
Abstract
Introduction
In the cardiovascular outcome trials (CVOT) EMPA-REG OUTCOME, TECOS and SAVOR-TIMI 53, empagliflozin [sodium/glucose cotransporter 2 (SGLT2) inhibitor], sitagliptin and saxagliptin [both dipeptidyl peptidase 4 (DPP4) inhibitors] + standard of care (SoC) were compared to SoC in patients with type 2 diabetes and established cardiovascular disease (CVD). This study assessed the cost-effectiveness (CE) of empagliflozin + SoC in comparison to sitagliptin + SoC and saxagliptin + SoC based on the respective CVOT.
Methods
The IQVIA Core Diabetes Model (CDM) was calibrated to reproduce the CVOT outcomes. EMPA-REG OUTCOME baseline characteristics and CVOT specific treatment effects on risk factors for cardiovascular disease [glycated haemogloblin A1c (HbA1c), body mass index (BMI), blood pressure, lipids] were applied. Three-year observed cardiovascular events of empagliflozin + SoC versus sitagliptin + SoC and saxagliptin + SoC were derived from EMPA-REG OUTCOME and an indirect treatment comparison. Relative risk (RR) adjustments to calibrate the CDM were estimated after consecutive attempts of running the model until the observed and CDM-predicted outcomes matched closely. The drug-specific treatment effects were considered up until treatment switch (when HbA1c reached 8.5%), after which, the United Kingdom Prospective Diabetes Study (UKPDS) 82 risk equations predicted events based on co-existing risk factors and treatment intensification to basal-bolus insulin were applied. The analysis was conducted from the perspective of the UK National Health Service. Costs and quality of life data were derived from UK national sources and published literature. A 50-year time horizon and discount rate of 3.5% were applied.
Results
The CDM projected quality-adjusted life years (QALYs) of 6.408, 5.917 and 5.704 and total costs of 50,801 GBP, 47,627 GBP and 48,071 GBP for empagliflozin + SoC, sitagliptin + SoC and saxagliptin + SoC, respectively. The incremental CE ratio (ICER) of empagliflozin + SoC versus sitagliptin + SoC and saxagliptin + SoC was 6464 GBP/QALY and 3878 GBP/QALY, respectively. One-way and probabilistic sensitivity analyses demonstrated the robustness of the results.
Conclusion
Results suggest that empagliflozin + SoC is cost-effective compared to sitagliptin + SoC and saxagliptin + SoC at a willingness to pay threshold of 20,000 GBP/QALY.
Funding
Boehringer Ingelheim International GmbH.
Similar content being viewed by others

Introduction
The goal of type 2 diabetes (T2D) management is to achieve and maintain target glycaemic control [haemogloblin A1c (HbA1c)] in order to avoid or delay the onset of costly macro and microvascular complications [1]. Glycaemic treatment targets should be individualized on the basis of patient preference and goals, patient characteristics and risk of adverse events associated with therapies (hypoglycaemia and weight gain) [1].
Considering that patients living with diabetes have an increased risk of cardiovascular disease (CVD), in 2008, the US Food and Drugs Administration (FDA) determined that concerns about cardiovascular (CV) risk should be more thoroughly addressed during drug development, requiring the industry to conduct post marketing cardiovascular outcome trials (CVOT) [2]. This recommendation was later followed by the European Medicines Agency (EMA). The EMA established that all new therapies for diabetes must undergo a rigorous assessment of CV safety through large-scale CVOT [3].
Schnell et al. reported that CVOT completed after 2008 demonstrated that new glucose-lowering drugs such as the dipeptidyl peptidase 4 (DPP4) inhibitors (saxagliptin, alogliptin and sitagliptin) and the glucagon-like peptide 1 (GLP-1) receptor agonists (lixisenatide and liraglutide) do not increase the CV risk in patients with T2D compared to standard of care (SoC) [4].
Regardless of the CV safety of all glucose-lowering drugs tested, in the SAVOR-TIMI 53 study, which assessed saxagliptin versus placebo, a significantly higher risk for heart failure (HF) was found in the active treatment group [5]. In the EXAMINE trial that compared alogliptin versus placebo as add-on to SoC, the same trend was found [5]. In contrast, there were no such concerns in the TECOS trial, in which sitagliptin was assessed versus placebo [5], and in the CARMELINA trial, in which linagliptin versus placebo added on top of SoC was evaluated [6]. As a result, the FDA issued a safety warning on DPP4 increasing the risk of heart failure (HF), particularly in patients who already have heart or kidney disease [7].
In the case of GLP-1 receptor agonists, in ELIXA, lixisenatide when added to SoC did not significantly alter the rate of major CV events or of HF hospitalisation [8]. In the LEADER trial, liraglutide compared to placebo in combination with SoC reduced the CV risk as shown by the composite outcome which included CV death, myocardial infarction (MI) and stroke. In this study, a non-significant reduction in hospitalisation for HF was observed [9].
With regards to sodium-glucose cotransporter 2 (SGLT2), the EMPA-REG OUTCOME trial [10] showed that empagliflozin significantly reduced CV risk compared to placebo as shown by the composite primary endpoint (death from CV causes, non-fatal MI or non-fatal stroke) and secondary endpoints all-cause mortality and hospitalisation for HF [11] and by the composite outcome of HF hospitalisation and CV death [12]. The DECLARE TIMI trial, which compared dapagliflozin versus placebo, did not show differences in the composite endpoint of CV death, MI or ischemic stroke but did result in a lower rate of CV death and hospitalisation for HF. In the CANVAS trial, canagliflozin also reduced the risk of CV death and HF hospitalisation compared to placebo [13].
Given the clinical evidence provided by these trials, the American Diabetes Association (ADA), American Heart Association (AHA) and the American College of Cardiology (ACC) recommend adding an agent that has proven CV benefit to reduce major adverse CV events and CV mortality in patients with long-standing suboptimally controlled T2D and established CVD [14, 15]. The aim of the current study is to assess the cost-effectiveness (CE) of empagliflozin versus sitagliptin and saxagliptin in combination with SoC in patients with T2D and established CVD.
Methods
The CE analysis was performed utilising the IQVIA Core Diabetes Model (CDM). The CDM is a web-based computer simulation model used to project the long-term health and economic outcomes of therapies commonly considered in the management of T2D. The basic structure of the CDM was described by Palmer et al. [16] and the most recent validation was published by McEwan et al. [17]. CDM version 9.0 was used in the current analysis and updated information and description of the structure are available online (http://www.core-diabetes.com/). Outcomes of analyses are life expectancy (LY), quality-adjusted life expectancy (QALY), cumulative incidence of diabetes-related complications, time to onset of diabetes-related complications and related costs. This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors. The CDM development is based on previously published data.
For this study, the standard version of the CDM (CDM v9.0) was redesigned and calibrated to reflect the clinical efficacy and outcomes observed in the CVOT. This calibration process involved fine-tuning the newly incorporated relative risk (RR) adjustments that are applied to the transition probabilities that predict diabetes-related complications. The steps to calibrate the model are described hereunder and summarized in an infographic (Fig. S1).
Firstly, the annual progression of diabetes-related complication risk factors [HbA1c, systolic blood pressure (SBP), diastolic blood pressure (DBP), low-density lipoprotein cholesterol (LDL), high-density lipoprotein cholesterol (HDL), hypoglycaemia rates, body mass index change, heart rate] per treatment arm were programmed in the CDM for the follow-up period of each trial and available endpoints. These input data were taken from the EMPA-REG OUTCOME study for empagliflozin, from the TECOS [18] study for sitagliptin and from the SAVOR-TIMI 53 [19] study for saxagliptin.
In the CDM, the risk equations (RE) to predict the risk of macrovascular complications are informed by risk factor progression values. In CDM v9.0, many REs are available: United Kingdom Prospective Diabetes Study (UKPDS) 68 [15], UKPDS 82 [20], Advance [21], Swedish National Diabetes Register [22, 23], ARIC study [24], Fremantle [25] and PROCAM [26]. The different RE were tested and the UKPDS 82 RE was found to be the best fit to predict the 3-year clinical outcomes of the EMPA-REG OUTCOME trial [20].
The second step of the calibration was an iterative process to identify the best set of RR adjustments required to align the UKPDS 82 RE predictions to match the observed 3-year clinical outcomes. The outcomes used to calibrate the predictions were primary and secondary MI, primary and secondary stroke, hospitalisation for angina, hospitalisation for heart failure, CV death, non-CV death, microalbuminuria (MAU) and end-stage renal disease (ESRD).
Three-year event rates for empagliflozin + SoC were taken directly from the EMPA-REG OUTCOME trial. Event rates for sitagliptin and saxagliptin were calculated using the RR from the indirect treatment comparison (ITC) reported by Balijepalli et al. (Tables 1, 2) [27]. In order to match the endpoints specified in the EMPA-REG OUTCOME trial and those reported by ITC with the CDM endpoints, some assumptions were made (Table S1). It should also be noted that renal outcomes were not included in the ITC, whereas in the EMPA-REG OUTCOME study a clear treatment effect on renal function is observed. Given that neither sitagliptin nor saxagliptin CVOT assessed the treatment benefit on the renal function, the observed outcomes on microalbuminuria and end-stage renal disease for both drugs were assumed to be equal to the placebo arm in the EMPA-REG OUTCOME study [18, 19, 28]. On the basis of this calibration, RR adjustments for outcomes were estimated (Table 1).
Modelling Cohort
The cohort of patients at the start of the analysis represents the EMPA-REG OUTCOME trial population. Baseline characteristics are presented in Table S2. Mean duration of diabetes was 9 years and determined by the time since diagnosis data available in the trial.
Clinical Data
Treatment Effects
The impacts of treatments on risk factors which were tracked in each trial were applied in the CDM in two steps. First-year benefit was programmed as change from baseline in the treatment settings, together with the associated adverse event rates of each treatment (Table 3). For the following years, progression tables of risk factors were populated with post 1st-year annual treatment effects up to the end of the study period. The data was taken from the respective trial publications [11, 18, 19, 28]. Of note, the treatment effects collected reflect the impact of both main therapy and the intensification of SoC, as specified in the different study protocols [11, 18, 19].
Treatment Duration/Switch to 2nd Line
The EMPA-REG OUTCOME and other CVOT were designed to assess the effect of the investigational drug versus placebo as an add-on to background therapy. This background therapy is a combination of other glucose-lowering drugs (such as sulfonylureas, metformin, GLP-1, glitazone, glinide and insulin) that were already administered at study initiation or escalated over the study duration. The background therapy was therefore diverse and evolving over time in the CVOT (Fig. 1).

Lifetime survival of all therapies
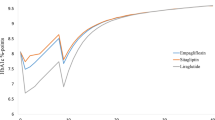
Nevertheless, as the time horizon for this analysis was 50 years, it would be inappropriate and unrealistic to apply the reported combined therapies throughout the time horizon. As such, treatment switch was programmed when an 8.5% HbA1c threshold was reached. As background insulin regimen was already provided to half of patients at week 164 of the EMPA-REG OUTCOME trial (50% in emplagliflozin vs. 58% in placebo), basal-bolus insulin therapy was considered to be the most plausible treatment escalation regimen. HbA1c progression was programmed to be exactly as described by the different CVOT and their follow-up period. After that, it was linearly extrapolated (see the evolution of HbA1c per treatment arm in Fig. 2). Treatment effects on risk factors and dose of basal bolus insulin were taken from Riddle et al. [29]. High dose mealtime insulin (57 IU) was combined with high dose glargine (94 IU).

Progression of HbA1c over time
Before treatment escalation, mortality, CV and renal outcomes in the CDM are predicted as described in the calibration process. Thus, they matched the event rates as reported in the EMPA-REG OUTCOME trial and those determined with relative treatment efficacy derived from indirect comparison. After treatment escalation, the standard UKPDS 82 RE [20] were applied to predict mortality and CV outcomes. The default CDM probabilities for developing renal disease are also applied.
Management Data
Clinical management data include the proportion of patients on preventive medications such as aspirin, statins and angiotensin-blocking agents, undergoing routine screening for diabetic complications and the sensitivity and specificity of the screening tests performed. These data are UK specific, and were sourced from published literature (Table S3).
Cost Data
This analysis was conducted from the UK healthcare payer perspective and as such only include direct costs.
Treatment costs comprised drug cost, needle cost (where applicable) and the costs associated with self-monitoring of blood glucose (SMBG). Unit costs were extracted from the British National Formulary (BNF) [30] and Monthly Index of Medical Specialties (MIMS) [31]. Annual treatment cost per arm accounted for the main therapy and the background insulin therapy including administration costs. Over the trial period, the proportion of patients receiving the different therapies changed. In some cases, it decreased, but in most cases it increased. As per trial protocol, physicians were asked to intensify SoC to keep patients’ glycaemic targets under control. The background treatment costs for insulin regimen were included as it was the main adjuvant treatment commonly present in all trials and it is associated with a considerable cost (unlike metformin or sulfonylurea).
Table 4 outlines the first and subsequent years’ treatments cost of each alternative therapy under comparison plus the cost of treatment intensification on basal bolus insulin regimen.
Costs of diabetes-related complications, with either acute events (event costs) or chronic conditions of an ongoing disease (health states), were obtained from published literature (Table S3). Management cost data include preventative medications and screening tests for diabetic complications. All costs were updated to 2018 using the Hospital & Community Health Service (HCHS) Index from the Personal Social Services Research Unit (PSSRU, 2017) [32].
Utility Data
The default CDM utility data associated with events and health states derived from Beaudet et al. [33] were applied (Table S4). A minimum approach is applied to estimate utilities, meaning that where a patient has a history of MI and stroke, the lowest utility value (i.e. associated with the stroke event) was used.
The impact of utility on BMI changes was estimated through the inclusion of a disutility of − 0.0061 per unit gain in BMI for over 25 kg/m2 based on Bagust and Beale [34]. It was assumed that BMI remained constant over time in all arms after treatment.
Mortality Data
The UK age-gender specific life tables were used to predict non-specific mortality [35]. These mortality rates represent the risk of death not covered in the complication and adverse event sub-models of the CDM.
Analysis Plan
Base Case
A 50-year time horizon was applied unless otherwise indicated. Lifetime clinical and economic outcomes (mean and standard deviation) were calculated within the model using a non-parametric bootstrapping approach. This process simulated the lifetime progression of diabetes in cohorts of 1000 hypothetical patients on the basis of the baseline characteristics described above and the process was repeated 1000 times. This produces 1000 mean values of clinical effectiveness and lifetime costs that were used to estimate the likelihood of the intervention being cost-effective versus the alternatives.
Costs and effects were discounted at a rate of 3.5% annually, as recommended by the National Institute for Health and Care Excellence (NICE) [36].
To assess the impact of changing key input variables on the outcomes, several scenario analyses and a probabilistic sensitivity analysis (PSA) were conducted.
Scenarios
The scenario analyses involved a series of simulations, in which key inputs and assumptions were varied to assess their impact on clinical and cost outcomes. The scenarios were excluding insulin cost from treatment costs, applying direct CVOT outcomes for 3 years only followed by the standard UKPDS82 RE for prediction of events, time horizon equal to 5 years with direct CVOT outcomes applied up to 5 years, and changing the HbA1c threshold for treatment switching from 8.5% to 9%.
Probabilistic Sensitivity Analysis
The CDM uses Monte Carlo simulations with a non-parametric bootstrapping approach to capture parameter uncertainty through the model so that the imprecision of CE results can be assessed. This process involves sampling with replacement of input parameters by distribution in each bootstrap iteration of the analysis. Cohort baseline values (age, duration of diabetes and baseline physiological parameter levels), the treatment effects on physiological parameter levels, transition probabilities for CV events, health state utilities and event disutilities, and direct costs are subject to random sampling. PSA outcomes are reported in a CE scatter plot and the CE acceptability curve.
Results
Calibration
The observed (in EMPA-REG OUTCOME and calculated via indirect comparison) and CDM-predicted outcomes are presented in Table 1 along with the RR adjustments per outcome that were used to calibrate the CDM. Differences between the observed and expected outcomes were well below 5% for CV, renal and mortality outcomes at year 3, implying that the model has been calibrated to an acceptable degree of accuracy.
Base Case Analysis
Empagliflozin + SoC provided additional life years (LYs—0.766 and 1.053) and quality-adjusted life years (QALYs—0.491 and 0.704) at an additional cost of 3174 GBP and 2730 GBP compared to sitagliptin + SoC and saxagliptin + SoC, respectively. The resulting incremental CE ratios (ICER) of empagliflozin + SoC compared to sitagliptin + SoC and saxagliptin + SoC were 6464 GBP/QALY and 3878 GBP/QALY, respectively [36] (Table 5). These results indicated that empagliflozin + SoC is cost-effective and below the NICE recommended willingness to pay (WTP) threshold of 20,000 GBP per QALY.
Compared to empagliflozin + SoC, both alternatives have lower HbA1c and BMI reduction in the first year (Fig. 2). On the comparators side, HbA1c and BMI progressed faster, leading to an increased risk of diabetes complications and a decline of survival (Fig. 1). Lower LYs and QALYs were observed in both comparator arms.
In the long term, a greater number of patients remained alive in the empagliflozin + SoC arm compared with sitagliptin + SoC and saxagliptin + SoC. As a result, a higher number of these patients are at risk of developing diabetes complications (survival paradox). Consequently, they have slightly higher lifetime cumulative incidence of stroke, peripheral vascular disease, unstable angina, diabetic retinopathy, foot ulcer, amputations and neuropathy, but a lower incidence of HF, MI and end-stage renal disease compared to sitagliptin + SoC and saxagliptin + SoC (Table 6). This is also reflected in terms of costs. Patients on empagliflozin + SoC have higher CVD and eye disease complications costs compared to both comparators. Additionally, management costs are higher because of longer survival (Table 7). On the contrary, as empagliflozin + SoC slows down the progression of renal disease, this arm demonstrated lower costs associated with the treatment renal disease complications (Table 6).
Scenario Analyses
Excluding the cost of insulin therapy led to lower treatment costs. Since the use of insulin was highest in the EMPA-REG OUTCOME study, excluding the cost of insulin resulted in a lower ICER.
When a 5-year time horizon was explored, empagliflozin + SoC provides added LYs (0.132 and 0.179) and QALYs (0.099 and 0.151), with additional incremental costs 790 GBP and 256 GBP, which results in an ICER of 7977 GBP/QALY and 1695 GBP/QALY compared to sitagliptin + SoC and saxagliptin + SoC respectively. Patients receiving empagliflozin + SoC have the lowest CV-related or non-CV-related mortality (Table 8).
When applying the direct CVOT outcomes up to 3 years (time horizon of the EMPA-REG OUTCOME study [11]) and keeping treatment escalation after an HbA1c of 8.5% is reached, empagliflozin + SoC continues to provide additional LYs (0.346 and 0.467) and QALYs (0.243 and 0.353) and is more costly compared to sitagliptin + SoC and saxagliptin + SoC. Empagliflozin + SoC continues to be a cost-effective therapy against these comparators (Table 8). Changing the treatment switch from HbA1c 8.5% to 9% leads to a higher ICER but the conclusions remain the same (Table 8).
PSA showed that empagliflozin + SoC is a cost-effective therapy in 96% of simulations versus sitagliptin + SoC and in 99% simulations versus saxagliptin + SoC at a WTP threshold of 20,000 GBP per QALY (Figs. 3, 4).

Cost-effectiveness scatter plot of empagliflozin versus sitagliptin (a) and saxagliptin (b)

Cost-effectiveness acceptability curve of empagliflozin versus sitagliptin and saxagliptin
Discussion
The current analysis based on the CDM and CVOT evidence showed that, in patients with an established risk of CVD, empagliflozin + SoC is cost-effective versus sitagliptin + SoC and saxagliptin + SoC. It showed that empagliflozin + SoC provides additional LYs and QALYs, for an additional cost below 3200 GBP. The incremental CE ratios were below the NICE WTP threshold of 20,000 GBP per QALY (Fig. 4).
A number of key assumptions were incorporated into the model. First, despite the fact that treatment guidelines recommend HbA1c thresholds of 7% or 7.5% for treatment switching, a threshold of 8.5% was applied. The reason for this is that the baseline HbA1c value was 8.07% and the treatment effect with empagliflozin was only 0.58%; therefore, testing a lower threshold would not be inappropriate because patients would switch to the next treatment line before the 3-year therapy of the trial was complete. Additionally, in the trial, there were several treatment adaptations that are taken into account in our HbA1c progression and costing.
Second, as previously discussed, patients were receiving a broad mix of therapies in CVOT, continuous adaptations to treatment were required to keep HbA1c low according local guidelines, and thus reflected in the CVO outcomes. Given the combined therapies administered to patients in the CVOT, a high dose of basal-bolus insulin was chosen as the next line therapy.
Thirdly, the extent of CV benefit beyond the study follow-up period remains uncertain. In the base case analysis, the benefit is applied over 8 years as a consequence of the 8.5% HbA1c switch threshold. To evaluate the impact on results, separate scenario analyses were conducted: evaluating the effects of treatments at a shorter time horizon (= 5 years), applying CVOT benefits for 3 years only (= trial follow-up period), applying treatment intensification at an HbA1c threshold of 9%; and excluding insulin costs from therapy costs. All analyses confirmed the cost-effectiveness profile of emplagliflozin + SoC against sitagliptin and saxagliptin as add-on to SoC.
PSA confirmed the robustness of results, as the majority of the simulations (at least 90%) showed that empagliflozin + SoC was a cost-effective therapy against both comparators for a WTP of 20,000 GBP per QALY.
The short-term clinical benefit of empagliflozin was demonstrated in avoiding death due to CV and other non-CV causes and in reducing the risk of HF, MI, eye disease, micro and macroalbuminuria, and ESRD. However, the life-long analyses showed that these benefits were partly offset by the extended life expectancy and prolonged time exposure to diabetes complications (survival paradox) (Table 6). As a result, complication, treatment and management costs were greater in the empagliflozin + SoC arm compared to sitagliptin + SoC and saxagliptin + SoC.
To conduct this analysis a customized version of the CDM was prepared to predict the results of the EMPA-REG OUTCOME trial [11], and to use the relative efficacy data of the different comparators produced by the ITC. We opted for the UKPDS 82 RE to predict lifetime health outcomes, also after treatment switch. The rationale to choose this equation was based on the calibration process which indicated that this RE predicted most closely the EMPA-REG OUTCOME trial outcomes. After calibration the CDM predicted the trials’ outcomes accurately at 3 years.
As a result of a lack of available head to head trials, the relative efficacy of the comparators were obtained via an ITC [27]. Some limitations concerning its methodology were reported by the authors of the ITC and which are also applicable to the current economic analyses. The included randomised controlled trials differed with respect to their sample sizes, follow-up periods, and on some important baseline patient and design characteristics (e.g. not all trials targeted only patients with established CVD). There were notable differences across the trials in the proportions of patients receiving various background medications for their CVD risk. Formal methods to control for these disparities were not feasible, because the evidence base consisted of only one study per treatment. Thus, these limitations may add some level of redundancy to estimates that are not possible to quantify.
The methodology applied in this study can also be (and was already) applied to other populations as for example to patients with type 2 diabetes free of a history of cardiovascular disease [37]. The requisite is of course that clinical evidence on impact on cardiovascular outcomes is available. Since clinical trials are ongoing in other indications, like hypertension on top of type 2 diabetes, we could also study the clinical and economic benefits of empagliflozin in those indications [38].
Other health economic studies based on the EMPA-REG OUTCOME study were published. Similar findings were found showing that empagliflozin is a cost-effective therapy in patients with diabetes and established CVD [39,40,41,42,43,44].
Conclusion
This study demonstrates that empagliflozin + SoC is a cost-effective treatment compared to sitagliptin + SoC and saxagliptin + SoC in the management of patients with T2D and an established risk of CVD from the UK NHS perspective. The scenario and sensitivity analyses confirmed the robustness of the findings.
References
Davies MJ, D’Alessio DA, Fradkin J, et al. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2018;41(12):2669–701.
Food and Drug Administration. Guidance for industry—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. Diabetes Mellit US Dep Health Hum Serv Wash DC 2008. 2008;8. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/diabetes-mellitus-evaluating-cardiovascular-risk-new-antidiabetic-therapies-treat-type-2-diabetes.
European Medicines Agency. Guideline on clinical investigation of medicinal products in the treatment or prevention of diabetes mellitus. Comm Med Prod Hum Use CHMP CPMPEWP108000 Rev 12012; 2012. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-medicinal-products-treatment-prevention-diabetes-mellitus-revision_en.pdf.
Schnell O, Rydén L, Standl E, Ceriello A, On behalf of the D&CVD EASD Study Group. Current perspectives on cardiovascular outcome trials in diabetes. Cardiovasc Diabetol. 2016;15(1):139.
Schnell O, Rydén L, Standl E, Ceriello A. Updates on cardiovascular outcome trials in diabetes. Cardiovasc Diabetol. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5637292/. Accessed 2018 Aug 28.
McGuire DK, Alexander JH, Johansen OE, et al. Linagliptin effects on heart failure and related outcomes in individuals with type 2 diabetes mellitus at high cardiovascular and renal risk in CARMELINA. Circulation. 2019;139(3):351–61.
Food and Drug Administration. Diabetes medications containing saxagliptin and alogliptin: drug safety communication—risk of heart failure. 2016. https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-adds-warnings-about-heart-failure-risk-labels-type-2-diabetes. Accessed 2016 Apr 13.
Pfeffer MA, Claggett B, Diaz R, et al. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med. 2015;373(23):2247–57.
Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375(4):311–22.
Zinman B, Inzucchi SE, Lachin JM, et al. Rationale, design, and baseline characteristics of a randomized, placebo-controlled cardiovascular outcome trial of empagliflozin (EMPA-REG OUTCOME™). Cardiovasc Diabetol. 2014;19(13):102.
Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117–28.
Fitchett D, Zinman B, Wanner C, et al. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: results of the EMPA-REG OUTCOME® trial. Eur Heart J. 2016;37(19):1526–34.
Rådholm K, Figtree G, Perkovic V, et al. Canagliflozin and heart failure in type 2 diabetes mellitus. Circulation. 2018;138(5):458–68.
American Diabetes Association. Introduction: standards of Medical Care in Diabetes-2018. Diabetes Care. 2018;41:S1–2.
American College of Cardiology, American Heart Association. Expert consensus decision pathway on novel therapies for cardiovascular risk reduction in patients with type 2 diabetes and atherosclerotic cardiovascular disease. A report of the American College of Cardiology Task Force on Expert Consensus Decision Pathways. J Am Coll Cardiol. 2018. http://www.onlinejacc.org/content/early/2018/11/29/j.jacc.2018.09.020. Accessed 2019 Feb 18.
Palmer AJ, Roze S, Valentine WJ, et al. The CORE Diabetes Model: projecting long-term clinical outcomes, costs and cost-effectiveness of interventions in diabetes mellitus (types 1 and 2) to support clinical and reimbursement decision-making. Curr Med Res Opin. 2004;20(Suppl 1):S5–26.
McEwan P, Foos V, Palmer JL, Lamotte M, Lloyd A, Grant D. Validation of the IMS CORE diabetes model. Value Health. 2014;17(6):714–24.
Green JB, Bethel MA, Armstrong PW, et al. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373(3):232–42.
Scirica BM, Bhatt DL, Braunwald E, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369(14):1317–26.
Hayes AJ, Leal J, Gray AM, Holman RR, Clarke PM. UKPDS outcomes model 2: a new version of a model to simulate lifetime health outcomes of patients with type 2 diabetes mellitus using data from the 30 year United Kingdom Prospective Diabetes Study: UKPDS 82. Diabetologia. 2013;56(9):1925–33.
Kengne AP, Patel A, Marre M, et al. Contemporary model for cardiovascular risk prediction in people with type 2 diabetes. Eur J Cardiovasc Prev Rehabil. 2011;18(3):393–8.
Ahmad Kiadaliri A, Clarke PM, Gerdtham U-G, et al. Predicting changes in cardiovascular risk factors in type 2 diabetes in the post-UKPDS Era: longitudinal analysis of the Swedish National Diabetes Register. J Diabetes Res. 2013;2013:241347.
Cederholm J, Eeg-Olofsson K, Eliasson B, Zethelius B, Nilsson PM, Gudbjörnsdottir S. Risk prediction of cardiovascular disease in type 2 diabetes. Diabetes Care. 2008;31(10):2038–43.
Folsom AR, Chambless LE, Duncan BB, Gilbert AC, Pankow JS, Atherosclerosis Risk in Communities Study Investigators. Prediction of coronary heart disease in middle-aged adults with diabetes. Diabetes Care. 2003;26(10):2777–84.
Davis WA, Knuiman MW, Davis TME. An Australian cardiovascular risk equation for type 2 diabetes: the Fremantle Diabetes Study. Intern Med J. 2010;40(4):286–92.
Assmann G, Cullen P, Schulte H. Simple scoring scheme for calculating the risk of acute coronary events based on the 10-year follow-up of the prospective cardiovascular Münster (PROCAM) study. Circulation. 2002;105(3):310–5.
Balijepalli C, Shirali R, Kandaswamy P, et al. Cardiovascular safety of empagliflozin versus dipeptidyl peptidase-4 (DPP-4) inhibitors in type 2 diabetes: systematic literature review and indirect comparisons. Diabetes Ther. 2018;9(4):1491–500.
Wanner C, Inzucchi SE, Lachin JM, et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med. 2016;375(4):323–34.
Riddle MC, Bolli GB, Ziemen M, et al. New insulin glargine 300 units/mL versus glargine 100 units/mL in people with type 2 diabetes using basal and mealtime insulin: glucose control and hypoglycemia in a 6-month randomized controlled trial (EDITION 1). Diabetes Care. 2014;37(10):2755–62.
BNF Publications. BNF online. 2018. https://www.bnf.org/products/bnf-online/. Accessed 2018 Mar 29.
Database of prescription and generic drugs, clinical guidelines | MIMS online. 2017. https://www.mims.co.uk/. Accessed 2017 May 2.
Personal Social Services Research Unit JE. Unit costs of health and social care 2017; 2017. p. 122.https://www.pssru.ac.uk/project-pages/unit-costs/unit-costs-2017/.
Beaudet A, Clegg J, Thuresson P-O, Lloyd A, McEwan P. Review of utility values for economic modeling in type 2 diabetes. Value Health. 2014;17(4):462–70.
Bagust A, Beale S. Modelling EuroQol health-related utility values for diabetic complications from CODE-2 data. Health Econ. 2005;14(3):217–30.
Office for National Statistics. National life tables, UK: 2014 to 2016. 2018. https://www.ons.gov.uk/releases/nationallifetablesuk2014to2016. Accessed 2018 Aug 29.
NICE. Developing NICE guidelines: the manual | Guidance and guidelines. 2014. https://www.nice.org.uk/process/pmg20/chapter/introduction-and-overview. Accessed 2018 Aug 29.
Lamotte M, Salem A, Mettam SR, Ustyugova AV, Zhang E, Ramos M. 1292-P: projected long-term clinical benefit and cost-effectiveness of empagliflozin compared with glimepiride in patients with type 2 diabetes in China. Diabetes. 2019;68(Suppl 1):1292.
McGill JB. The SGLT2 inhibitor empagliflozin for the treatment of type 2 diabetes mellitus: a bench to bedside review. Diabetes Ther. 2014;5(1):43–63.
Daacke I, Kandaswamy P, Tebboth A, Kansal A, Reifsnider O. Cost-effectiveness of empagliflozin (Jardiance) in the treatment of patients with type 2 diabetes mellitus (T2DM) in the UK based on Empa-Reg outcome data. Value Health. 2016;19(7):A673.
Gourzoulidis G, Tzanetakos C, Ioannidis I, et al. Cost-effectiveness of empagliflozin for the treatment of patients with type 2 diabetes mellitus at increased cardiovascular risk in Greece. Clin Drug Investig. 2018;38(5):417–26.
Iannazzo S, Mannucci E, Reifsnider O, Maggioni AP. Cost-effectiveness analysis of empagliflozin in the treatment of patients with type 2 diabetes and established cardiovascular disease in Italy, based on the results of the EMPA-REG OUTCOME study. Farmeconomia Health Econ Ther Pathw. 2017. https://journals.seedmedicalpublishers.com/index.php/FE/article/view/1332. Accessed 2018 Aug 29.
Kansal A, Reifsnider O, Lee J, et al. Cost-effectiveness analysis of empagliflozin compared with canagliflozin or standard of care (SoC) in patients with T2DM and established cardiovascular (CV) Disease. Diabetes. 2018;67(Suppl 1):1294.
Mettam SR, Bajaj H, Kansal AR, Kandaswamy P. Cost effectiveness of empagliflozin in patients with T2DM and high CV risk in Canada. Value Health. 2016;19(7):A674.
Nguyen E, Coleman CI, Nair S, Weeda ER. Cost-utility of empagliflozin in patients with type 2 diabetes at high cardiovascular risk. J Diabetes Complicat. 2018;32(2):210–5.
Acknowledgements
We thank Emma Gray, who at the time of submission was an employee of IQVIA, for her native English review.
Funding
This study and journal’s Rapid Service Fee were sponsored by Boehringer Ingelheim International GmbH.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contributions
All authors are responsible for the content of the manuscript, including the data collection and analyses. MR, VF, ML, AU contributed to the conception and design of the study, acquisition of data, analyses and interpretation of data. MR and ML: drafted the manuscript. VF, AU, NH, PG revised it.
Prior Presentations
The study results have been presented at two conferences in 2018, Europe ISPOR, held in Barcelona and at 54th EASD Annual Meeting 2018 in Berlin.
Disclosures
Mafalda Ramos is an employee of IQVIA which received consulting fees for the adaptation of the CDM. Volker Foos was a full-time employee of IQVIA which received consulting fees for the adaptation of the CDM. Mark Lamotte is an employee of IQVIA at the time the study was conducted, which received consulting fees for the adaptation of the CDM. Volker Foos is now a full-time employee of HEOR Germany, Düren, Germany. Anastasia Ustyugova is a full-time employee of Boehringer Ingelheim. Nikco Hau is a full-time employee of Boehringer Ingelheim. Pranav Gandhi was a full-time employee of Boehringer Ingelheim at the time the study was conducted and is now a full-time employee of Allergan plc, New Jersey, USA.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
The IQVIA CDM is available online to license holders only. The programming code of the IQVIA CDM is not publicly available. Permission to receive a Word copy of the settings used to run the analyses can be requested to anastasia.ustyugova@boehringer-ingelheim.com.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.9896354.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Ramos, M., Foos, V., Ustyugova, A. et al. Cost-Effectiveness Analysis of Empagliflozin in Comparison to Sitagliptin and Saxagliptin Based on Cardiovascular Outcome Trials in Patients with Type 2 Diabetes and Established Cardiovascular Disease. Diabetes Ther 10, 2153–2167 (2019). https://doi.org/10.1007/s13300-019-00701-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-019-00701-3