrance. Some of this variability can be accounted for by subtle epigenetic differences. In FSHD1, there is an inverse correlation between residual repeat size and disease severity, with carriers of 1 to 6 D4Z4 unit repeats being more severely affected [18]. This is likely due to changes in the chromatin structure associated with larger contractions. Carriers with a range of 7 to 10 unit repeats show the highest clinical variability and nonpenetrance is more common [15, 19]. Family studies show that affected compared to nonaffected carriers tend to have a greater degree of D4Z4 hypomethylation than might be expected based on the sizes of the D4Z4 arrays, suggesting a greater epigenetic susceptibility and less impact of repeat size on penetrance and disease severity in individuals with 7 to 10 repeats [15]. SMCHD1 mutations do not only seem to play a critical role in FSHD2, but also have been identified as a disease modifier in FSHD1: Patients with both an FSHD1 allele and an SMCHD1 mutation were more severely affected than affected family members with only 1 of the 2 genetic mutations [20, 21]. The modifier role of SMCHD1 on disease severity has also been studied in a mouse model by crossbreeding D4Z4-2.5 mice with mice haploinsufficient for SMCHD1 which resulted in an exacerbated phenotype [22]. It will be important to learn more about how SMCHD1 variants affect the D4Z4 structure and how this influences FSHD disease variability and penetrance. In addition, mutations in another gene, the FAT1 gene on chromosome 4q is postulated to function as an epigenetic modifier of the D4Z4 repeats [23, 24].
In FSHD2, both the D4Z4 repeat array size and the nature of the SMCHD1 mutation have shown to have an impact on D4Z4 hypomethylation and disease severity [15]. The repeat size contributes to variability within a family, while the type of mutation is responsible for variability between families. A permissive allele carrying a smaller sized D4Z4 repeat along with a SMCHD1 mutation that preserves the open reading frame generally results in greater disease severity compared to longer repeats or SMCHD1 mutations disrupting the open reading frame [15]. Some patients with FSHD2 carry 2 A alleles instead of 1 A and 1 B allele. Having 2 hypomethylated A alleles compared to 1 did not seem to influence the phenotype [14].
The discovery of the digenic form of FSHD did not only extend the list of pathogenic mutations, but also provided a new avenue of research, to quantify and characterize methylation status and examine its relationship to phenotypic variability in FSHD.
Molecular Pathomechanism: DUX4 Toxicity
Both genetic mechanisms of FSHD1 and FSHD2 converge at the level of chromatin relaxation, transcription of DUX4 mRNA, and inappropriate expression of DUX4 protein in myonuclei, in the setting of a permissive 4qA allele [12, 25]. In myotube cultures, DUX4 is expressed only within a few myonuclei, but within those in substantial amounts, and similar findings are seen in the iDUX4pA mouse model [25, 26]. There is consensus that DUX4 expression is toxic to skeletal muscle and causes FSHD. DUX4 is normally expressed early in development, in the germline and pluripotent stem cells [25]. DUX4 is also reported to be expressed in the thymus [9], and keratinocytes [27], but is epigenetically suppressed in most somatic tissues including muscle [25]. The role of DUX4 in the human germline is not well established, but seems to play a role in promotion of embryonal transcription [28]. Understanding the toxic effects of DUX4 protein on skeletal muscle and FSHD pathophysiology is still subject of ongoing research efforts, but several mechanisms have been proposed, including activating expression of stem cell genes, suppression of the innate immune response [29] and nonsense-mediated RNA decay (NMD) pathways [30], altering RNA processing with accumulation of aberrant and double-stranded RNAs [31], inhibition of myogenesis and muscle regeneration, and induction of cell death [32, 33]. In search of potential treatment targets, several factors responsible for DUX4 repression have been postulated, such as involvement of MYC-mediated apoptotic pathways [31], the nucleosome remodeling deacetylase (NuRD), and chromatin assembly factor 1 (CAF-1) complexes [34].
Expression of DUX4 has been reported in thymus and keratinocytes [27], suggesting that DUX4 may have a function outside the germline [22]. Hence, future research will need to clarify whether therapeutic repression of DUX4 might have detrimental side effects and whether treatments will need to be tissue selective. In addition, DUX4 has been seen in muscle tissue of genetically diagnosed asymptomatic FSHD subjects and at low levels in genetically unaffected subjects, leading back to the question of whether DUX4 is tolerated in muscle in certain situations at low levels and how epigenetic modifiers play a role in DUX4 repression, disease onset, and progression [35].
The Role of the Stage of Cell Development and DUX4 Expression
While there is consensus about the mechanisms associated with incomplete epigenetic repression of DUX4, other epigenetic factors and the selective tissue involvement are not yet well understood. It has been shown that DUX4 mRNA is only present in a small subset of nuclei, which produce a relatively large amount of DUX4 mRNA and protein. In addition, somatic expression of DUX4 mRNA per se is not pathogenic as it can be detected at lower levels in cultures of healthy myogenic cells. Furthermore, it has been shown that SMCHD1 protein levels decrease during muscle cell differentiation, correlating with DUX4 expression. Hence, differentiated muscle cells might be particularly prone to incomplete D4Z4 repression [36].
These findings suggest that DUX4 might be expressed in different cells at different times, due to the state of the cell, indicating that levels and/or timing of somatic DUX4 expression influences disease [15, 35, 37, 38].
Differences Between FSHD 1 and 2
FSHD1 and 2 have the same downstream disease mechanism, DUX4 derepression, yet there are some unique epigenetic responses with respect to the underlying genetic mechanism upstream. In FSHD2 but not in FSHD1, PRC2-mediated H3K27 trimethylation of D4Z4 seems to play a role in the disease [36]. This might have implications for drug development, depending on which target is chosen.
Animal Models
The identification of DUX4 derepression as the unifying disease mechanism in both FSHD1 and 2 led to the development of DUX4 expressing animal and cell models, to further study the pathology and test therapeutic interventions. The human DUX4 gene is not found in mice and therefore transgenic models are necessary. A mouse model which integrates a pathogenic FSHD1 D4Z4 repeat size of 2.5 repeats including the distal polyadenylation site shows low levels of DUX4 mRNA and protein in skeletal muscle but lacks a muscle phenotype. One possible hypothesis for the failure of modeling the FSHD muscle phenotype was the complex spatial and temporal expression patterns of the transgene [22]. Nevertheless, since this model carries the D4Z4 repeats, it can still be used to study the epigenetic regulation of the D4Z4 repeat array involving modifiers that bind to D4Z4, such as SMCHD1. When levels of SMCHD1 are decreased, DUX4 mRNA is more abundant with a more severe skin phenotype, albeit without showing symptoms or signs involving the muscle [22].
An inducible iDUX4pA mouse model was created by knocking in a genomic fragment from the terminal D4Z4 repeat of an FSHD 4qA allele under the control of the doxycycline-inducible promoter into the X chromosome of the mouse. This allows for muscle-specific induction of DUX4 expression and the effects of DUX4 in the (male) mice, that are now able to survive past weaning until 4 months. DUX4 induction in these mice causes dystrophic changes as well as impaired regeneration. This could be a good mouse model to test DUX4 protein or mRNA-targeted therapeutic interventions [26].
The most recent mouse model, FLExDUX4, is a transgenic mouse that can be induced to produce mosaic expression patterns of DUX4 mRNA in a fraction of skeletal myonuclei resulting in a muscle phenotype, similar to the bursts of DUX4 mRNA expression seen in FSHD. This model will be useful for developmental and therapeutic studies, and studying DUX4 downstream pathways [38].
Symptoms and Signs
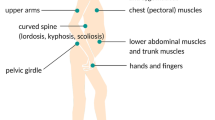
Patients with FSHD can present at any age and disease progression is slow. Patients often report problems with activities above their shoulders, difficulty whistling, sleeping with eyes open (reported by spouses or parents), catching their toes due to foot drop, and change in their appearance due to atrophy and muscle weakness with scapular winging and protuberant abdomen. Pain and fatigue are commonly experienced [39]. Neurological examination is characteristic: weakness of the periscapular muscles, specifically weakness of the lower trapezius muscle, results in winging and upward movement of the scapula with rounding of the shoulders and horizontal clavicles. While the deltoid muscle is often relatively spared early on, biceps, triceps, and pectoral muscles are typically affected, resulting in horizontal axillary folds. Asymmetric muscle weakness is more common than in other muscular dystrophies, but becomes less prominent in advanced disease. Abdominal muscle weakness is an early feature in FSHD and can be observed on examination as a protuberant abdomen, in supine position as a positive Beevor’s sign (an upward deflection of the umbilicus upon neck flexion due to weakness of the lower rectus abdominis muscle) or inability to do a sit up. Weakness of the paraspinal muscles can result in camptocormia, which in rare instances can be the presenting symptom, or in lumbar lordosis [40, 41].
Scapular winging in combination with weakness of facial muscles such as the orbicularis oculi and oris, with absent ptosis and spared extraocular muscles, along with a positive Beevor’s sign is highly suggestive and nearly pathognomonic of FSHD in the absence of atypical features. However, clinical diagnosis of milder or atypical presentations can be more challenging. Despite advanced disease with severe weakness, contractures do typically not occur. Extraocular and bulbar muscles are usually not affected. Restrictive lung disease has been reported in about 10% of patients [42], with 1 to 8% requiring ventilatory support [43]. A recent study reported a higher prevalence of sleep-disordered breathing and respiratory involvement with reduced forced vital capacities in 38% of patients and 14% requiring noninvasive ventilation. Further studies are necessary to assess the prevalence of respiratory involvement across different patient cohorts [44]. Generally, patients with severe disease, weak hip flexion or wheelchair use, and kyphoscoliosis are at higher risk for restrictive lung disease, which often times is asymptomatic [45].
Extramuscular Manifestations
FSHD does not affect the cardiac muscle. Mild, typically asymptomatic conduction abnormalities have been reported including asymptomatic right bundle branch block [46]. Rare complications in patients with large D4Z4 contractions include high-frequency hearing loss and about 0.8% of patients develop an exudative retinopathy (Coats’ syndrome) [47].
Disease Progression
FSHD is a slowly progressive disease. Due to its high clinical variability, the degree of severity and rate of progression vary. Patients with very large contractions typically have earlier onset and more severe disease with faster progression. Earlier age at onset of facial weakness in patients with early onset FSHD (symptoms occurring within the first 10 years of life) has been associated with greater disease severity [48, 49]. Men have been reported as more severely affected than women [19, 50], although this was not observed in a study assessing patients with early disease onset [48]. On a molecular basis, a gender effect on methylation levels was not seen [15]. A recent study proposed an estrogen receptor as a potential disease modifier by interfering with DUX4 transcriptional activity [51], but further studies are necessary to investigate gender-specific disease effects.
As a disease affecting shoulder and facial muscles first, the 6-year risk of wheelchair use has been described as 24.0%, with a peak in the second decade associated with large D4Z4 contractions, followed by an age-related increase in risk [5].
Therapeutic Approaches
Current Available Treatments
There are currently no pharmacological disease-modifying treatments in FSHD. Prior therapeutic trials were either directed at increasing muscle strength or to halt disease progression.
Given the inflammatory changes often seen on muscle biopsy, an anti-inflammatory approach similar to Duchenne muscular dystrophy seemed promising, but a pilot trial of 8 patients treated with 12 weeks of prednisone did not show a benefit on strength or muscle mass [52]. β2-adrenergic agonists, because of their known anabolic effects, have been tested in several randomized controlled trials in FSHD. There have been limited effects such as improvement of grip strength and lean muscle mass but no effect on the primary outcome, a change in global strength by maximum voluntary isometric contraction testing [53]. Other studies showed a positive effect in some but not all tested muscles [54], no effect with periodic use [55], and no effect on pain or fatigue [56]. It is of interest, given these prior trials, that recent studies in FSHD cell cultures show that β2-adrenergic agonists suppress the expression of DUX4 mRNA and decrease DUX4 expression [57]. A phase I/II trial tested MYO-029, a neutralizing antibody to myostatin, which is a negative regulator of muscle growth, with a tolerable safety profile, but no effects on muscle strength or function [58]. A drug interacting with myostatin and injected intramuscularly is currently subject of a phase 2 clinical trial (NCT02927080). An open-label pilot trial treating 19 patients with diltiazem for 24 weeks did not result in significant improvement in strength, function, or muscle mass [59]. Oxidative stress has been proposed as a downstream effect of DUX4 [60, 61]. A randomized, double-blind, placebo-controlled pilot trial tested the effects of vitamin C, vitamin E, zinc gluconate, and selenomethionine on physical performance. One of the primary outcome measures, the 2-minute-walk test, did not improve, while maximal voluntary contraction and endurance limit time showed some benefit [62]. A current trial is evaluating safety and efficacy of testosterone and rHGH in FSHD (NCT03123913).
There are several nonpharmaceutical interventions that can be offered to patients with FSHD. A recent review provided an evidence-based guideline summary for evaluation, diagnosis, and management of FSHD [63]. Ankle foot orthoses for patients with ankle dorsiflexor weakness can improve mobility and prevent falls. Patients with knee extension weakness might benefit from an ankle–knee–foot orthosis. Stabilizing the shoulder with braces has limited utility in patients with FSHD as it is often not well tolerated, but can be used for short time periods for certain activities. Surgical scapular fixation can improve elevation of the upper extremities in selected patients with preserved proximal strength and who gain strength by manual fixation of the scapula, carefully assessing risks of loss of mobility and possible surgical complications [64,65,66]. Abdominal binders and posture braces can be beneficial in certain individuals with truncal weakness. Given recent reports of higher prevalence of restrictive lung disease, obtaining a baseline FVC is indicated. Continued monitoring of FVC is recommended for patients with abnormal baseline testing or symptoms, moderate to severely affected patients, patients with marked truncal weakness, wheelchair-bound patients, or with kyphoscoliosis. Regular cardiac monitoring is not indicated unless patients experience symptoms. Patients with large deletions should be monitored for hearing loss and retinal vascular disease as they have a higher risk for systemic extramuscular features [5].
Pain is common in patients with FSHD [39, 56], mostly thought to be musculoskeletal, although a contributing inflammatory component has been discussed, specifically for periodically occurring pains [56]. Physical therapists can help to elucidate the mechanism of musculoskeletal pain, often times originating from scapular instability or truncal weakness. There are no specific recommendations for medical treatment but generally nonsteroidal anti-inflammatory medications can be useful for acute pain, while antidepressants or antiepileptics for chronic musculoskeletal pain [63].
One study tested the effect of albuterol and dynamic and isometric exercises of elbow flexors and ankle dorsiflexors with a progressive overload scheme using weights [54]. The exercises were safe and improved dynamic strength. Exercises did not result in increased pain [56]. Another study tested the effects of 12 weeks of low-intense aerobic exercise on a cycle ergometer. Exercise was well tolerated with no evidence of muscle damage (measured as a change of CK levels and muscle histology) and improved maximal oxygen uptake and workload with improved self-reported strength, endurance, and activity level [67]. A multicenter, assessor-blinded, randomized controlled trial showed a positive effect of 16 weeks of aerobic exercise training and cognitive behavioral therapy on severe chronic fatigue in FSHD patients. Both interventions showed sustained increase of physical activity in both groups and improved social participation following cognitive behavioral therapy [68].
Future Perspectives for Targeted Treatment
The feasibility of therapeutic approaches are guided by accumulating knowledge about the pathomechanism of FSHD. Although FSHD1 and FSHD2 are genetically distinct, they converge on the same downstream disease mechanism of DUX4 expression. Consequently, similar therapeutic approaches can potentially target both forms of FSHD (Fig. 2).

This figure demonstrates several different approaches for targeted treatment: modifying epigenetic repression of DUX4, targeting DUX4 mRNA, DUX4 protein, or cellular downstream effects of DUX4 expression
-
1)
Enhance the epigenetic repression of the D4Z4
-
a)
Modulating SMCHD1: In both types of FSHD myotube cultures, 2- to 3-fold overexpression of SMCHD1 resulted in a 70 to 90% reduction in DUX4 mRNA levels [36]. A 1.5- to 3-fold increase in SMCHD1 protein levels led to a significant decrease in DUX4 levels and that of its target genes. This demonstrated that the derepression of DUX4 in FSHD muscle cells is a reversible process that can be rescued by increasing SMCHD1 levels.
-
b)
Modulating other DUX4 repressive proteins/pathways: Regulators of the D4Z4 repeat were characterized by an engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) method followed by mass spectrometry (MS) proteomics (enChIP-MS). This method allowed for the identification of D4Z4-associated factors including SMCHD1, as well as many of the components of the nucleosome remodeling deacetylase (NuRD) complex and chromatin assembly factor 1 (CAF-1) complex. Components shared by these complexes were found to mediate D4Z4 repeat repression. Promoting the activity of such complexes with the goal of silencing the DUX4 gene could be a potential treatment strategy [34]. Chemical and pharmacon libraries are used in screening tests on patient myotubes to look for molecules that inhibit DUX4 expression, monitored by the levels of DUX4 target genes. These screening tests have identified inhibitors of the bromodomain and extraterminal (BET) family of proteins and agonists of the β2-adrenergic receptor as potentially promising therapeutic candidates [57]. Previous trials testing albuterol have not shown an overall improvement of functional outcome measures. However, outcome measures and biomarkers have been refined and might be more sensitive and other β2-adrenergic agents such as formoterol or clenbuterol might be more potent [57].
-
a)
-
2)
Targeting the DUX4 mRNA: For example, by altering splicing or polyadenylation and preventing mRNA from making DUX4 protein. Preclinical studies are underway assessing adenoassociated viral (AAV) vectors carrying microRNAs targeting the DUX4 mRNA with the goal to silence the gene [69]. Antisense oligonucleotides have been tested targeting DUX4 mRNA in myotube cultures [70].
-
3)
Blocking the activity of the DUX4 protein or inhibiting the DUX4-induced processes downstream, which lead to pathology: When targeting DUX4 protein, it is important to understand how much DUX4 protein is tolerated by the muscle, whether DUX4 protein is essential in other healthy tissues and, consequently, whether treatment needs to be tissue specific. Targeting DUX4 protein-induced downstream effects is the most challenging approach at this point, as, although some of the downstream effects are known, it is not clear which of these multiple mechanisms is the primary cause of the underlying dystrophy.
Trial Preparedness
To monitor treatment effects, patient-relevant outcome measures and disease-relevant and sensitive biomarkers are necessary [71, 72]. In FSHD, a spectrum of molecular, imaging, and electrodiagnostic biomarkers are being developed in addition to functional and patient-reported outcome measures. DUX4 and its target genes measured in muscle tissue are currently being evaluated as a biomarker [73]. Exploratory studies have taken first steps in evaluating potential serum biomarkers [74, 75]. MR imaging parameters of the muscle correlate with clinical outcome measures and severity of disease. MRI is useful in identifying affected muscles and assessing the degree of fatty infiltration of the muscle. In addition, STIR sequences (T2-weighted sequences with nulling of the fat signal) detect inflammation of the muscle, potentially reflecting a more active phase of the disease [72, 76, 77]. This correlation between STIR signal and active disease is of particular interest as those muscles might be at greatest risk of degeneration and therefore an ideal target for treatment and monitoring of treatment effects. Longitudinal studies are underway to assess the sensitivity of MRI as a biomarker of disease progression over time. Electrical impedance myography (EIM) uses the resistance to current flow through a particular muscle to assess changes in muscle structure. While this technology has demonstrated reliable measurements and correlation to functional outcomes [78], it did not demonstrate sensitivity to disease progression over 12 months in a preliminary study with a relatively small number of patients [79].
A recently developed functional facioscapulohumeral muscular dystrophy composite outcome measure (FSHD-COM) combines several assessments of patient-identified areas of functional burden [80]. The FSHD-COM correlates well with disease severity, duration, and strength. However, the FSHD-COM still needs to be validated in multicenter trials and demonstrate sensitivity to disease change [80]. A patient-reported outcome tool, the FSHD Health Index (FSHD-HI), is currently being evaluated in a prospective study [81]. Overall, there has been marked progress in approaching trial preparedness in FSHD, with the foundation of national and international networks and collaborations, and patient engagement through registries, all of which are vital to successfully study this rare disease.
References
Flanigan KM, Coffeen CM, Sexton L, et al. Genetic characterization of a large, historically significant Utah kindred with facioscapulohumeral dystrophy. Neuromuscul Disord. 2001;11(6–7):525–529.
Tawil R, van der Maarel SM, Tapscott SJ. Facioscapulohumeral dystrophy: the path to consensus on pathophysiology. Skelet Muscle. 2014;4:12.
Deenen JC, Arnts H, van der Maarel SM, et al. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology. 2014;83(12):1056–1059.
van der Maarel SM, Deidda G, Lemmers RJ, et al. De novo facioscapulohumeral muscular dystrophy: frequent somatic mosaicism, sex-dependent phenotype, and the role of mitotic transchromosomal repeat interaction between chromosomes 4 and 10. Am J Hum Genet. 2000;66(1):26–35.
Statland JM, McDermott MP, Heatwole C, et al. Reevaluating measures of disease progression in facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2013;23(4):306–312.
Wijmenga C, Frants RR, Brouwer OF, et al. Location of facioscapulohumeral muscular dystrophy gene on chromosome 4. Lancet. 1990;336(8716):651–653.
Wijmenga C, Padberg GW, Moerer P, et al. Mapping of facioscapulohumeral muscular dystrophy gene to chromosome 4q35-qter by multipoint linkage analysis and in situ hybridization. Genomics. 1991;9(4):570–575.
van Deutekom JC, Wijmenga C, van Tienhoven EA, et al. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum Mol Genet. 1993;2(12):2037–2042.
Das S, Chadwick BP. Influence of Repressive Histone and DNA Methylation upon D4Z4 Transcription in Non-Myogenic Cells. PLoS One. 2016;11(7):e0160022.
Lemmers RJ, de Kievit P, Sandkuijl L, et al. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat Genet. 2002;32(2):235–236.
Lemmers RJ, Wohlgemuth M, van der Gaag KJ, et al. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2007;81(5):884–894.
Lemmers RJ, van der Vliet PJ, Klooster R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329(5999):1650–1653.
van Overveld PG, Lemmers RJ, Sandkuijl LA, et al. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet. 2003;35(4):315–317.
de Greef JC, Lemmers RJ, Camano P, et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology. 2010;75(17):1548–1554.
Lemmers RJ, Goeman JJ, van der Vliet PJ, et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum Mol Genet. 2015;24(3):659–669.
Lemmers RJ, Tawil R, Petek LM, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;44(12):1370–1374.
van den Boogaard ML, Lemmers R, Balog J, et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am J Hum Genet. 2016;98(5):1020–1029.
Lunt PW, Harper PS. Genetic counselling in facioscapulohumeral muscular dystrophy. J Med Genet. 1991;28(10):655–664.
Ricci G, Scionti I, Sera F, et al. Large scale genotype-phenotype analyses indicate that novel prognostic tools are required for families with facioscapulohumeral muscular dystrophy. Brain. 2013;136(Pt 11):3408–3417.
Sacconi S, Lemmers RJ, Balog J, et al. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am J Hum Genet. 2013;93(4):744–751.
Larsen M, Rost S, El Hajj N, et al. Diagnostic approach for FSHD revisited: SMCHD1 mutations cause FSHD2 and act as modifiers of disease severity in FSHD1. Eur J Hum Genet. 2015;23(6):808–816.
de Greef JC, Krom YD, den Hamer B, et al. Smchd1 haploinsufficiency exacerbates the phenotype of a transgenic FSHD1 mouse model. Hum Mol Genet. 2018;27(4):716–731.
Park HJ, Lee W, Kim SH, et al. FAT1 Gene Alteration in Facioscapulohumeral Muscular Dystrophy Type 1. Yonsei Med J. 2018;59(2):337–340.
Mariot V, Roche S, Hourde C, et al. Correlation between low FAT1 expression and early affected muscle in facioscapulohumeral muscular dystrophy. Ann Neurol. 2015;78(3):387–400.
Snider L, Geng LN, Lemmers RJ, et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6(10):e1001181.
Bosnakovski D, Chan SSK, Recht OO, et al. Muscle pathology from stochastic low level DUX4 expression in an FSHD mouse model. Nat Commun. 2017;8(1):550.
Gannon OM, Merida de Long L, Saunders NA. DUX4 Is Derepressed in Late-Differentiating Keratinocytes in Conjunction with Loss of H3K9me3 Epigenetic Repression. J Invest Dermatol. 2016;136(6):1299–1302.
De Iaco A, Planet E, Coluccio A, et al. DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat Genet. 2017;49(6):941–945.
Geng LN, Yao Z, Snider L, et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell. 2012;22(1):38–51.
Feng Q, Snider L, Jagannathan S, et al. A feedback loop between nonsense-mediated decay and the retrogene DUX4 in facioscapulohumeral muscular dystrophy. Elife. 2015;4.
Shadle SC, Zhong JW, Campbell AE, et al. DUX4-induced dsRNA and MYC mRNA stabilization activate apoptotic pathways in human cell models of facioscapulohumeral dystrophy. PLoS Genet. 2017;13(3):e1006658.
Kowaljow V, Marcowycz A, Ansseau E, et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul Disord. 2007;17(8):611–623.
Wallace LM, Garwick SE, Mei W, et al. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann Neurol. 2011;69(3):540–552.
Campbell AE, Shadle SC, Jagannathan S, et al. NuRD and CAF-1-mediated silencing of the D4Z4 array is modulated by DUX4-induced MBD3L proteins. Elife. 2018;7.
Jones TI, Chen JC, Rahimov F, et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis. Hum Mol Genet. 2012;21(20):4419–4430.
Balog J, Thijssen PE, Shadle S, et al. Increased DUX4 expression during muscle differentiation correlates with decreased SMCHD1 protein levels at D4Z4. Epigenetics. 2015;10(12):1133–1142.
Jones TI, King OD, Himeda CL, et al. Individual epigenetic status of the pathogenic D4Z4 macrosatellite correlates with disease in facioscapulohumeral muscular dystrophy. Clin Epigenetics. 2015;7:37.
Jones T, Jones PL. A cre-inducible DUX4 transgenic mouse model for investigating facioscapulohumeral muscular dystrophy. PLoS One. 2018;13(2):e0192657.
Moris G, Wood L, FernaNdez-Torron R, et al. Chronic pain has a strong impact on quality of life in facioscapulohumeral muscular dystrophy. Muscle Nerve. 2018;57(3):380–387.
Ghosh PS, Milone M. Camptocormia as presenting manifestation of a spectrum of myopathic disorders. Muscle Nerve. 2015;52(6):1008–1012.
Jordan B, Eger K, Koesling S, Zierz S. Camptocormia phenotype of FSHD: a clinical and MRI study on six patients. J Neurol. 2011;258(5):866–873.
Scully MA, Eichinger KJ, Donlin-Smith CM, Tawil R, Statland JM. Restrictive lung involvement in facioscapulohumeral muscular dystrophy. Muscle Nerve. 2014;50(5):739–743.
Wohlgemuth M, van der Kooi EL, van Kesteren RG, van der Maarel SM, Padberg GW. Ventilatory support in facioscapulohumeral muscular dystrophy. Neurology. 2004;63(1):176–178.
Moreira S, Wood L, Smith D, et al. Respiratory involvement in ambulant and non-ambulant patients with facioscapulohumeral muscular dystrophy. J Neurol. 2017;264(6):1271–1280.
Wohlgemuth M, Horlings CGC, van der Kooi EL, et al. Respiratory function in facioscapulohumeral muscular dystrophy 1. Neuromuscul Disord. 2017;27(6):526–530.
van Dijk GP, van der Kooi E, Behin A, et al. High prevalence of incomplete right bundle branch block in facioscapulohumeral muscular dystrophy without cardiac symptoms. Funct Neurol. 2014;29(3):159–165.
Statland JM, Sacconi S, Farmakidis C, et al. Coats syndrome in facioscapulohumeral dystrophy type 1 Frequency and D4Z4 contraction size. Neurology. 2013;80(13):1247–1250.
Mah JK, Feng J, Jacobs MB, et al. A multinational study on motor function in early-onset FSHD. Neurology. 2018.
Goselink RJM, Voermans NC, Okkersen K, et al. Early onset facioscapulohumeral dystrophy - a systematic review using individual patient data. Neuromuscul Disord. 2017;27(12):1077–1083.
Tonini MM, Passos-Bueno MR, Cerqueira A, et al. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul Disord. 2004;14(1):33–38.
Teveroni E, Pellegrino M, Sacconi S, et al. Estrogens enhance myoblast differentiation in facioscapulohumeral muscular dystrophy by antagonizing DUX4 activity. J Clin Invest. 2017;127(4):1531–1545.
Tawil R, McDermott MP, Pandya S, et al. A pilot trial of prednisone in facioscapulohumeral muscular dystrophy. FSH-DY Group. Neurology. 1997;48(1):46–49.
Kissel JT, McDermott MP, Mendell JR, et al. Randomized, double-blind, placebo-controlled trial of albuterol in facioscapulohumeral dystrophy. Neurology. 2001;57(8):1434–1440.
van der Kooi EL, Vogels OJ, van Asseldonk RJ, et al. Strength training and albuterol in facioscapulohumeral muscular dystrophy. Neurology. 2004;63(4):702–708.
Payan CA, Hogrel JY, Hammouda EH, et al. Periodic Salbutamol in Facioscapulohumeral Muscular Dystrophy: A Randomized Controlled Trial. Archives of Physical Medicine and Rehabilitation. 2009;90(7):1094–1101.
van der Kooi EL, Kalkman JS, Lindeman E, et al. Effects of training and albuterol on pain and fatigue in facioscapulohumeral muscular dystrophy. J Neurol. 2007;254(7):931–940.
Campbell AE, Oliva J, Yates MP, et al. BET bromodomain inhibitors and agonists of the beta-2 adrenergic receptor identified in screens for compounds that inhibit DUX4 expression in FSHD muscle cells. Skelet Muscle. 2017;7(1):16.
Wagner KR, Fleckenstein JL, Amato AA, et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. 2008;63(5):561–571.
Elsheikh BH, Bollman E, Peruggia M, et al. Pilot trial of diltiazem in facioscapulohumeral muscular dystrophy. Neurology. 2007;68(17):1428–1429.
Bosnakovski D, Xu Z, Gang EJ, et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 2008;27(20):2766–2779.
Dmitriev P, Bou Saada Y, Dib C, et al. DUX4-induced constitutive DNA damage and oxidative stress contribute to aberrant differentiation of myoblasts from FSHD patients. Free Radical Biology and Medicine. 2016;99:244–258.
Passerieux E, Hayot M, Jaussent A, et al. Effects of vitamin C, vitamin E, zinc gluconate, and selenomethionine supplementation on muscle function and oxidative stress biomarkers in patients with facioscapulohumeral dystrophy: a double-blind randomized controlled clinical trial. Free Radic Biol Med. 2015;81:158–169.
Tawil R, Kissel JT, Heatwole C, et al. Evidence-based guideline summary: Evaluation, diagnosis, and management of facioscapulohumeral muscular dystrophy: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology. 2015;85(4):357–364.
Le Hanneur M, Saint-Cast Y. Long-term results of Letournel scapulothoracic fusion in facioscapulohumeral muscular dystrophy: A retrospective study of eight cases. Orthop Traumatol Surg Res. 2017;103(3):421–425.
Cooney AD, Gill I, Stuart PR. The outcome of scapulothoracic arthrodesis using cerclage wires, plates, and allograft for facioscapulohumeral dystrophy. J Shoulder Elbow Surg. 2014;23(1):e8–e13.
Orrell RW, Copeland S, Rose MR. Scapular fixation in muscular dystrophy. Cochrane Database Syst Rev. 2010(1):CD003278.
Olsen DB, Orngreen MC, Vissing J. Aerobic training improves exercise performance in facioscapulohumeral muscular dystrophy. Neurology. 2005;64(6):1064–1066.
Voet N, Bleijenberg G, Hendriks J, et al. Both aerobic exercise and cognitive-behavioral therapy reduce chronic fatigue in FSHD: an RCT. Neurology. 2014;83(21):1914–1922.
Wallace LM, Saad NY, Pyne NK, et al. Pre-clinical Safety and Off-Target Studies to Support Translation of AAV-Mediated RNAi Therapy for FSHD. Mol Ther Methods Clin Dev. 2018;8:121–130.
Chen JC, King OD, Zhang Y, et al. Morpholino-mediated Knockdown of DUX4 Toward Facioscapulohumeral Muscular Dystrophy Therapeutics. Mol Ther. 2016;24(8):1405–1411.
Tawil R, Shaw DW, van der Maarel SM, Tapscott SJ. Clinical trial preparedness in facioscapulohumeral dystrophy: outcome measures and patient access: 8–9 April 2013, Leiden, The Netherlands. Neuromuscul Disord. 2014;24(1):79–85.
Tawil R, Padberg GW, Shaw DW, et al. Clinical trial preparedness in facioscapulohumeral muscular dystrophy: Clinical, tissue, and imaging outcome measures 29-30 May 2015, Rochester, New York. Neuromuscul Disord. 2016;26(2):181–186.
Yao Z, Snider L, Balog J, et al. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum Mol Genet. 2014;23(20):5342–5352.
Petek LM, Rickard AM, Budech C, et al. A cross sectional study of two independent cohorts identifies serum biomarkers for facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul Disord. 2016;26(7):405–413.
Statland J, Donlin-Smith CM, Tapscott SJ, van der Maarel S, Tawil R. Multiplex Screen of Serum Biomarkers in Facioscapulohumeral Muscular Dystrophy. J Neuromuscul Dis. 2014;1(2):181–190.
Mul K, Vincenten SCC, Voermans NC, et al. Adding quantitative muscle MRI to the FSHD clinical trial toolbox. Neurology. 2017;89(20):2057–2065.
Frisullo G, Frusciante R, Nociti V, et al. CD8(+) T cells in facioscapulohumeral muscular dystrophy patients with inflammatory features at muscle MRI. J Clin Immunol. 2011;31(2):155–166.
Statland JM, Heatwole C, Eichinger K, et al. Electrical impedance myography in facioscapulohumeral muscular dystrophy. Muscle Nerve. 2016;54(4):696–701.
Mul K, Heatwole C, Eichinger K, et al. Electrical impedance myography in facioscapulohumeral muscular dystrophy: A 1-year follow-up study. Muscle Nerve. 2018.
Eichinger K, Heatwole C, Iyadurai S, et al. Facioscapulohumeral muscular dystrophy functional composite outcome measure. Muscle Nerve. 2018.
Johnson NE, Quinn C, Eastwood E, Tawil R, Heatwole CR. Patient-identified disease burden in facioscapulohumeral muscular dystrophy. Muscle Nerve. 2012;46(6):951–953.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(PDF 1224 kb)
Rights and permissions
About this article

Cite this article
Hamel, J., Tawil, R. Facioscapulohumeral Muscular Dystrophy: Update on Pathogenesis and Future Treatments. Neurotherapeutics 15, 863–871 (2018). https://doi.org/10.1007/s13311-018-00675-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-018-00675-3