
Abstract
Although hyperglycemia is a key therapeutic focus in the management of patients with type 2 diabetes mellitus (T2DM), many patients experience sub-optimal glycemic control. Current glucose-lowering agents involve the targeting of various body organs. Sodium glucose co-transporter type 2 (SGLT2) inhibitors target the kidney, reduce renal glucose reabsorption, and increase urinary glucose elimination, thus lowering glucose blood levels. This review examines some of the key efficacy and safety data from clinical trials of the main SGLT2 inhibitors approved or currently in development, and provides a rationale for the use of SGLT2 inhibitors in the treatment of T2DM.
Similar content being viewed by others


Introduction
Cardiovascular disease (CVD) is the major cause of death in patients with diabetes mellitus (DM); however, microvascular complications (e.g., retinopathy, nephropathy, and neuropathy) cause significant morbidity and disability, such as visual impairment/blindness, progressive renal impairment, and non-traumatic amputations. Hyperglycemia increases the risk of microvascular complications, and improved control of hyperglycemia reduces the risk of microvascular complications. Adiposopathy (i.e., positive caloric balance leading to adipocyte hypertrophy, visceral fat accumulation, “lipotoxicity”, and subsequent pathogenic adipocyte and adipose tissue endocrine and immune responses) is often the initial promoter of insulin resistance and, therefore, of hyperglycemia [1]. However, once elevated glucose levels are present, chronic hyperglycemia itself may worsen glucose control by further promoting insulin resistance and impairing pancreatic beta-cell function (via a reduced beta-cell survival and mass, decreased insulin gene transcription, and decreased insulin synthesis and secretion) [2, 3], through a process often termed glucotoxicity. Hyperglycemia may also promote macrovascular complications via direct and indirect effects on vasculature similar to those observed in atherosclerosis [4–7]. Finally, hyperglycemia may further worsen the adiposopathic dyslipidemia often associated with type 2 DM (T2DM) [8–11].
DM is defined by hyperglycemia and, given the proven health benefits of reducing hyperglycemia, glucose control remains a key therapeutic focus for the treatment of DM [12–19], with glycosylated hemoglobin A1c (HbA1c) being a commonly used measure of longer-term glycemic control. Some studies are inconclusive in determining the efficacy of intensive versus standard glycemic control in reducing macrovascular disease in T2DM [20–23]. However, one interpretation of the existing data is that the potential benefit of intensive versus less intensive (or “standard”) glucose control is dependent on the mechanism of action of the antidiabetes agent, as well as the speed and extent by which glucose lowering is achieved [24]. The greatest potential for macrovascular CVD benefit seems to be achieved with antidiabetes agents having the most favorable effects on CVD risk factors and the least potential to promote hypoglycemia, as well as when aggressive therapy is implemented early in the disease process in younger individuals with limited comorbidities.
The recommended HbA1c target of the American Diabetes Association, the European Association for the Study of Diabetes, and the International Diabetes Federation is <7.0% (53 mmol/mol) [25–27], which is applicable to many non-pregnant adults with DM [25]. However, it is increasingly recognized that the best health outcomes are often achieved via individualization of DM treatment objectives [26]. Less stringent HbA1c goals (such as <8.0%) may be appropriate for some patient groups, such as DM patients with hypoglycemia unawareness, as well as individuals with repeated bouts of severe hypoglycemia, comorbid conditions, and advanced microvascular/macrovascular complications [25]. Conversely, if significant hypoglycemia or other treatment side effects can reasonably be avoided, then more stringent HbA1c goals (such as 6.0–6.5%) might be considered in selected patients with short disease duration, minimal to no DM complications, and otherwise good health [25].
The key point is that improved glucose control in DM patients can reduce the risk of microvascular disease, and possibly reduce macrovascular disease in selected individuals. However, in clinical practice, glycemic control remains sub-optimal in many patients [28–32]. Data from the 2004 US National Health and Nutrition Examination Survey revealed that approximately 43% of DM patients had HbA1c >7.0% [33]. Reasons for the failure to achieve glycemic targets are multifactorial, and may include issues relating to the health-care provider (e.g., failure to sufficiently instruct on lifestyle changes, reluctance to intensify antidiabetes drugs, complexity of antidiabetes drug management, or lack of expertise) [34], and to the patient (e.g., non-adherence to favorable lifestyle habits and other therapies, lack of attendance at clinic, lack of understanding of the disease, reluctance to use insulin when required, longer duration of DM, or younger age [<40 years]) [30, 31, 35, 36].
Lifestyle modifications, such as nutritional and physical activity interventions, remain important toward improving both the glucose levels and overall health of DM patients. However, the reality is that the majority of patients with DM are managed with glucose-lowering therapeutic agents. Some examples of target organs for agents that lower glucose levels in DM include the pancreas, liver, muscle, adipose tissue, gastrointestinal system, and central nervous system. Due to the potential DM complication of nephropathy, the kidney has historically been regarded solely as a “victim” in DM management. With the development of the sodium glucose co-transporter type 2 (SGLT2) inhibitors, the kidney is now recognized as a potential “ally” in the management of DM [37]. Specifically, SGLT2 inhibitors reduce renal glucose reabsorption and promote urinary glucose excretion, thus lowering glucose blood levels. This supports the concept of the kidney as a target organ in the treatment of DM.
This review examines kidney glucose management and literature supporting SGLT2 inhibitors as a therapeutic approach to treating hyperglycemia.
Methods
The literature search involved review and original articles published up to July 11, 2013 using PubMed, with key search terms including SGLT2 inhibitors, sodium glucose co-transporter 2 inhibitors, glucose and kidney, and the individual drug names (dapagliflozin or BMS-512148; canagliflozin or JNJ-24831754; empagliflozin or BI10773; luseogliflozin or TS-071; tofogliflozin or CSG452; ipragliflozin or ASP1941; LX4211; EGT0001442; and ertugliflozin or PF04971729). Other sources of information for this review included abstracts from the American Diabetes Association (2010–2013) and the European Association for the Study of Diabetes (2010–2012), and clinical trial listings of SGLT2 inhibitors posted on ClinicalTrials.gov.
Glucose Reabsorption in the Kidney
Overview of Renal Structure and Function
The anatomy of the kidney is shown in Fig. 1. The main structural and functional unit of the kidney is the nephron. A normal human kidney contains approximately 1 million nephrons, with the majority located in the renal cortex and the remainder situated near the cortico-medullary junction. Each nephron consists of a glomerulus, containing afferent and efferent capillaries, and a renal tubule, which includes the glomerular (or Bowman’s) capsule, proximal convoluted tubule, loop of Henle, distal convoluted tubule, and the collecting duct. Higher positive pressure in the glomerular blood vessels forces fluid and solutes from the plasma into the glomerular capsule (filtration), and this filtrate then flows through the renal tubule. Much of this glomerular filtrate undergoes reabsorption into capillary blood via the proximal convoluted tubule. Nitrogenous and other waste products largely remain in the filtrate and pass into the collecting duct, eventually leading to urinary excretion. Other substances (e.g., hydrogen ions, potassium ions, ammonia, and drugs) undergo transport from peritubular capillaries into the renal tubule cells, and then into the filtrate for ultimate urinary excretion via the ureter, bladder, and urethra.

Renal anatomy. Nephrons are predominantly located in the renal cortex, with the remainder at the cortico-medullary junction. Each nephron consists of a glomerulus, containing afferent and efferent capillaries, and a renal tubule, including proximal and distal sections and a collecting duct
Physiology of Renal Glucose Transport
Key kidney functions that help achieve glucose homeostasis involve renal gluconeogenesis, glucose uptake from the circulation, and glucose reabsorption from the glomerular filtrate [38]. Given an average plasma glucose concentration of approximately 100 mg/dL (5.5 mmol/L) and a normal glomerular filtration rate of approximately 180 L/day, healthy individuals filter in the region of 180 g/day of glucose. Virtually all glucose is reabsorbed in the proximal convoluted tubule and returned to the circulation, so that effectively no glucose is excreted in the urine of an otherwise healthy individual. This system is highly efficient and allows conservation of glucose, which is a valuable energy source. Given the figure of 180 g/day of glucose reabsorbed, and the fact that the kidneys produce 15–55 g/day of glucose via gluconeogenesis and metabolize 25–35 g/day, renal absorption is a primary mechanism by which the kidney influences glucose homeostasis [38].
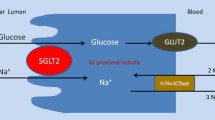
To retrieve glucose in the filtrate, the kidney utilizes two types of membrane-bound carrier proteins: SGLTs (sometimes described as symporters because they transport both glucose and sodium) and the facilitated glucose transporters (GLUTs, sometimes described as uniporters because they only transport glucose) [39, 40]. Details of the SGLT and GLUT families are given in Table 1 [40, 41]. Reabsorption of glucose from the glomerular filtrate is mediated by SGLTs in the proximal convoluted tubule (Fig. 2), in a process that is independent of insulin. Approximately 90% of filtered renal glucose is reabsorbed in the first segment (S1) of the proximal convoluted tubule by SGLT2, a low-affinity high-capacity transporter, and the remaining 10% is removed in the distal segment (S3) by SGLT1, a high-affinity low-capacity transporter [39, 40]. In the kidney, SGLT2 and SGLT1 are located on the luminal surface of epithelial cells lining the proximal convoluted tubule [40]. SGLT2 is expressed to a lower extent in other organs, including the liver, while SGLT1 is extensively expressed in the small intestine, where it has a significant role in glucose absorption [40]. SGLTs actively transport glucose against its concentration gradient via coupling to the electrochemical sodium gradient, using energy from a sodium/potassium adenosine triphosphatase pump [39, 40]. Glucose is released from the proximal convoluted tubule and returned to the bloodstream via GLUT2 in the S1/S2 segment and via GLUT1 in the S3 segment of the proximal convoluted tubule [39, 40]. This is a passive process requiring no energy (adenosine triphosphatase) input.

Glucose transporters in the renal proximal tubule. Data suggest approximately 90% of filtered glucose is reabsorbed in the first part (S1) of the proximal tubule and is mediated by SGLT2. The remaining 10% is reabsorbed in the distal (S2/S3) part of the tubule and this is mediated by SGLT1. This process is extremely efficient and virtually no glucose escapes into the urine of a healthy individual. Glucose is returned to the bloodstream via GLUT2 in the S1/S2 segment and via GLUT1 in the S3 segment of the proximal tubule
The amount of glucose filtered in the kidney increases linearly with increasing plasma glucose concentration until the transport maximum for glucose is reached (abbreviated as TMG and often expressed as mg glucose/min). Beyond the level of the TMG, the glucose transport system becomes saturated; therefore, any excess glucose remains in the filtrate and is excreted in the urine (i.e., glucosuria). In healthy, glucose-tolerant individuals, TMG is equivalent to a filtration rate of 260–350 mg/min [42]. The plasma glucose concentration at which TMG is reached is called the renal threshold, and occurs at approximately 200 mg/dL (11.0 mmol/L) [43]. This threshold may vary between individual nephrons due to variation in their activity and actual reabsorption capacity, which may be below the TMG level; the difference between the theoretical and actual renal thresholds is called “splay” [44].
Renal Glucose Handling in T2DM
In T2DM patients, glucose handling by the kidney may be altered, with an increase in TMG and urinary glucose excretion (UGE; i.e., glucosuria) at more elevated plasma glucose levels [38]. Mean TMG may increase to up to 20% or higher in those with DM, compared with healthy individuals [45]. Furthermore, SGLT2 and GLUT2 expression may be up-regulated in T2DM [46, 47]. These processes might be considered maladaptive in that they attenuate glucosuria, resulting in enhanced glucose reabsorption and further worsening hyperglycemia [37]. Inhibiting this cycle would be expected to increase glucose excretion in the urine, and thus reduce plasma glucose concentrations.
Targeting Renal Glucose Reabsorption with Sglt2 Inhibitors
Inhibiting SGLT2 provides an attractive addition to the DM treatment armamentarium. SGLT2 inhibitors reduce the TMG, so less glucose is reabsorbed in the proximal convoluted tubule; they also lower the renal threshold, so UGE occurs at a lower plasma glucose concentration (Fig. 3 [48]). The net result is increased UGE and decreased hyperglycemia. In addition to potentially improving hyperglycemic symptoms and DM disease complications, normalization of plasma glucose concentration may improve the adverse effects of glucotoxicity, which may contribute to DM itself, by reducing insulin resistance, decreasing hepatic gluconeogenesis, and potentially improving pancreatic beta-cell function [49].

Renal glucose handling before and after SGLT2 inhibition. SGLT2 inhibition reduces the transport maximum for glucose (TMG), which decreases glucose reabsorption in the proximal renal tubule, and lowers the renal threshold so that urinary glucose excretion (i.e., glucosuria) occurs at a lower plasma glucose concentration (reproduced with permission from [48])
Genetic models can often provide insight into what might be expected with therapeutic interventions. Individuals with familial renal glucosuria (FRG) have mutations in the gene encoding SGLT2 that cause loss of function. FRG is characterized by UGE, varying from a few grams to >200 g/day, depending on the presence of a homozygous or heterozygous mutation, in the presence of normal plasma glucose concentrations and without evidence of renal tubular dysfunction [50]. Most individuals affected by FRG have no symptoms and only rarely suffer from hypoglycemia or hypovolemia [50]. The lack of adverse events experienced by individuals with FRG due to persistent UGE suggests that long-term UGE induced by pharmacologic inhibition of SGLT2 may also be well tolerated. However, the number of FRG patients studied to date is small and these patients typically did not have DM. Therefore, while intriguing, long-term safety data for any therapeutic agents based upon this mechanism (i.e., SGLT2 inhibitors) are required before any conclusions can be made regarding the potential value of such agents in the clinical management of DM.
SGLT2 Inhibitors
Phlorizin is a naturally occurring glucoside found in various plants, such as the root bark of apple and other fruit trees, and was the prototype SGLT2 inhibitor. The structure of phlorizin is shown in Fig. 4. First isolated in the 1800s, research into phlorizin provided the evidence that altered renal glucose excretion could improve glycemic control [51, 52]. Studies from the 1950s revealed that phlorizin blocked sugar transport in several tissues, including the kidney and small intestine [53], and this was subsequently found to be due to inhibition of SGLT proteins. Phlorizin was ultimately found to be a competitive inhibitor of SGLT1 and SGLT2, but with a greater affinity for SGLT2 [40, 51]. In the 1980s, investigators found that phlorizin-induced UGE was effective in reducing hyperglycemia via an insulin-independent mechanism, without causing hypoglycemia [54, 55]. Animal studies also supported the use of phlorizin in improving insulin sensitivity without affecting insulin action in healthy control animals, with hyperglycemia and insulin resistance both returning after phlorizin discontinuation [54].

SGLT2 inhibitors in late phase clinical development
Unfortunately, phlorizin was unsuitable for clinical development as a therapeutic agent for a number of reasons. Firstly, phlorizin has a low selectivity for SGLT2 versus SGLT1, resulting in the inhibition of SGLT1 as well as SGLT2. As SGLT1 is primarily expressed in the small intestine, where it is responsible for the absorption of glucose and galactose from the diet, SGLT1 inhibition can result in gastrointestinal side effects such as severe diarrhea, dehydration, and malabsorption [40]. Secondly, phlorizin has a low oral bioavailability and is metabolized to phloretin by glucosidase enzymes in the gut, which means it must be given parenterally. Lastly, the phlorizin metabolite phloretin is a potent inhibitor of GLUT1 [51], which may lead to interference with glucose uptake in various tissues (e.g., the central nervous system [41]).
Nonetheless, phlorizin served as a model demonstrating how SGLT2 inhibition may become a therapeutic target for hyperglycemia [51]. Subsequent pharmacology research focused on phlorizin derivatives that possess increased stability, better bioavailability, more potent SGLT2 selectivity, and which were more suitable for once daily oral dosing with acceptable tolerability. Such investigations have included both O- and C-glucoside compounds. The first reported SGLT2 inhibitor was T-1095, an orally administered O-glucoside pro-drug that was metabolized in the liver into its active form, T-1095A [56]. Although T-1095 demonstrated increased SGLT2 selectivity and a dose-dependent glucosuric effect in preclinical studies [56], its non-selective SGLT1 inhibition led to discontinuation. Other O-glucoside compounds, such as sergliflozin, were discontinued during phase 2 studies. Attention then turned to the C-glucoside compounds, which had the advantage of increased metabolic stability (see below) [57].
SGLT2 inhibitor compounds in clinical development are shown in Table 2 and the chemical structures of those in phase 3 trials are shown in Fig. 4. Dapagliflozin was approved in the European Union in 2012, and is awaiting the outcome of resubmission of an application to the US Food and Drug Administration (FDA) following a complete response letter. The FDA approved canagliflozin in March 2013 and the submission made to the European Medicines Agency (EMA) is still under consideration. Marketing applications for empagliflozin were submitted to the FDA and EMA in March 2013, while marketing applications for ipragliflozin, luseogliflozin, and tofogliflozin were also recently submitted to the Japanese regulatory body.
Dapagliflozin is the most advanced of the SGLT2 inhibitors in terms of clinical development and, thus, has the largest amount of published clinical data. Publications for the other SGLT2 inhibitors are more limited and largely consist of data presented in scientific abstracts. A summary of key efficacy data from completed phase 3 trials and larger phase 2 trials using SGLT2 inhibitors developed in the US/Europe are presented in Table 3.
Dapagliflozin Overview
Dapagliflozin 1–50 mg orally once daily was evaluated as monotherapy in previously untreated patients with T2DM [58–60], or as add-on combination therapy with metformin [59, 61, 62], other oral anti-hyperglycemic agents [63–65], or insulin-based therapy [66–68]. Dapagliflozin significantly reduced HbA1c and fasting plasma glucose (FPG) levels (Table 3), with longer-term extension studies (≥100 weeks) supporting maintained efficacy [61, 63, 68]. Dapagliflozin monotherapy (2.5–50 mg/day for 12 weeks) in T2DM patients resulted in the urinary excretion of 52–85 g/day glucose at the end of the study period, compared with a loss of 6 g/day with placebo or metformin [69]. Dapagliflozin also reduced body weight, with an approximate 2 kg loss versus placebo after 12 weeks [66] or 24 weeks [58, 61], 1–2 kg loss versus comparator after 24 weeks [59], and 4 kg loss versus comparator after 52 weeks [63]. Although body weight increased when dapagliflozin was co-administered with pioglitazone, the increase was smaller than that of the placebo plus pioglitazone group (0.69–1.35 kg vs. 2.99 kg, respectively) [65].
In terms of safety and tolerability, dapagliflozin was associated with a small increase in the incidence of minor hypoglycemic events (0–10.0%) compared with the control group (placebo/comparator, 0.7–9.0%), although this was not statistically significant [59, 63–65]. A trial using dapagliflozin in combination with insulin (with/without ≤2 oral anti-hyperglycemic agents) reported slightly higher rates of hypoglycemic events (dapagliflozin [total groups] 56.6% vs. placebo 51.8%), but major hypoglycemic episodes were comparable between groups (dapagliflozin [total groups] 1.3% vs. placebo 1.0%) [67]. In another trial report, patients receiving dapagliflozin added to metformin experienced significantly fewer hypoglycemic events (3.5%) compared with glipizide plus metformin (40.8%; P < 0.0001) [63]. A safety analysis of 12 pooled placebo-controlled trials (n > 4,500) reported that hypoglycemia was more common with dapagliflozin than with placebo (10.7–16.3% vs. 8.0%, respectively), and that imbalances in individual studies were only observed when dapagliflozin was combined with a sulfonylurea or insulin [70, 71].
Dapagliflozin reduced systolic blood pressure (SBP) by up to 5 mmHg, with no significant increase in heart rate or occurrence of orthostatic hypotension [58, 61–65, 67].
Rates of hypotension, dehydration, and hypovolemia were similar in dapagliflozin groups (1–2%) to those in the placebo/comparator groups (0–1%) [58, 67, 70]. Dapagliflozin treatment was not associated with an increased risk of acute renal toxicity or deterioration of renal function [72]. The dapagliflozin Summary of Product Characteristics advises against its use in patients receiving loop diuretics or who are volume depleted, and recommends appropriate monitoring if volume depletion is likely to occur [73].
Symptoms suggestive of genital infection, such as cutaneous fungal infections, and lower urinary tract infection (UTI) were common adverse events with dapagliflozin and were reported more frequently compared with placebo/comparator. Genital infection occurred in 2–13% of patients receiving dapagliflozin compared with 0–5% of those receiving placebo/comparator, with women affected more commonly than men [58, 61–65, 67]. Most cases were not severe and responded well to standard therapy. Lower UTIs also occurred more frequently with dapagliflozin (3.0–12.5%) than with placebo/comparator (0–9.0%) [58, 61–65, 67]. None of these events were serious, and all cases resolved with standard antibiotic therapy. The pooled safety analysis (n = 4,545) reported that genital infections and UTIs were more common with dapagliflozin than placebo, and between-group differences were less marked for UTIs (genital infection: 4.1–5.7% dapagliflozin vs. 0.9% placebo; UTIs: 3.6–5.7% dapagliflozin vs. 3.7% placebo) [74, 75].
Canagliflozin Overview
Canagliflozin 50–300 mg once daily and 300 mg twice daily was evaluated as monotherapy in previously untreated patients with T2DM [76], or as add-on combination therapy with metformin [77–79], other oral anti-hyperglycemic agents [80–83], or insulin-based therapy [84, 85]. Canagliflozin significantly reduced HbA1c and FPG levels from baseline in studies of 12–52 weeks’ duration, as shown in Table 3, and modestly reduced body weight (up to 2.9 kg compared with control groups) [76, 77, 84]. Reductions in SBP with canagliflozin, when used as monotherapy and in combination, ranged from −0.8 to −6.8 mmHg [76, 78, 79, 82]. A recent pooled analysis of six phase 3 studies (n = 4,158; treatment duration not stated) revealed that canagliflozin produced modest reductions in SBP (−3.3 and −4.5 mmHg for 100 and 300 mg, respectively) relative to placebo [86].
The overall incidence of hypoglycemia was low and rates were similar across canagliflozin (2–6%), placebo (2–3%), and comparator (5%) groups [76, 77, 84]; however, canagliflozin groups (4.9–5.6%) reported lower rates of hypoglycemia compared with glimepiride (34.2%) [79]. Prescribing information for canagliflozin states that rates of hypoglycemia were higher when canagliflozin was administered with insulin or sulfonylureas [87]. Genital mycotic infections were higher with canagliflozin (3–15%) versus placebo/comparator (0–6%); these events were mild to moderate in severity and none led to study discontinuation [76, 77, 79–81]. As with dapagliflozin, genital mycotic infections were more common in women. Genital mycotic infections with canagliflozin were also assessed in a pooled analysis of four 26-week phase 3 studies (n = 2,313) [88]. Genital mycotic infections were more common in canagliflozin groups than placebo, occurring in 11% of women and 4% of men, versus 3% and 1% in the placebo groups, respectively. These events were generally mild or moderate in severity and were managed with standard treatments; in addition, few such events led to study discontinuation [10 cases, canagliflozin groups (6 cases 100 mg, 4 cases 300 mg)]. In a larger data set of eight phase 3 studies (n = 9,439) with longer mean exposure (68 weeks of canagliflozin, 64 weeks of placebo), the rate of male genital mycotic infection was higher (8% canagliflozin, 2% placebo) and was more common in uncircumcised men (11% vs. 3% in circumcised men) [88]. Reported UTI events showed a similar trend. Higher rates of UTI occurred in the canagliflozin groups (2.3–12.0%) versus the placebo/comparator groups (2.1–8.0%); the events were mild to moderate in severity and responded to standard treatment [76–82]. The pooled analysis of four 26-week phase 3 studies (n = 2,313) stated that UTIs occurred in 5.1% of patients receiving canagliflozin (100 mg plus 300 mg groups) and in 4.0% of those receiving placebo [89].
Empagliflozin Overview
Empagliflozin 1–50 mg once daily was evaluated as monotherapy in previously untreated patients with T2DM [90, 91], or as add-on combination therapy with metformin [92–95], other oral anti-hyperglycemic agents [96], or insulin-based therapy [97]. As shown in Table 3, empagliflozin significantly lowered HbA1c, reduced FPG, and decreased body weight (up to 2 kg vs. placebo). Empagliflozin 10 and 25 mg produced placebo-corrected reductions in SBP of approximately 2–5 mmHg after 24 weeks [91, 94, 95].
The rate of hypoglycemia was low with empagliflozin monotherapy (0.4–1.8%), and was comparable to placebo (0.4%) and comparator (sitagliptin monotherapy 0.4%, metformin monotherapy 7.1%) [91, 93]. The rate of hypoglycemia was higher when empagliflozin was given in combination therapy, particularly in regimens containing sulfonylurea or insulin (empagliflozin + metformin 2.4–3.6% vs. sitagliptin + metformin 5.4% [93]; empagliflozin + metformin 1.4–1.8% vs. placebo + metformin 0.5% [95]; empagliflozin + metformin + sulfonylurea 11.5–16.1% vs. placebo + metformin + sulfonylurea 8.4% [94]; and empagliflozin + insulin 36.1% vs. placebo + insulin 35.3% [97]).
UTI events after 24 weeks were reported in 8.3–10.3% of empagliflozin patients versus 8.0% of those on placebo, and the rates of genital infection events were 2.3–2.7% for empagliflozin and 0.9% for placebo [94]. A pooled analysis of safety data from four 24-week phase 3 trials (n = 2,477) examined the effect of empagliflozin on UTIs and genital infections [98]. The percentage of patients with events consistent with a UTI was similar across all groups (7.5% and 9.3% for empagliflozin 10 and 25 mg, respectively, vs. 8.2% for placebo); however, more patients receiving empagliflozin reported events consistent with genital infection (4.2% and 3.6% for empagliflozin 10 and 25 mg, respectively, vs. 0.7% for placebo) [98]. Both types of event were more common in women than in men, and were more common in patients with a history of UTI or genital infection [98]. Nevertheless, of those who reported events consistent with UTI or genital infection, most experienced only one episode; the episodes were generally mild in severity, and very few led to study discontinuation [4 cases of UTI (placebo: 1 case; empagliflozin: 2 cases, 10 mg and 1 case, 25 mg); 3 cases of genital infection (empagliflozin: 1 case, 10 mg; 2 cases, 25 mg)] [98].
Ipragliflozin Overview
Ipragliflozin is currently being developed in Japan (Table 2). Ipragliflozin 50–300 mg once daily was evaluated as monotherapy in previously untreated patients with T2DM [99, 100], or as add-on combination therapy with metformin [101] and other oral anti-hyperglycemic agents [102], and showed significant decreases in HbA1c and FPG versus placebo over periods of 12–24 weeks. Ipragliflozin monotherapy produced a placebo-corrected weight loss of −1.47 kg after 16 weeks [100]. After 12–16 weeks, SBP was reduced by −3.2 to −4.3 mmHg with ipragliflozin compared with placebo [100, 101].
Hypoglycemia was reported in 1.0–5.9% of ipragliflozin dose groups versus 0–3.0% in the placebo/comparator groups [101, 102]. During a 12-week study, UTIs were infrequent and were reported in all treatment groups (placebo 6.1% vs. ipragliflozin 1.4–6.9%) [101]. Genital infections occurred with greater frequency in the ipragliflozin versus placebo groups (3.0–4.3% vs. 1.5%, respectively) [101].
Other SGLT2 inhibitors in clinical development had few publications available at the time of this review.
Other Issues
Currently available information on outcomes such as stroke, heart attack, and other vascular complications is limited, but larger studies with cardiovascular end points are ongoing and will provide data in 2017 onwards [103, 104]. The Canagliflozin Cardiovascular Assessment Study (CANVAS; NCT01032629) has recruited more than 4,000 patients with T2DM and elevated risk of CVD, while the Empagliflozin Cardiovascular Outcome Event Trial (NCT01131676) has recruited an estimated 7,000 patients to date and the Dapagliflozin Effect on Cardiovascular Events (NCT01730534) study has recently begun recruitment (for further details of these trials see ClinicalTrials.gov).
The potential relationship between SGLT2 inhibitors and neoplasia is also being investigated. Although the overall proportion of patients with malignant or unspecified tumors was similar between those treated with dapagliflozin (1.43%) and placebo/comparator (1.30%), breast and bladder cancer events were more common with dapagliflozin [71]. The FDA regulatory submission for dapagliflozin stated that 9 cases of breast cancer were reported out of 4,287 patients receiving dapagliflozin compared with no cases out of 1,941 patients in the placebo/comparator group, and 7 cases of bladder cancer were reported out of 4,310 patients receiving dapagliflozin compared with no cases out of 1,962 patients in the placebo/comparator group [105]. Hematuria was documented before exposure to dapagliflozin in 4 of the patients later found to have bladder cancer, and the patients with breast cancer had received dapagliflozin for <1 year (2/9 cases were diagnosed within 6 weeks of dapagliflozin treatment initiation). Whether these are chance findings or clinically relevant concerns requires further study.
The incidence of breast or bladder tumor events was low for canagliflozin and occurred at a similar rate across treatment groups (breast cancer 0.41% vs. 0.39% and bladder cancer 0.07% vs. 0.11% for canagliflozin vs. non-canagliflozin groups, respectively) [106]. No data on cancer cases from other SGLT2 inhibitor studies are currently available.
This overview was limited to major data from late phase and/or large trials of those compounds that are the most advanced along the drug development pathway. Due to the emerging nature of this field, full text journal publications are limited for many of these agents.
Conclusions
SGLT2 inhibitors represent a therapeutic approach in the treatment of T2DM that is independent of insulin secretion and activity. Clinical trials have supported the efficacy of SGLT2 inhibitors as add-on therapy with metformin, sulfonylureas, thiazolidinediones, dipeptidyl peptidase-4 (DPP-4) inhibitors, or insulin. SGLT2 inhibitors may also have a role as monotherapy in patients who are intolerant to metformin (e.g., due to gastrointestinal side effects), as well as to potentially facilitate the use of triple combinations of oral anti-hyperglycemic agents (e.g., metformin + DPP-4 inhibitor + SGLT2 inhibitor).
SGLT2 inhibitors improve glycemic control in T2DM, reducing HbA1c and FPG levels, and are somewhat effective in reducing body weight and blood pressure, which are also CVD treatment targets for many patients with T2DM. SGLT2 inhibitors are generally well tolerated with few serious adverse events reported to date. When evaluated versus comparator groups, the hypoglycemic episodes associated with SGLT2 inhibitors were mostly mild in severity and not statistically significant. Among the more common adverse events of these agents is an increased risk of genital infections, which appears to be more common in women.
Some data regarding SGLT2 inhibitors are lacking. For example, little data exist on the use of SGLT2 inhibitors in debilitated older patients (especially those with central nervous system dysfunction, decreased cognition, and/or impaired thirst mechanisms) who may be at risk of volume depletion, hypotension, and electrolyte disturbances. Additional studies of interest would also include patients with varying degrees of renal impairment, given that the action of these drugs depends upon mechanisms involving glomerular filtration rate and renal function, and data from several such studies have been published or presented at congresses [72, 81, 107].
References
Bays HE. Adiposopathy, diabetes mellitus, and primary prevention of atherosclerotic coronary artery disease: treating “sick fat” through improving fat function with antidiabetes therapies. Am J Cardiol. 2012;110:4B–12B.
Del Prato S. Role of glucotoxicity and lipotoxicity in the pathophysiology of Type 2 diabetes mellitus and emerging treatment strategies. Diabet Med. 2009;26:1185–92.
Kaiser N, Leibowitz G, Nesher R. Glucotoxicity and beta-cell failure in type 2 diabetes mellitus. J Pediatr Endocrinol Metab. 2003;16:5–22.
Tanaka J, Qiang L, Banks AS, et al. Foxo1 links hyperglycemia to LDL oxidation and endothelial nitric oxide synthase dysfunction in vascular endothelial cells. Diabetes. 2009;58:2344–54.
Zheng Z, Chen H, Li J, et al. Sirtuin 1-mediated cellular metabolic memory of high glucose via the LKB1/AMPK/ROS pathway and therapeutic effects of metformin. Diabetes. 2012;61:217–28.
El-Osta A, Brasacchio D, Yao D, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205:2409–17.
Villeneuve LM, Reddy MA, Lanting LL, Wang M, Meng L, Natarajan R. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc Natl Acad Sci USA. 2008;105:9047–52.
Verges B. Abnormal hepatic apolipoprotein B metabolism in type 2 diabetes. Atherosclerosis. 2010;211:353–60.
Caron S, Verrijken A, Mertens I, et al. Transcriptional activation of apolipoprotein CIII expression by glucose may contribute to diabetic dyslipidemia. Arterioscler Thromb Vasc Biol. 2011;31:513–9.
Lee SJ, Campos H, Moye LA, Sacks FM. LDL containing apolipoprotein CIII is an independent risk factor for coronary events in diabetic patients. Arterioscler Thromb Vasc Biol. 2003;23:853–8.
Bays HE, Toth PP, Kris-Etherton PM, et al. Obesity, adiposity, and dyslipidemia: a consensus statement from the National Lipid Association. J Clin Lipidol. 2013;7:304–83.
Reichard P, Nilsson BY, Rosenqvist U. The effect of long-term intensified insulin treatment on the development of microvascular complications of diabetes mellitus. N Engl J Med. 1993;329:304–9.
Diabetes Control and Complications Trial Research (DCCT) group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–86.
Ohkubo Y, Kishikawa H, Araki E, et al. Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes Res Clin Pract. 1995;28:103–17.
UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:837–53.
UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:854–65.
Stratton IM, Adler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000;321:405–12.
Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560–72.
Perkovic V, Heerspink HL, Chalmers J, et al. Intensive glucose control improves kidney outcomes in patients with type 2 diabetes. Kidney Int. 2013;83:517–23.
Turnbull FM, Abraira C, Anderson RJ, et al. Intensive glucose control and macrovascular outcomes in type 2 diabetes. Diabetologia. 2009;52:2288–98.
Boussageon R, Bejan-Angoulvant T, Saadatian-Elahi M, et al. Effect of intensive glucose lowering treatment on all cause mortality, cardiovascular death, and microvascular events in type 2 diabetes: meta-analysis of randomised controlled trials. BMJ. 2011;343:d4169.
Hemmingsen B, Lund SS, Gluud C, et al. Intensive glycaemic control for patients with type 2 diabetes: systematic review with meta-analysis and trial sequential analysis of randomised clinical trials. BMJ. 2011;343:d6898.
Avitabile NA, Banka A, Fonseca VA. Glucose control and cardiovascular outcomes in individuals with diabetes mellitus: lessons learned from the megatrials. Heart Fail Clin. 2012;8:513–22.
Ray KK, Seshasai SR, Wijesuriya S, et al. Effect of intensive control of glucose on cardiovascular outcomes and death in patients with diabetes mellitus: a meta-analysis of randomised controlled trials. Lancet. 2009;373:1765–72.
American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care. 2013;36(Suppl. 1):S11–66.
Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35:1364–79.
International Diabetes Federation (IDF). Global guidelines for type 2 diabetes. 2012. Available at: https://www.idf.org/guidelines. Accessed 28 February 2013.
Fox KM, Gerber RA, Bolinder B, Chen J, Kumar S. Prevalence of inadequate glycemic control among patients with type 2 diabetes in the United Kingdom general practice research database: a series of retrospective analyses of data from 1998 through 2002. Clin Ther. 2006;28:388–95.
Spann SJ, Nutting PA, Galliher JM, et al. Management of type 2 diabetes in the primary care setting: a practice-based research network study. Ann Fam Med. 2006;4:23–31.
Ali MK, Bullard KM, Imperatore G, Barker L, Gregg EW; Centers for Disease Control and Prevention (CDC). Characteristics associated with poor glycemic control among adults with self-reported diagnosed diabetes—National Health and Nutrition Examination Survey, United States, 2007–2010. Morb Mortal Wkly Rep. 2012;61(Suppl.):32–7.
Juarez DT, Sentell T, Tokumaru S, Goo R, Davis JW, Mau MM. Factors associated with poor glycemic control or wide glycemic variability among diabetes patients in Hawaii, 2006–2009. Prev Chronic Dis. 2012;9:120065.
Stark Casagrande S, Fradkin JE, Saydah SH, Rust KF, Cowie CC. The prevalence of meeting A1C, blood pressure, and LDL goals among people with diabetes, 1988–2010. Diabetes Care. 2013;36:2271–9.
Hoerger TJ, Segel JE, Gregg EW, Saaddine JB. Is glycemic control improving in US adults? Diabetes Care. 2008;31:81–6.
Wallace TM, Matthews DR. Poor glycaemic control in type 2 diabetes: a conspiracy of disease, suboptimal therapy and attitude. QJM. 2000;93:369–74.
Grant R, Adams AS, Trinacty CM, et al. Relationship between patient medication adherence and subsequent clinical inertia in type 2 diabetes glycemic management. Diabetes Care. 2007;30:807–12.
Khan H, Lasker SS, Chowdhury TA. Exploring reasons for very poor glycaemic control in patients with Type 2 diabetes. Prim Care Diabetes. 2011;5:251–5.
Bays H. From victim to ally: the kidney as an emerging target for the treatment of diabetes mellitus. Curr Med Res Opin. 2009;25:671–81.
Gerich JE. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabet Med. 2010;27:136–42.
Hediger MA, Rhoads DB. Molecular physiology of Sodium–glucose cotransporters. Physiol Rev. 1994;74:993–1026.
Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev. 2011;91:733–94.
Thorens B, Mueckler M. Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab. 2010;298:E141–5.
Zelikovic I. Aminoaciduria and glycosuria. In: Avner ED, Harmon WE, Niaudet P, editors. Pediatric Nephrology. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2004. p. 701–28.
Moe OW, Wright SH, Palacín M. Renal handling of organic solutes. In: Brenner BM, editor. Brenner and Rector’s The Kidney. Philadelphia: Saunders Elsevier; 2008. p. 214–47.
Silverman M, Turner JR. Glucose transport in the renal tubule. In: Windhager EE, editor. Handbook of Physiology. New York: Oxford University Press; 1992. pp. 2017–38.
Mogensen CE. Maximum tubular reabsorption capacity for glucose and renal hemodynamics during rapid hypertonic glucose infusion in normal and diabetic subjects. Scand J Clin Lab Invest. 1971;28:101–9.
Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes. 2005;54:3427–34.
Vestri S, Okamoto MM, de Freitas HS, et al. Changes in sodium or glucose filtration rate modulate expression of glucose transporters in renal proximal tubular cells of rat. J Membr Biol. 2001;182:105–12.
DeFronzo RA, Davidson JA, Del Prato S. The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab. 2012;14:5–14.
Rossetti L, Giaccari A, DeFronzo RA. Glucose toxicity. Diabetes Care. 1990;13:610–30.
Santer R, Kinner M, Lassen CL, et al. Molecular analysis of the SGLT2 gene in patients with renal glucosuria. J Am Soc Nephrol. 2003;14:2873–82.
Ehrenkranz JR, Lewis NG, Kahn CR, Roth J. Phlorizin: a review. Diabetes Metab Res Rev. 2005;21:31–8.
White JR Jr. Apple trees to sodium glucose co-transporter inhibitors: a review of SGLT2 inhibition. Clin Diabetes. 2010;28:5–10.
Alvarado F, Crane RK. Phlorizin as a competitive inhibitor of the active transport of sugars by hamster small intestine, in vitro. Biochim Biophys Acta. 1962;56:170–2.
Rossetti L, Smith D, Shulman GI, Papachristou D, DeFronzo RA. Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. J Clin Invest. 1987;79:1510–5.
Rossetti L, Shulman GI, Zawalich W, DeFronzo RA. Effect of chronic hyperglycemia on in vivo insulin secretion in partially pancreatectomized rats. J Clin Invest. 1987;80:1037–44.
Oku A, Ueta K, Arakawa K, et al. T-1095, an inhibitor of renal Na+-glucose cotransporters, may provide a novel approach to treating diabetes. Diabetes. 1999;48:1794–800.
Hardman TC, Dubrey SW. Development and potential role of type-2 Sodium–glucose transporter inhibitors for management of type 2 diabetes. Diabetes Ther. 2011;2:133–45.
Ferrannini E, Ramos SJ, Salsali A, Tang W, List JF. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care. 2010;33:2217–24.
Henry RR, Murray AV, Marmolejo MH, Hennicken D, Ptaszynska A, List JF. Dapagliflozin, metformin XR, or both: initial pharmacotherapy for type 2 diabetes, a randomised controlled trial. Int J Clin Pract. 2012;66:446–56.
Bailey CJ, Iqbal N, T’Joen C, List JF. Dapagliflozin monotherapy in drug-naive patients with diabetes: a randomized-controlled trial of low-dose range. Diabetes Obes Metab. 2012;14:951–9.
Bailey CJ, Gross JL, Pieters A, Bastien A, List JF. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:2223–33.
Bolinder J, Ljunggren O, Kullberg J, et al. Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J Clin Endocrinol Metab. 2012;97:1020–31.
Nauck MA, Del Prato S, Meier JJ, et al. Dapagliflozin versus glipizide as add-on therapy in patients with type 2 diabetes who have inadequate glycemic control with metformin: a randomized, 52-week, double-blind, active-controlled noninferiority trial. Diabetes Care. 2011;34:2015–22.
Strojek K, Yoon KH, Hruba V, Elze M, Langkilde AM, Parikh S. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with glimepiride: a randomized, 24-week, double-blind, placebo-controlled trial. Diabetes Obes Metab. 2011;13:928–38.
Rosenstock J, Vico M, Wei L, Salsali A, List JF. Effects of dapagliflozin, an SGLT2 inhibitor, on HbA(1c), body weight, and hypoglycemia risk in patients with type 2 diabetes inadequately controlled on pioglitazone monotherapy. Diabetes Care. 2012;35:1473–8.
Wilding JP, Norwood P, T’Joen C, Bastien A, List JF, Fiedorek FT. A study of dapagliflozin in patients with type 2 diabetes receiving high doses of insulin plus insulin sensitizers: applicability of a novel insulin-independent treatment. Diabetes Care. 2009;32:1656–62.
Wilding JP, Woo V, Soler NG, et al. Long-term efficacy of dapagliflozin in patients with type 2 diabetes mellitus receiving high doses of insulin: a randomized trial. Ann Intern Med. 2012;156:405–15.
Wilding JP, Woo V, Rohwedder K, Sugg J, Parikh S. Dapagliflozin in patients with type 2 diabetes receiving high doses of insulin: efficacy and safety over 2 years. Diabetes Obes Metab. 2013. doi:10.1111/dom.12187.
List JF, Woo V, Morales E, Tang W, Fiedorek FT. Sodium–glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care. 2009;32:650–7.
Ptaszynska A, Johnsson KM, Apanovitch A-M, Sugg J, Parikh S, List J. Safety of dapagliflozin in clinical trials for T2DM. Diabetes. 2012;61(Suppl.):A258 (Abstract 1011-P).
Bristol-Myers Squibb and AstraZeneca EEIG. Assessment report: Forxiga (dapagliflozin). European Medicines Agency (18 September 2012). EMA/689976/2012. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002322/WC500136024.pdf. Accessed 17 July 2013.
Ptaszynska A, Chalamandaris AG, Sugg JE, Johnsson KM, Parikh S, List JL. Effect of dapagliflozin on renal function Diabetes. 2012;61(Suppl.):A283 (Abstract 1098-P).
Bristol Myers Squibb and AstraZeneca EEIG. Summary of Product Characteristics: Forxiga 5 mg & 10 mg film coated tablets. Electronic Medicines Compendium 2012. Available at: http://www.medicines.org.uk/emc/medicine/27188/SPC/Forxiga+5 mg++%26+10+mg+film+coated+tablets/. Accessed 17 July 2013.
Johnsson KM, Ptaszynska A, Schmitz B, Sugg J, Parikh SJ, List JF. Vulvovaginitis and balanitis in patients with diabetes treated with dapagliflozin. J Diabetes Complicat. 2013;27:479–84.
Johnsson KM, Ptaszynska A, Schmitz B, Sugg J, Parikh SJ, List JF. Urinary tract infections in patients with diabetes treated with dapagliflozin. J Diabetes Complicat. 2013;27:473–8.
Stenlof K, Cefalu WT, Kim KA, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab. 2013;15:372–82.
Rosenstock J, Aggarwal N, Polidori D, et al. Dose-ranging effects of canagliflozin, a sodium–glucose cotransporter 2 inhibitor, as add-on to metformin in subjects with type 2 diabetes. Diabetes Care. 2012;35:1232–8.
Wilding J, Mathieu C, Deng L, et al. Canagliflozin, a sodium glucose co-transporter 2 inhibitor, improves glycaemia in subjects with type diabetes inadequatley controlled with metformin plus sulphonylurea. Diabetologia. 2012;55(Suppl. 1):S315 (Abstract 766).
Cefalu WT, Leiter LA, Yoon KH, et al. Efficacy and safety of canagliflozin versus glimepiride in patients with type 2 diabetes inadequately controlled with metformin (CANTATA-SU): 52 week results from a randomised, double-blind, phase 3 non-inferiority trial. Lancet. 2013;382:941–50.
Schernthaner G, Gross JL, Rosenstock J, et al. Canagliflozin compared with sitagliptin for patients with type 2 diabetes who do not have adequate glycemic control with metformin plus sulfonylurea: a 52-week randomized trial. Diabetes Care. 2013;36:2508–15.
Yale JF, Bakris G, Cariou B, et al. Efficacy and safety of canagliflozin in subjects with type 2 diabetes and chronic kidney disease. Diabetes Obes Metab. 2013;15:463–73.
Bode B, Stenlöf K, Sullivan D, Fung A, Usiskin K. Efficacy and safety of canagliflozin treatment in older subjects with type 2 diabetes mellitus: a randomized trial. Hosp Pract (1995). 2013;41(2):72–84.
Forst T, Guthrie R, Goldenberg R, et al. Efficacy and safety of canagliflozin (CANA) in subjects with type 2 diabetes mellitus (T2DM) on metformin (MET) and pioglitazone (PIO) over 52 weeks. Diabetes. 2013;62(Suppl. 1):A284 (Abstract 1098-P).
Devineni D, Morrow L, Hompesch M, et al. Canagliflozin improves glycaemic control over 28 days in subjects with type 2 diabetes not optimally controlled on insulin. Diabetes Obes Metab. 2012;14:539–45.
Matthews DR, Fulcher G, Perkovic V, et al. Efficacy and safety of canagliflozin (CANA), an inhibitor of sodium glucose co-transporter 2 (SGLT2), added-on to insulin therapy ± oral agents in type 2 diabetes. Diabetologia. 2012;55(Suppl. 1):S314 (Abstract 764).
Weir MR, Januszewicz A, Gilbert RE, Lavalle Gonzalez FJ, Meininger G. Lower blood pressure (BP) with canagliflozin (CANA) in subjects with type 2 diabetes mellitus (T2DM). Diabetes. 2013;62(Suppl. 1):A278 (Abstract 1077-P).
Janssen Pharmaceuticals Inc. INVOKANA™ (canagliflozin) tablets, for oral use. Prescribing information (issued March 2013). 2013. Available at: http://www.invokanahcp.com/prescribing-information.pdf. Accessed 17 July 2013.
Nyirjesy P, Sobel J, Fung A, Gassmann-Meyer C, Ways K, Usiskin K. Genital mycotic infections with canagliflozin (CANA) in subjects with type 2 diabetes mellitus (T2DM). Diabetes. 2013;62(Suppl. 1):A276 (Abstract 1069-P).
Nicolle LE, Capuano G, Fung A, Usiskin K. Urinary tract infection (UTI) with canagliflozin (CANA) in subjects with type 2 diabetes mellitus (T2DM). Diabetes. 2013;62(Suppl. 1):A296 (Abstract 1139-P).
Ferrannini E, Seman L, Seewaldt-Becker E, Hantel S, Pinnetti S, Woerle H. A phase IIb, randomised, placebo-controlled study of the SGLT2 inhibitor empagliflozin in patients with type 2 diabetes. Diabetes Obes Metab. 2013;15:721–8.
Roden M, Weng J, Eilbracht J, et al. Empagliflozin monotherapy with sitagliptin as an active comparator in patients with type 2 diabetes: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2013. doi:10.1016/S2213-8587(13)70084-6.
Rosenstock J, Seman LJ, Jelaska A, et al. Efficacy and safety of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, as add-on to metformin in type 2 diabetes with mild hyperglycaemia. Diabetes Obes Metab. 2013. doi: 10.1111/dom.12185.
Woerle HJ, Ferrannini E, Berk A, Manun’ebo M, Pinnetti S, Broedl UC. Safety and efficacy of empagliflozin as monotherapy or add-on to metformin in a 78-week open-label extension study in patients with type 2 diabetes. Diabetes. 2012;61(Suppl.):LB13 (Abstract 49-LB).
Häring HU, Merker L, Seewaldt-Becker E, et al. Empagliflozin as add-on to metformin plus sulfonylurea in patients with type 2 diabetes: a 24-week randomized, double-blind, placebo-controlled trial. Diabetes Care. 2013. doi: 10.2337/dc12-2673.
Häring HU, Merker L, Seewaldt-Becker E, Weimer M, Meinicke T. Empagliflozin as add-on to metformin for 24 weeks improves glycemic control in patients with type 2 diabetes (T2DM). Diabetes. 2013;62(Suppl. 1):A282 (Abstract 1092-P).
Kovacs CS, Seshiah V, Swallow R, et al. Empagliflozin improves glycaemic and weight control as add-on therapy to pioglitazone or pioglitazone plus metformin in patients with type 2 diabetes: a 24-week, randomized, placebo-controlled trial. Diabetes Obes Metab. 2013. doi:10.1111/dom.12188.
Rosenstock J, Jelaska A, Kim G, Broedl UC, Woerle HJ. Empagliflozin as add-on to basal insulin for 78 weeks improves glycemic control with weight loss in insulin-treated type 2 diabetes (T2DM). Diabetes. 2013;62(Suppl. 1):A285 (Abstract 1102-P).
Kim G, Gerich JE, Salsali A, et al. Empagliflozin (EMPA) increases genital infections but not urinary tract infections (UTIs) in pooled data from four pivotal phase III trials. Diabetes. 2013;62(Suppl. 1):LB21 (Abstract 74-LB).
Schwartz SL, Akinlade B, Klasen S, Kowalski D, Zhang W, Wilpshaar W. Safety, pharmacokinetic, and pharmacodynamic profiles of ipragliflozin (ASP1941), a novel and selective inhibitor of sodium-dependent glucose co-transporter 2, in patients with type 2 diabetes mellitus. Diabetes Technol Ther. 2011;13:1219–27.
Kashiwagi A, Takinami Y, Kazuta K, Yoshida S, Utsuno A, Nagase I. Ipragliflozin improved glycaemic control with additional benefits of reductions of body weight and blood pressure in Japanese patients with type 2 diabetes mellitus: BRIGHTEN Study. Diabetologia. 2011;54(Suppl. 1):S68 (Abstract 149).
Wilding JP, Ferrannini E, Fonseca VA, Wilpshaar W, Dhanjal P, Houzer A. Efficacy and safety of ipragliflozin in patients with type 2 diabetes inadequately controlled on metformin: a dose-finding study. Diabetes Obes Metab. 2013;15:403–9.
Kashiwagi A, Shiga T, Akiyama N, et al. Ipragliflozin reduced HbA1c and body weight in Japanese type 2 diabetes patients who have inadequate glycaemic control on sulfonylurea or pioglitazone alone. Diabetologia. 2012;55(Suppl. 1):S302 (Abstract 739).
Basile JN. The potential of sodium glucose cotransporter 2 (SGLT2) inhibitors to reduce cardiovascular risk in patients with type 2 diabetes (T2DM). J Diabetes Complicat. 2013;27:280–6.
Foote C, Perkovic V, Neal B. Effects of SGLT2 inhibitors on cardiovascular outcomes. Diab Vasc Dis Res. 2012;9:117–23.
Bristol Myers Squibb-AstraZeneca. NDA 202293, Dapaglifozin tablets, 5 and 10 mg. FDA Briefing Document (2011, July 19), Endocrinologic and Metabolic Drugs Advisory Committee. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/ucm262994.pdf. Accessed 17 July 2013.
Janssen Research & Development LLC. Canagliflozin as an adjunctive treatment to diet and exercise alone or co-administered with other antihyperglycemic agents to improve glycemic control in adults with type 2 diabetes mellitus. JNJ-28431754 (Canagliflozin) NDA 204042. FDA Briefing Document (2013, January 10), Endocrinologic and Metabolic Drugs Advisory Committee. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM334551.pdf. Accessed 7 August 2013.
Barnett AH, Mithal A, Manassie J, Rattunde H, Woerle HJ, Broedl UC. Empagliflozin in patients with type 2 diabetes mellitus and renal impairment. Diabetes. 2013;62(Suppl. 1):A286 (Abstract 1104-P).
Lavalle Gonzalez FJ, Januszewicz A, Davidson JA, Qiu R, Tong C, Meininger G. Canagliflozin (CANA) compared with sitagliptin (SITA) in subjects with type 2 diabetes mellitus (T2DM) on Metformin (MET) over 52 weeks. Diabetes. 2013;62(Suppl. 1):A61 (Abstract 238-OR).
Acknowledgments
Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Debra Brocksmith, MB ChB, PhD, of Envision Scientific Solutions during the preparation of this manuscript. Boehringer Ingelheim was given the opportunity to check the data used in the manuscript for factual accuracy only. Boehringer Ingelheim funded the article processing charges for this review. Dr H Bays is the guarantor for this article, and takes responsibility for the integrity of the work as a whole.
Conflict of interest
HB has served as a consultant and/or speaker to Amarin, Amgen, Catabasis, Daiichi Sankyo, Eisai Merck, VIVUS, and WPU, and his research site has received research grants from Amarin, Amgen, Ardea, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, California Raisin Board, Catabasis, Eisai, Elcelyx, Eli Lilly, Esperion, Essentialis, Forest, Gilead, Given, GlaxoSmithKline, High Point Pharmaceuticals, LLC, Hoffman LaRoche, Home Access, Merck, Micropharma Limited, Necktar, Novartis, Novo Nordisk, Omthera, Orexigen, Pfizer, Regeneron, Stratum Nutrition, Takeda, TIMI, Transtech Pharma, Trygg, TWI Bio, VIVUS, WPU, and Xoma.
Compliance with ethical guidelines
This article does not contain any studies with human or animal subjects that were performed by the author.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Bays, H. Sodium Glucose Co-transporter Type 2 (SGLT2) Inhibitors: Targeting the Kidney to Improve Glycemic Control in Diabetes Mellitus. Diabetes Ther 4, 195–220 (2013). https://doi.org/10.1007/s13300-013-0042-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-013-0042-y