
Abstract
Progress in the treatment of adult high-grade gliomas (HGG), including chemoradiation with concurrent and adjuvant temozolomide for glioblastoma, has not translated into significant therapeutic advances for pediatric HGG, where overall survival has plateaued at 15% to 20%, especially when considering specialized pediatric treatment in tertiary care centers, maximal safe neurosurgical resection, optimized delivery of involved field radiation, and improvements in supportive care. However, recent advances in our understanding of pediatric HGG, including the application of next-generation sequencing and DNA methylation profiling, have identified mutations in the histone variant H3.3 and canonical H3.1 genes, respectively. These mutations are relatively specific to neuroanatomic compartments (cortex, midline structures, thalamus, brainstem) and are often associated with other mutations, especially in specific growth factor receptor tyrosine kinases. Targeting epigenetic pathways affected by these histone mutations, alone or in combination with small molecule inhibitors of growth factor receptor signaling pathways, will inform new treatment strategies for pediatric HGG and should be incorporated into novel cooperative group clinical trial designs.
Similar content being viewed by others
Introduction: Diagnosis and Current Management Strategies
Brain tumors are the most common solid tumors of childhood and have overtaken leukemia as the most common cause of cancer-related mortality in children [1]. Gliomas are tumors arising from the glial cell lineage (astrocytic and /or oligodendroglial) and were historically classified into low (low grade glioma; grade I–II) or high grade [high grade glioma (HGG); grade III–IV] based on histological appearance, biological behavior, and clinical course [2]. The majority of gliomas in children (~50%) are low grade and astrocytic in origin [3]. HGG constitute 3% to 7% of all childhood brain tumors, are less common in children than in adults, and include a heterogeneous group of tumors (anaplastic astrocytoma, glioblastoma (GBM), giant cell GBM, anaplastic oligodendroglioma, etc.) [2]. However, the current revised classification of HGG (and gliomas in general) incorporates characteristic genomic abnormalities (genetic and epigenetic) along with the clinicopathological features (diffuse, cell lineage, anatomical location, etc.) to define distinct tumor entities. This creates the potential for therapies directed against specific clinical and molecular profiles [4]. One such distinct group of tumors occurs primarily in children and is characterized by somatic driver mutations (K27M) in the histone H3 genes (H3F3A, HIST1H3B), midline location (thalamus, brain stem, spinal cord), and diffuse growth pattern on histology [4]. This group is classified as diffuse midline glioma, H3 K27M-mutant and includes previously defined entities such as brain stem gliomas [diffuse intrinsic pontine gliomas (DIPG)]. Another distinct group of diffuse astrocytic tumors seen in children (<20 years) arise in the cerebral hemispheres and are characterized by G34R/V mutations that are restricted to H3F3A (H3.3-G34R/V) [5].
Diffuse midline glioma involving the brain stem constitutes ~12% of all pediatric brain tumors (0–14 years) [1] and > 80% of them are localized to the pons and were formerly known as DIPG [1]. The median age of diagnosis of diffuse midline gliomas is 10 years with pontine tumors occurring earlier (median ~7 years) than the thalamic tumors (median ~11 years) and they occur equally in both sexes [6, 7]. Patients with pontine tumors have rapid onset of symptoms (<2–3 months from the time of diagnosis) and often present with cranial nerve palsies, long tract signs, and ataxia [8]. Thalamic gliomas present with features of raised intracranial pressure, motor weakness/hemiparesis, and gait disturbances [7]. Depending on the location of the hemispheric tumors, the presenting symptoms can vary from developmental delay, failure to thrive, raised intracranial pressure (headache, nausea, vomiting), hemiparesis, seizures, and so on [9]. The diagnosis of diffuse midline pontine gliomas has been based on the clinical presentation with characteristic radiological features and until recently there has been no need for a histological diagnosis in the majority of cases [8]. This “no biopsy” practice has hindered scientific advances in DIPG when compared with other pediatric brain tumors such as medulloblastomas and, to a lesser extent, ependymomas. Diagnostic image-guided stereotactic biopsy of DIPG tumors is now considered to be safe, and provides a means to identify actionable therapeutic targets and risk stratify patients for both therapy and prognosis (as discussed in [8]). The identification of histone mutations (H3.3 and H3.1 K27M) by immunohistochemistry in diffuse midline gliomas may avoid misdiagnosis because of sampling bias due to histological heterogeneity seen in these tumors [10]. Specifically, there is spatial and temporal homogeneity of these histone mutations in the primary and metastatic tumors [11, 12], which is similar to the subgroup stability seen in medulloblastoma across disease sites [13].
Currently, fractionated radiation therapy (RT) directed to the tumor (54–60 Gy, 30 fractions) alleviates symptoms in 60% to 80% of the patients with DIPG and stabilizes disease, albeit on a temporary basis. However, the tumor progresses within the first year of diagnosis with overall survival of 8 to 14 months. Several strategies involving radiation (hyper- or hypofractionation), radiation sensitization, chemotherapy (neoadjuvant, adjuvant, combination with RT), targeted therapy, and agents that alter blood–brain barrier (BBB) permeability have not had any impact on patient survival (event-free and overall survival) [8]. Re-irradiation at the time of tumor progression may add a few months of survival and is tolerated well [8]. The current management of hemispheric diffuse gliomas (Children’s Oncology Group-ACNS0423) includes maximal surgical resection (if possible) followed by involved field RT with concurrent temozolomide (TMZ) and subsequent adjuvant chemotherapy (TMZ with lomustine) [14, 15]. Diffuse midline thalamic gliomas have a poor prognosis similar to DIPG and high-grade histology is associated with short overall survival in these patients [16].
Chromatin Biology and Histone Mutations in Pediatric Diffuse Gliomas
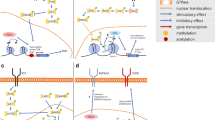
Histone proteins form the core of the nucleosome, which is the fundamental repeating unit of eukaryotic chromatin [17]. Each nucleosome consists of 2 copies of 4 core histone proteins (H3, H4, H2A, and H2B) to form a histone octamer that is wrapped around its radial surface by 147 base pairs of DNA (Fig. 1) [18]. Histone H3 has three isoforms (H3.1, H3.2, and H3.3) with only limited differences: H3.1 differs from H3.2 by a single amino acid, while H3.3 differs at 4 positions [17]. Histones H3.1 and 3.2 are the canonical H3 forms, and are incorporated into DNA during S phase by chromatin assembly factor 1 and at DNA repair sites in a replication-dependent manner [17]. In contrast, incorporation of the noncanonical histone H3.3 is independent of replication and can occur in nondividing cells [17]. H3.3 is deposited at actively transcribed and heterochromatic subtelomeric regions by the DAXX/ATRX complex and at pericentromeric regions by the HIRA chaperone (reviewed in [17]). Histone H3.1 is encoded by 10 genes clustered on chromosome 6 and H3.2 is encoded by 3 genes (HIST2H3A, HIST2H3C, and HIST2H3D) on chromosome 1 [17]. The noncanonical histone H3.3 is encoded by 2 isolated genes (H3F3A on chromosome 1 and H3F3B on chromosome 17) that produce identical proteins [17].

An overview of epigenetic targets in pediatric high-grade gliomas. The nucleosome is depicted as an octamer of core histone (H2A, H2B, H3, and H4). Glioma-associated mutations occurring in canonical histone H3 (H3.1) or the H3.3 variant are indicated and the H3K27M to reduce levels of histone H3-lysine 27 methylation (H3K27me3). Representative histone modifications (methylation and acetylation) are shown on the N-terminal histone H3 tails, together with DNA methylation (me), which is catalyzed by DNA methyltransferase (DNMT) enzymes. These modifications are: 1) placed by histone methyltransferase (HMT) or histone acetyltransferase (HAT) writers (W); 2) bound by readers (R) containing a chromo- or bromodomain that can recruit additional regulatory proteins; and 3) removed by erasers (E) that include histone demethylase (HDM) or histone deacetylase (HDAC) enzymes. Therapeutics undergoing preclinical or clinical testing that target these activities are indicated, as well as those that inhibit DNA methylation. Combinations with either chemotherapy or radiation therapy (yellow icons) are noted in the text
Histone proteins undergo a wide range of covalent modifications predominantly at the amino (N-) and carboxy (C-) terminal tails, including acetylation, methylation, phosphorylation, adenosine diphosphate ribosylation, and ubiquitylation to name a few [18]. These modifications (colloquially referred to as histone marks) are mediated by writers, erasers, and readers.
Writers are enzymes that modify specific substrates by adding chemical groups (i.e., histone acetyl transferases, histone methyl transferases). Erasers are proteins that remove specific groups [i.e., histone deacetylases (HDACs), histone demethylases]. Readers are proteins that recognize specific histone modifications (i.e., histone acetyl groups by bromodomains, histone methyl groups by chromodomains) [18]. Histone modifications are associated with various nuclear functions, such as gene transcription or silencing, mitosis, DNA replication/repair, and chromatin assembly [17]. For instance, acetylation of histone tails results in a relaxed chromatin structure (euchromatin) that facilitates gene transcription, whereas methylation can be associated with a range of functions depending on the residue modified and number of groups added, which can involve mono- (me1), di- (me2), or trimethylation (me3) [18]. One such important mark is the trimethylation of histone H3 on lysine 27 (H3K27me3), which is placed by the enhancer of zeste homolog-1/2 writers (EZH1/2) and erased by the JMJD3/KDM6B and UTX/KDM6A enzymes to control stem cell and oncogenic gene expression programs [19, 20].
Over the past several years, chromatin defects have become central to the pathogenesis of HGG in children when compared with adults. Next-generation sequencing of pediatric HGGs has identified recurrent somatic “oncohistone” mutations in H3 histone genes (summarized in Table 1) [5, 6, 10, 17, 49, 55–57]. These mutations include: 1) missense mutations in H3F3A that lead to substitution of lysine-27 with methionine (H3.3-K27M) and, on rare occasions, to isoleucine (H3.3-K27I) in diffuse midline gliomas; 2) glycine 34 to arginine (H3.3-G34R) or, less frequently, glycine 34 to valine (H3.3-G34V) substitutions in cortical tumors; and 3) mutations in the canonical histone H3 genes HIST1H3B and HIST1H3C that encode H3.1-K27M or H3.2-K27M, respectively, in tumors of the pontine region. Strikingly, these mutations are very location specific and occur in distinct age groups (as discussed below), as well as having considerable diagnostic, therapeutic, and prognostic significance (Table 1) [5, 10, 49, 55].
Co-existing Genetic Mutations in Pediatric Diffuse Gliomas
A number of obligate partner mutations are frequently associated with the H3 histone alterations in pediatric HGGs (Table 1) [5, 16, 21, 49, 55–57, 64]. As for the H3 mutations themselves, these are specific to the anatomic location and also serve as potential therapeutic targets. For example, the H3.3-K27M mutation in the pons is associated with loss of function mutations in TP53 (60%) and gain-of-function mutations or amplifications in platelet-derived growth factor receptor alpha (PDGFRA; 40%) [65, 66]. The H3.1-K27M mutations in the pons are associated with recurrent gain of function somatic mutations in the activin A receptor type 1 (ACVR1; 20%) [6, 21, 56, 57]. In contrast, thalamic gliomas may undergo mitogen-activated protein kinase (MAPK) pathway activation via fibroblast growth factor receptor 1 (FGFR1) hotspot mutations or the BRAF-V600E mutation [16]. The mutations in these growth factor receptors (PDGFRA, ACVR1, and FGFR1) are not seen concurrently. Moreover, mutations in ACVR1 and FGFR1 are mutually exclusive of alterations of TP53 [64], and FGFR1 and BRAF mutations do not co-occur [16]. Hemispheric tumors harboring H3.3-G34R/V mutations are associated with TP53 mutations (100%) in conjunction with mutations in the DAXX/ATRX complex (50%) [65, 66]. These tumors also exhibit MYCN upregulation when compared with H3F3A wild-type tumors [22]. Fusion genes involving the kinase domain of the neurotrophin receptors are seen in both hemispheric and midline diffuse gliomas [21], suggesting a common pathogenic mechanism. In patients with wild-type H3F3A, a small group display mutations in isocitrate dehydrogenase-1 (3%) [37] and SET domain containing 2 (15%), which is a histone methyltransferase that catalyzes di- and trimethylation of lysine 36 on histone H3 [37, 67]. These tumors further define a class that is localized to the frontal and temporal lobes and has a relatively better prognosis. In each of these examples, the mutations that occur in conjunction with those in histone H3 are not chosen randomly and may provide a survival advantage, at least in part, by increasing the levels of mutant histone H3 in tumor cells. For example, co-occurrence of the H3.1-K27M and ACVR1 mutations (a growth factor that induces cell division) result in increased synthesis of both wild-type and mutant H3.1 proteins [11]. Similarly, the simultaneous presence of H3.3-K27M and TP53 mutations is thought to allow evasion of cell death and contribute to driving gliomagenesis [11, 50].
Mechanisms of Tumorigenesis in Pediatric Diffuse Gliomas
Through concerted research efforts, the neuro-oncology community is beginning to understand the role of histone H3 mutations in the initiation and maintenance of pediatric diffuse gliomas. The human embryonic stem-cell model of H3.3-K27M diffuse glioma established by Funato et al. [50] revealed the following salient points: 1) the K27M mutation is mitogenic in neural progenitor cells (NPCs) but not in undifferentiated embryonic stem cells or astrocytes, suggesting developmental timing and cell specificity are important determinants of their pathogenic activity; 2) histone H3 mutations alone are not sufficient to initiate tumors in vivo and require cooperating mutations (i.e., PDGFRA overexpression and TP53 attenuation)—notably, NPCs with these 3 genetic alterations (P5K cells) give rise to slow growing tumors in vivo that display low-grade histology (lack of necrosis and angiogenesis), supporting that additional mutations or cellular context enable HGG development; 3) the histone mutations upregulate the expression of stem cell-related genes (LIN28B, PLAG1, and PLAGL1) and knockdown of these genes inhibits tumor cell proliferation in vitro, suggesting that the K27M mutation acts partly by promoting de-differentiation of NPCs to a more primitive stem-cell state [50, 68]. To this end, Lewis et al. [69] demonstrated that H3K27M is a gain-of-function mutation that inhibits EZH2 methyltransferase activity resulting in a global reduction in H3K27 di- (H3K27me2) and tri-methylation (H3K27me3). Nevertheless, Chan et al. [70] showed that a subset of genes exhibited elevated H3K27me3 and EZH2 occupancy in the presence of H3K27M mutations, together driving transcriptional reprogramming and cancer development. In contrast to the global effects of H3K27M mutations, G34R/V mutant tumors only show reduced H3K36me2/me3 on nucleosomes bearing these mutations and do not affect total cellular levels of H3K36me2/me3 [22, 70]. This leads to discrete changes in gene expression that suppress differentiation (i.e., downregulation of OLIG1/2) and promote growth through upregulation of proto-oncogenes (i.e., MYCN) implicated in pediatric HGGs [5, 22, 71].
Potential Epigenetic/Genetic Therapies in Pediatric Diffuse Gliomas
Epigenetic mutations in combination with obligate genetic alterations drive the formation and maintenance of pediatric diffuse gliomas. As noted, the spatial/temporal conservation of these driver mutations is clinically relevant and creates attractive therapeutic targets [11, 12]. To date, conventional cytotoxic chemotherapy either alone or in combination with RT have failed to improve clinical outcome in diffuse midline gliomas in children (reviewed in [72]). Therefore, there is an urgent need for innovative biologically driven (with biopsy-proven molecular targets) clinical trials that use targeted therapies in combination with conventional treatments [73]. Disruption of epigenetic regulation is the cornerstone of pediatric HGGs and these epigenetic modifications can be reversed by epigenetic-based therapies.
Epigenetic Targets
Erasers (Histone Demethylases)
The gain-of-function H3K27M mutations leads to inactivation of polycomb repressive complex 2 (PRC2) methyltransferase and results in global hypomethylation of H3K27 and transcriptional derepression of potential oncogenes [69]. Hashizume et al. [38] hypothesized that inhibition of H3 demethylation and restoration of physiological H3K27 methylation could contribute to the treatment of H3.3-K27M diffuse pontine gliomas. Treatment of patient-derived H3.3-K27M mutant cell lines with the JMJD3 H3K27 histone demethylase inhibitor GSKJ4 resulted in increased H3K27 methylation (me2/me3) together with decreased cell proliferation and viability in vitro [38]. The authors also showed that specific inhibition of JDJM3 by GSKJ4 and not by UTX (ubiquitously transcribed X chromosome tetraicopeptide repeat protein) resulted in decreased proliferation and increased survival. Importantly, using the same mutant cell lines in an orthotopic murine model, the authors showed that GSKJ4 reduced tumor growth, increased animal survival, and increased levels of H3K27me3 in tumor cells. In this context, it was established that the drug crossed the BBB and was present in the brainstem. The activity of GSKJ4 appears specific to H3K27M tumors as this drug had no effect on tumors bearing wild-type H3.3 or H3.3-G34R/V mutants either in vitro or in vivo. As a result, other formulations of GSKJ4 suitable for use in humans are poised to enter clinical trials in H3 K27M-mutant diffuse midline gliomas and could be offered at diagnosis in combination with other targeted therapies and RT.
Erasers (HDACs)
The global H3 demethylation in histone H3.3-K27M mutation is associated with increased H3K27 acetylation, which may itself play a role in gliomagenesis [69, 74]. A high-throughput drug screen using patient-derived DIPG cell cultures showed substantial sensitivity to HDAC inhibitors (HDACi) [39]. Treatment with panobinostat (a pan-HDACi) showed decreased proliferation and viability of these cell cultures, which was associated with increased H3 acetylation and partial rescue of H3K27 trimethylation [39]. In vivo validation of panobinostat using a murine orthotopic patient-derived xenograft model showed increased survival and decreased tumor growth with the drug crossing the BBB. In testing for the efficacy of combination therapy, when used in conjunction with GSKJ4, panobinostat showed synergism against DIPG cell cultures in vitro [39]. Based on this promising preclinical success, panobinostat is currently offered in a phase I trial as a single agent in treatment of recurrent/progressive DIPG and HGG (PBTC-047).
Similar combinations of HDAC and demethylase inhibitors have shown preclinical activity both in vitro and in vivo in patient-derived xenografts [40]. Using a combination of vorinostat (HDACi) and tranylcypromine, a KDM1A/lysine-specific demethylase-1 inhibitor, Singh et al. [40] showed extended survival of mice in an orthotopic GBM model. In a retrospective analysis of children with HGGs (grade III–IV), valproic acid (an HDACi) in combination with chemoradiotherapy was well tolerated and sometimes demonstrated better clinical responses [75]. However, abundant caution should be exercised when selecting candidate drugs for combination use in epigenetic therapy as seen by the emergence of MGMT resistance when TMZ and vorinostat were combined [76]. In this case, vorinostat treatment of a TMZ-sensitive GBM cell line resulted in increased H3K9ac, decreased H3K9me2, and upregulation of MGMT to confer acquired TMZ resistance [76].
Writers (Histone Methyltransferases)
In a human embryonic stem cell model of H3.3-K27M gliomas, Funato et al. [50] carried out a drug screen with compounds targeting epigenetic regulators and discovered menin inhibitor 2 (MI-2) as their top hit. Menin inhibitor 2 treatment of NPC-P5K cells resulted in decreased proliferation and astrocytic differentiation in vitro, and was associated with decreased tumor growth in vivo using an orthotopic tumor model [50]. Menin (encoded by MEN1) is a member of the trithorax family histone methyltransferase complex and acts as an oncogenic cofactor in MLL-mediated leukemogenesis [51]. Current models suggest that blocking menin–MLL interactions disrupts MLL-mediated histone methylation and may therefore help in the treatment of H3.3-K27M mutant and hemispheric tumors [21, 51, 52].
Readers (Bromodomain Inhibitors)
Bromodomain and extraterminal domain (BET) proteins are epigenetic readers that bind to acetylated lysine residues in histone tails and regulate biological processes that include DNA damage repair, DNA replication, and transcriptional regulation. The H3.3-K27M mutation causes increased H3K27ac and increased occupancy of the bromodomain-containing proteins bromodomain (BRD) containing 1 and bromodomain containing 4 in the mutant nucleosomes [74]. Additionally, inhibition of bromodomain containing 4 using the BET inhibitor JQ1 in c-Myc- or MYCN-amplified tumors blocked MYCN-related transcription [23] and c-Myc downregulation [24]. Therefore, BET inhibitors could be especially useful in the H3.3-G34R/V tumors when there is MYCN upregulation, and when used in combination with panobinostat (HDACi), could synergize and prevent resistance in H3.3-K27M tumors [41]. Although MYCN is not considered to be a direct druggable target owing to its biochemical structure (there is no ligand binding site), small-molecule targeting of proteins (i.e., checkpoint kinase 1 and aurora kinase A) that stabilize MYCN levels provides a novel therapeutic strategy in the treatment of G34R/V tumors (Table 1) [22].
Isocitrate Dehydrogenase Mutant HGGs: DNA Methyl Transferase Inhibitors
Mutations in the isocitrate dehydrogenase (IDH) 1 and 2 enzymes lead to loss of normal enzymatic activity and increased production of the oncometabolite R-2-hydroxyglutarate (R-2-HG) [77]. Excessive accumulation of R-2-HG competitively inhibits α-ketoglutarate-dependent enzymes such as histone demethylases (JMJD2A, JMJD2C, and FBXL11) and the TET 5-methylcytosine hydroxylases [77]. This R-2-HG accumulation results in increased trimethylation of multiple histone lysine residues, DNA hypermethylation resulting in the glioma CpG island methylation phenotype, and blockage of differentiation in IDH mutant gliomas [77]. In this context, targeting DNA methylation may provide therapeutic benefit. This process is carried out by DNA methyltransferases that catalyze the transfer of a methyl group from S-adenosyl-methionine to cytosine residues in DNA [78]. Notably, the DNA methyltransferases inhibitor 5-azacytidine (Vidaza) and its deoxy analog 5-aza-deoxycytidine (Decitabine) have been used in clinical trials for patients with acute myeloid leukemia and myelodysplastic syndromes [31]. Preclinical validation of both 5-azacytidine and 5-aza-deoxycytidine in IDH1 mutant gliomas showed significant reduction of flank tumor growth, glial differentiation, and reduced DNA promoter methylation [31, 32].
Genetic Targets
There are distinct genetic abnormalities associated with histone mutations that coincide with specific anatomic locations (Table 1). Activation of the receptor tyrosine kinase, phosphatidylinositol 3-kinase/Akt, and mitogen-activated protein kinase pathways by mutations in the PDGFRA, FGFR, and MET receptors (10%) are potentially actionable and could be used in combination with conventional and other targeted therapies in order to provide synergy and prevent resistance [42, 43, 53]. Presence of the BRAF-V600E mutation should be routinely tested for in diffuse gliomas of all locations owing to the availability of small-molecule inhibitors and excellent clinical responses [44–47]. The Trk family of neurotrophin receptor fusion mutations can be specifically targeted by lestaurtinib (currently used in clinical trials in acute myeloid leukemia/myelodysplastic syndromes) in combination with BET inhibitors in the treatment of infantile hemispheric tumors [25, 26]. ACVR1 mutations leading to ligand-independent signaling of the bone morphogenetic protein (BMP) pathway are almost exclusively seen in H3.1-K27M pontine tumors and without inhibitors in clinical trials [58], there are further opportunities for therapeutic development.
Summary
There has been an explosion of knowledge in the understanding of the biology of pediatric HGGs in the past decade. The identification of driver histone mutations in specific anatomic locations has led to a new clinicopathological classification of these tumors. The identification of obligate partner mutations in nonhistone genes and their interactions with histone mutations requires further elucidation. The spatial and temporal homogeneity or conservation of the histone/nonhistone mutations in both primary and metastatic tumors suggests “epigenetic addiction,” validates biopsy-guided therapy, and supports epigenetic mutations as attractive therapeutic targets. Availability of elegant preclinical models will help us to identify and validate epigenetic targets and companion therapies. Using this strategy, clinical trials with drugs that inhibit histone modifications (writers, erasers and readers) are already underway. There is also a need to evaluate the efficacy of targeted therapies upfront when used with conventional genotoxic therapies (chemotherapy and radiation) rather than in the post-treatment relapse situation. Finally, using drug combinations that inhibit multiple targets may increase efficacy, provide synergy, and possibly avoid development of resistance to therapy.
References
Ostrom QT, de Blank PM, Kruchko C, et al. CBTRUS statistical report: ALSF infant and childhood primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol 2015;17(Suppl. 1):v1–35.
Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007; 114(2):97–109.
Kaderali Z, Lamberti-Pasculli M, Rutka JT. The changing epidemiology of paediatric brain tumours: a review from the Hospital for Sick Children. Childs Nerv Syst 2009; 25:787–793.
Louis DN, Perry A, Reifenberger G et al. The 2016 World Health Organization classification of tumors of the central nervous system. Acta Neuropathol 2016; 131:803–820.
Sturm D, Witt H, Hovestadt H, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012; 22: 425–437.
Buczkowicz P, Hoeman C, Rakopoulus P, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 2014; 46:451–456.
Kramm CM, Butenhoff S, Rausche U, et al. Thalamic high-grade gliomas in children: a distinct clinical subset? Neuro Oncol 2011; 13:680–689.
Vanan MI, Eisenstat DD. DIPG in children—what can we learn from the past. Front Oncol 2015; 5:237:1–17.
Grundy R, Walker D. Brain and spinal tumors: contemporary challenges in clinical practice. Pediatr Child Health 2010; 20:117–122.
Buczkowicz P, Bartels U, Bouffet E, Becher O, Hawkins C. Histopathological spectrum of pediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications. Acta Neuropathol 2014; 128:573–581.
Nikbakht H, Panditharatna E, Mikael LG, et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat Commun 2016; 7:11185.
Hoffman LM, DeWire M, Ryall S, et al. Spatial genomic heterogeneity in diffuse intrinsic pontine and midline high-grade glioma: implications for diagnostic biopsy and targeted therapeutics. Acta Neuropathol Commun 2016; 4: 1–8.
Wang X, Dubuc AM, Ramaswamy V, et al. Medulloblastoma subgroups remain stable across primary and metastatic compartments Acta Neuropathol 2015; 129: 449–457.
Jakacki RI, Cohen KJ, Buxton A, et al. Phase 2 study of concurrent radiotherapy and temozolomide followed by temolozomide and lomustine in the treatment of children with high-grade glioma: a report of the Children’s Oncology Group ACNS0423 study. Neuro Oncol 2016; 18: 1442–1450.
Bouffet E, Ramaswamy V. Old chemotherapy makes a comeback: dual alkylator therapy for pediatric high-grade glioma. Neuro Oncol 2016; 18: 1333–1334.
Ryall S, Krishnatry R, Arnoldo A, et al. Targeted detection of genetic alterations reveal the prognostic impact of H3K27M and MAPK pathway aberrations in pediatric thalamic glioma. Acta Neuropathol Commun 2016; 4: 93–103.
Kallappagoudar S, Yadav RK, Lowe BR, Partridge JF. Histone H3 mutations—a special role for H3.3 in tumorigenesis? Chromosoma 2015; 124: 177–179.
Maze I, Noh KM, Allis CD. Histone regulation in the CNS: Basic principles of Epigenetic plasticity. Neuropsychopharmacology 2013; 38:3–22
Agger K, Cloos PAC, Christensen J, et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007; 449:731–734.
Shen X, Liu Y, Hsu Y, et al. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell 2008; 32: 491–502.
Wu G, Diaz AK, Paugh BS, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 2014; 46:444–450.
Bjerke L, Mackay A, Nandhabalan M, et al. Histone H3.3 mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov 2013; 3:512–519.
Puissant A, Frumm SM, Alexe G, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov 2013; 3: 308–323.
Cheng Z, Gong Y, Ma Y, et al. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin Cancer Res 2013; 19: 1748–1759.
Iyer R, Varela CR, Minturn JE, et al. AZ64 inhibits TrkB and enhances the efficacy of chemotherapy and local radiation in neuroblastoma xenografts. Cancer Chemother Pharmacol 2012; 70:477–486.
Iyer R, Evans AE, Qi X, et al. Lestaurtinib enhances the antitumor efficacy of chemotherapy in murine xenograft models of neuroblastoma. Clin Cancer Res 2010; 16:1478–1485.
Brockmann M, Poon E, Berry T, et al. Small molecule inhibitors of Aurora-A induce proteasomal degradation of N-Myc in childhood neuroblastoma. Cancer Cell 2013; 24:75–89.
Shimomura T, Hasako S, Nakatsure Y, et al. MK-5108, a highly selective Aurora-A kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol Cancer Ther 2010; 9:157–166.
Cole KA, Huggins J, Laquaglia M, et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc Natl Acad Sci U S A 2011; 108:3336–3341.
Wadhwa E, Nicolaides T. Bromodomain inhibitor review: bromodomain and extra-terminal Family Protein Inhibitors as a potential new therapy in central nervous system tumors. Cureus 2016; 8:e620.
Turcan S, Fabius AWM, Borodovsky A et al. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT inhibitor decitabine. Oncotarget 2013; 4: 1729–1736.
Borodovsky A, Salmasi V, Turcan S, et al. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget 2013; 4: 1737–1747.
Rohle D, Popovici-Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013; 340:626–630.
Wang F, Travins J, DeLaBarre B, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013; 340:622–626.
Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med 2016; 21:373–380.
Ferris SP, Goode B, Joseph NM et al. IDH1 mutation can be present in diffuse astrocytomas and giant cell glioblastomas of young children under 10 years of age Acta Neuropathol 2016; 132: 153–155.
Sturm D, Bender S, Jones DTW, et al. Paediatric and adult glioblastoma multiform (epi)genomic culprits emerge. Nat Rev Cancer 2014; 14:92–107.
Hashizume R, Andor N, Ihara Y, et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med 2014; 20: 1394–1396.
Grasso CS, Tang Y, Truffaux N, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med 2015; 21:555–559.
Singh MM, Johnson B, Venkatarayan A, et al. Preclinical activity of combined HDAC and KDM1A inhibition in glioblastoma. Neuro Oncol 2015; 17: 1463–1473.
Ramaswamy V, Remke M, Taylor MD. An epigenetic therapy for diffuse intrinsic pontine gliomas Nat Med 2014; 20:1378–1379.
Bender S, Gronych J, Warnatz HJ, et al. Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat Med 2016; 22:1314–1320.
Chi AS, Batchelor TT, Kwak EL, et al. Rapid radiographic and clinical improvement after treatment of MET-amplified recurrent glioblastoma with a mesenchymal-epithelial transition inhibitor. J Clin Oncol 2012; 30:e30-e33.
Nicolaides TP, Li H, Solomon DA, et al. Targeted therapy for BRAF-V600E malignant astrocytoma. Clin Cancer Res 2011; 17:7595–7604.
Belden S, Flaherty KT. MEK and RAF inhibitors for BRAF-mutated cancers. Expert Rev Mol Med 2012; 14:1–10.
Robinson GW, Orr BA, Gajjar A. Complete clinical regression of a BRAF V600E- mutant pediatric glioblastoma multiforme after BRAF inhibitor therapy. BMC Cancer 2014; 14: 258–262.
Preusser M, Bienkowski M, Birner P. BRAF inhibitors in BRAF-V600 mutated primary neuroepithelial brain tumors. Expert Opin Investig Drugs 2015; 25: 1–8.
Milde T, Kleber S, Korshunov A et al. A novel human high-risk ependymoma stem cell model reveals the differentiation inducing potential of the histone deacetylase inhibitor vorinostat. Acta Neuropathol 2011; 122: 637–650.
Buczkowicz P, Hawkins C. Pathology, molecular genetics and epigenetics of diffuse intrinsic pontine glioma Front Oncol 2015;5:147.
Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 2014; 346:1529–1533.
Grembecka J, He S, Shi A, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol 2012; 8: 277–284.
Ren J, Xu W, Tang L, et al. Design and synthesis of benzylpiperidine inhibitors targeting the menin-MLL1 interface. Bioorganic Med Chem Lett 2016; 26: 4472–4476.
Koschmann C, Zamler D, Mackay A, et al. Characterizing and targeting PDGFRA alterations in pediatric high-grade glioma. Oncotarget 2016 Oct 4 [Epub ahead of print].
Au K, Singh SK, Burrell K et al. A preclinical study demonstrating the efficacy of nilotinib in inhibiting the growth of pediatric high-grade glioma. J Neurooncol 2015; 122: 471–480.
Castel D, Phillipe C, Calmon R, et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol 2015; 130: 815–827.
Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet 2014; 46(5):462–466.
Taylor KR, Mackay A, Truffaux N et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma Nat Genet 2014; 46: 457–461.
Luo Y, Alsamarah A, Zhang K, Hao J. Development of new therapeutic agents for Fibrodysplasia ossificans Progressiva. Curr Mol Med 2016; 16:4–11.
Sanvitale CE, Kerr G, Chaikaud A, et al. A new class of small molecule inhibitor of BMP signaling. PLOS ONE 2013; 8:e62721.
Shimono K, Tung W, Macolino C, et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-g agonists. Nat Med 2011; 17:454–462.
Cannon JE, Upton PD, Smith JC, et al. Intersegmental vessel formation in zebrafish: requirement for VEGF but not BMP signaling revealed by selective and non-selective BMP antagonists. Br J Pharm 2010; 161:140–149.
Shankar GM, Lelic N, Gill CM, et al. BRAF alteration status and the histone H3F3A gene K27M mutation segregate spinal cord astrocytoma histology. Acta Neuropathol 2016; 131:147–150.
Solomon DA, Wood MD, Tihan T, et al. Diffuse midline gliomas with histone H3-K27M mutation: A series of 47 cases assessing the spectrum of morphologic variation and associated genetic alterations. Brain Pathol 2016; 26: 569–580.
Fontebasso AM, Gayden T, Nikbakht H, et al. Epigenetic dysregulation: a novel pathway of oncogenesis in pediatric brain tumors Acta Neuropathol 2014; 128: 615–627.
Khuong-Quang DA, Buczkowicz P, Rakopoulos P, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 2012; 124:439–447.
Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3.3 and chromatin remodeling genes in paediatric glioblastoma. Nature 2012; 482:226–231.
Fontebasso AM, Schwartzentruber J, Khuong-Quang DA, et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol 2013; 125:659–669.
Becher OJ, Wechsler-Reya RJ. For pediatric glioma, leave no histone unturned. Science 2014; 346:1458–1459.
Lewis PW, Müller MM, Koletsky MS, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013; 340, 857–861.
Chan KM, Fang H, Gan H, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 2013; 27:985–990.
Swartling FJ, Savov V, Persson AI, et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell 2012; 21: 601–613.
Vanan MI, Eisenstat DD. Management of high-grade gliomas in the pediatric patient: past, present and future. Neuro Oncol Pract 2014; 1:145–157.
Jones C, Karajannis MA, Jones DT, et al. Pediatric high-grade glioma: biologically and clinically in need of new thinking. Neuro Oncol 2016 Jun 9 [Epub ahead of print].
Herz HM, Morgan M, Gao X, et al. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 2014; 345:1065–1070.
Masoudi A, Elopre M, Amini E, et al. Influence of valproic acid on outcome of High-grade gliomas in children. Anticancer Res 2008; 28: 2437–2442.
Kitange GJ, Mladek AC, Carlson BL, et al. Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin Cancer Res 2012; 18: 4070–4079.
Waitkus MS, Diplas BH, Yan H. Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol 2016; 18:16–26.
Cheishvili D, Boureau L, Szyf M. DNA methylation and invasive cancer: implications for therapeutics. Br J Pharmacol 2015; 172: 2705–2715.
Acknowledgements
Dr. Issai Vanan is the Father Peter J. Mckenna St Baldrick’s Cancer Research Scholar, supported by the St. Baldrick’s Foundation (USA). Dr. Underhill holds the Mary Johnston Chair in Melanoma Research, Alberta Cancer Foundation and University of Alberta (Canada). Dr. Eisenstat holds the Muriel and Ada Hole Kids with Cancer Society Chair in Pediatric Oncology, University of Alberta (Canada).
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Vanan, M.I., Underhill, D.A. & Eisenstat, D.D. Targeting Epigenetic Pathways in the Treatment of Pediatric Diffuse (High Grade) Gliomas. Neurotherapeutics 14, 274–283 (2017). https://doi.org/10.1007/s13311-017-0514-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-017-0514-2