
Abstract
Introduction
This study examined the safety, tolerability, pharmacokinetics, and pharmacodynamics of empagliflozin, a potent and highly selective sodium glucose cotransporter 2 (SGLT2) inhibitor, in patients with type 2 diabetes mellitus (T2DM).
Methods
A total of 48 patients with T2DM were randomized to receive one of four doses of empagliflozin (2.5, 10, 25, or 100 mg qd) or placebo over 8 days. In every dose group, nine patients received active drug and three received placebo. The primary endpoint was safety and tolerability. Pharmacokinetic and pharmacodynamic parameters were measured as secondary endpoints.
Results
Empagliflozin was rapidly absorbed, reaching peak levels 1.5–3.0 h after dosing and showed a biphasic decline. The mean terminal elimination half-life ranged from 10 to 19 h. Increases in exposure (area under the plasma concentration–time curve [AUC] and maximum concentration of analyte in plasma [C max]) were approximately proportional with dose. Empagliflozin increased the rate and total amount of glucose excreted in urine compared to placebo. After administration of a single dose of empagliflozin, cumulative amounts of glucose excreted in urine over 24 h ranged from 46.3 to 89.8 g, compared with 5.84 g with placebo. Similar results were seen after multiple doses. Fasting plasma glucose levels decreased by 17.2–25.8% with empagliflozin and by 12.7% with placebo. The frequency of adverse events was 33.3–66.7% with empagliflozin and 41.7% with placebo. There were no changes in urine volume or micturition frequency under the controlled study conditions.
Conclusion
Overall, pharmacokinetic assessments demonstrated a dose-proportional increase in drug exposure and support once-daily dosing. Elevated urinary glucose excretion was observed with all doses. Multiple once-daily oral doses of empagliflozin (2.5–100 mg) reduced plasma glucose and were well tolerated in patients with T2DM. EudraCT (2007-000654-32).
Similar content being viewed by others

Introduction
Traditional approaches to the treatment of type 2 diabetes mellitus (T2DM) have focused on improving insulin secretion and insulin sensitivity with the aim of lowering blood glucose levels to meet glycosylated hemoglobin (HbA1c) targets while avoiding hypoglycemia [1, 2]. However, evidence suggests that only around half of patients achieve an HbA1c target of less than 7% [3]. Recent studies report limited benefits of intensive glucose lowering on mortality and cardiovascular outcomes in patients with T2DM [4–8]. Therefore, future management strategies should address features beyond glucose control [9, 10].
Sodium glucose cotransporter 2 (SGLT2) inhibitors are a promising new class of compounds in development that offer an alternative mechanism for control of hyperglycemia in T2DM by reducing glucose reabsorption in the kidney and thereby increasing urinary glucose excretion (UGE) [11–13]. Clinical studies of SGLT2 inhibitors have demonstrated glucose lowering efficacy, a favorable safety profile, and beneficial effects on weight, blood pressure, and other metabolic parameters [14–19].
Empagliflozin is an SGLT2 inhibitor in development as a treatment for T2DM. In in vitro studies, empagliflozin exhibited highly potent inhibition of SGLT2 (half maximal inhibitory concentration [IC50] of 3.1 nM [pIC50 (SE) 8.5 (0.02) nM]), with a >2,500-fold selectivity for SGLT2 over SGLT1 [20]. Single oral doses of empagliflozin were well tolerated in healthy volunteers resulting in dose-dependent UGE without inducing hypoglycemia [21]. The objective of this multiple ascending dose study, the first study of empagliflozin in patients with T2DM, was to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple rising doses of empagliflozin in this patient population.
Study Design
This was a multiple rising dose, randomized, double-blind trial with within-group placebo control. Patients were randomized upon admission to the trial center to one of four doses of empagliflozin (2.5, 10, 25, and 100 mg), which were tested sequentially in ascending order of dose. Within every dose group, patients were randomized 3:1 to receive active drug or placebo. Treatment allocation was carried out according to a randomized list of patient and medication numbers. Within each dose group, patients were randomized to receive placebo or active drug and patients and investigators were double blinded until the study had completed. Patients and investigators were aware of the dosing stage (but not if patients were receiving placebo).
Patients received a single dose on day 1 and once-daily dosing on days 3–9. The dosing on day 2 was skipped to allow estimation of the terminal elimination half-life after a single dose to compare with the steady state half-life. Study drug was administered at the same time every day with 240 mL of water. Patients were fasted overnight for 10 h on days −2, −1, 1, 8, and 9. An end-of-study examination was carried out in the 1-week post-treatment period (days 15–21). An oral glucose tolerance test (OGTT; administration of 75 g glucose solution after overnight fasting) was performed on days −1, 1, and 9.
Subjects
The trial was conducted at the Profil Institute for Metabolic Research, Neuss, Germany. Eligible subjects were patients with T2DM treated with diet and exercise only or ≤2 oral anti-diabetic agents with at least one agent taken at ≤50% of its maximum dose; HbA1c ≤8.5% at screening; and body mass index (BMI) 18.5–40 kg/m2. Patients on insulin or thiazolidinediones were excluded, as were patients with high fasting [>240 mg/dL (13.3 mmol/L)] or postprandial [>400 mg/dL (>22 mmol/L)] blood glucose and patients with clinically significant concomitant diseases or abnormalities in the screening laboratory.
Evaluations
The primary endpoint was safety and tolerability. Safety parameters, including adverse events (AEs), electrocardiogram (ECG), vital signs, physical examination, and laboratory parameters in blood and urine samples, were measured throughout the study. Meals and fluid intake were standardized during the in-house period of the study (from day −2 to day 9).
Secondary endpoints were pharmacokinetic and pharmacodynamic parameters. Pharmacokinetic endpoints included mean plasma concentration time profiles of empagliflozin, maximum concentration of analyte in plasma (C max), time to reach peak levels (t max), terminal elimination half-life (t 1/2), apparent terminal rate constant (λz), area under the plasma concentration–time curve (AUC), renal clearance (CLR) and the fraction of the dose that was excreted unchanged in urine (fe). Pharmacodynamic measurements included UGE, 8-point weighted mean daily glucose (MDG), and fasting plasma glucose (FPG). Samples for the determination of analyte plasma concentrations were drawn before drug administration on days −2, −1, 1, 2, 3, and 5–9, at time points up to 48 h after dosing on day 1, and up to 72 h after dosing on day 9. On days −2, −1, 1, 8, and 9 all urine voided over a period of 24 h (48 h for day 1 and 72 h for day 9) was collected for UGE measurements. Urine samples collected on days 1 and 9 were also used for pharmacokinetic measurements.
Empagliflozin concentrations in plasma and urine were analyzed using a validated high performance liquid chromatography, tandem mass spectrometry (HPLC–MS/MS) assay with a lower limit of quantification of 1.11 nmol/L (0.5 ng/mL) for plasma, or 4.44 nmol/L (2 ng/mL) for urine. Pharmacokinetic parameters were calculated using WinNonlin™ software (v5.2, Pharsight Corporation, Mountain View, CA, USA). Mean plasma concentration–time profiles of empagliflozin were calculated only if at least 2/3 of the individual patient concentrations at each time point were above the limit of quantification (1.1 nM empagliflozin). C max and t max values were directly determined from the plasma concentration–time profiles of each subject. The t 1/2 was calculated as the quotient of the natural log (ln)(2) and λz. The λz was estimated from a regression of ln(C) versus time over the terminal log-linear drug disposition portion of the concentration–time profiles. AUC to the last quantifiable time point was calculated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations. CLR was determined as the quotient of the amount of drug excreted unchanged in urine over AUC. The fe was determined by the quotient of the sum of drug excreted over all dosing intervals and the dose administered.
Mean cumulative UGE over 24 h (Ae0–24) was measured after single doses of empagliflozin with OGTT (day 1) and after multiple doses after fasting (day 8) or with OGTT (day 9). Weighted MDG was estimated by dividing the area under the 24-h glucose curve (AUEC0–24) by 24 h. Inhibition of glucose reabsorption was calculated by comparing renal tubular glucose reabsorption rate (TG) on day 8 with day −2 as baseline. TG was calculated as the difference between filtered load (FL) and glucose excretion rate (ER). The FL was calculated as the product of estimated glomerular filtration rate (eGFR) and plasma MDG. ER was estimated by dividing the cumulative amount of glucose excreted in urine (UGE) over a dosing interval by 24 h. MDG measurements were performed during the OGTT on days −1, 1, and 9, and under ‘normal’ trial conditions (standardized meals without OGTT) on days −2 and 8. The impact of empagliflozin on postprandial glucose (PPG) levels was analyzed from the area under the glucose concentration–time curve AUEC 1–5 h (AUEC1–5) after OGTT.
Statistical Analysis
Safety variables, pharmacokinetic and pharmacodynamic parameters were evaluated by descriptive statistical methods. Safety and pharmacodynamic analyses were based on the randomized set, which was identical to the treated set and included all 48 patients treated with active drug or placebo. Pharmacokinetic analyses were based on the pharmacokinetic set, which included all 36 patients treated with active drug. Attainment of steady state using trough concentrations between days 5 and 9 was analyzed using a repeated measures linear model on the logarithmic scale including ‘patient’ as a random effect and ‘time’ as a repeated effect. Dose proportionality was explored using a regression model applied to log-transformed data. UGE, weighted MDG, and FPG were analyzed daily for change from baseline using an analysis of covariance (ANCOVA) model. The effect ‘patient’ was considered random while the effect ‘treatment’ was considered fixed. The baseline value was included as a continuous covariate. Statistical analysis utilized SAS® statistics software version 8.2 (SAS Institute Inc. Cary, NC, USA).
Results
Patient Disposition
Seventy-four patients were screened and 48 (39 white males, 8 white females, 1 black female) were randomized (12 to each dose group, with 9 patients randomized to receive empagliflozin treatment and 3 to placebo). All randomized patients received at least 1 dose of study drug; 46 patients received 8 doses of empagliflozin or placebo over 8 days, 1 patient received 6 doses of placebo over 6 days, and 1 patient received 2 doses of empagliflozin over 2 days. Forty-six patients completed the study, with 2 patients discontinuing treatment prematurely due to adverse events. Baseline characteristics are summarized in Table 1.
Safety and Tolerability
In total, 24 (50.0%) patients experienced AEs during the treatment period. No AEs were reported in the 1-week post-treatment period. The frequency of AEs was 52.8% in patients receiving empagliflozin treatment and 41.7% with placebo. The most frequently reported AE was headache, reported by 4 patients (8.3%) during the treatment period, occurring in 0–2 patients per group (Table 2). There were no fatalities or serious AEs. A total of 16 patients (33.3%) experienced AEs that the investigator considered related to study medication, but there were no treatment-related differences in their frequency. The most common investigator-defined treatment-related AEs were headache, diarrhea, and hypoglycemia. Three patients in the empagliflozin treatment group experienced hypoglycemic events, defined as blood glucose levels <3.5 mmol/L (<63 mg/dL), but all of them occurred within 5 h of OGTT administration. These events were considered by the investigator to be moderate in intensity, but all patients recovered following therapy. Two patients experienced AEs that led to discontinuation of study treatment [1 patient in the placebo group had an increase in hepatic enzymes (maximum alanine transaminase level of 335 U/L) considered to be of severe intensity and 1 patient treated with empagliflozin experienced cellulitis (phlegmon of the lower left arm) considered to be of moderate intensity, which the investigator did not believe to be related to the study medication]. No urinary tract or genital tract infections were reported. There were no findings of clinical significance in the laboratory clinical evaluations (including electrolytes and lipid parameters; Table 3), 12-lead ECG, vital signs, weight, or waist circumference. No major differences in total urine volume were observed between the empagliflozin and placebo groups. On day 8, the mean volume of urine collected ranged from 2.36 to 4.08 L following empagliflozin dosing, compared with a mean of 3.23 L for the placebo group. Similarly, there were no changes to micturition frequency after empagliflozin administration compared with placebo under the controlled fluid intake conditions of the trial (Table 4). Creatinine clearance was similar in all dose groups except for the 2.5 mg empagliflozin dose group at 166 h (Table 5). In this group, two patients had high derived creatinine clearance values in urine fractions collected over a 2-h period pre-dose on day 8 (1,392 mL/min × 1.73 m2 and 1,124 mL/min × 1.73 m2 change from baseline), based on urinary creatinine concentrations of 251 mg/dL in a volume of 670 mL and 84.1 mg/dL in a volume of 2,115 mL of urine, respectively, and plasma creatinine concentrations of 0.68 mg/dL and 0.94 mg/dL, respectively. This resulted in a mean (SD) change from baseline in derived creatinine clearance of 360 (620) mL/min × 1.73 m2 in the 2.5 mg empagliflozin group at 166 h, compared with −12.1 (54.6) mL/min × 1.73 m2 for the placebo group (Table 5).
Pharmacokinetics
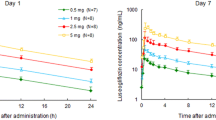
Empagliflozin was rapidly absorbed after oral administration, reaching peak levels between 1.5 and 3 h after a single dose, thereafter, plasma levels declined in a biphasic fashion with a rapid distribution phase and slower elimination phase (Fig. 1). Both AUC and C max increased approximately proportionally with dose over the dose range from 2.5 to 100 mg (Table 6). After repeated dosing, trough concentrations were similar on days 5–8, indicating that steady state was reached by day 5. At steady state, empagliflozin concentration–time profiles showed a trend similar to single dose profiles on day 1 (Fig. 1) [note, 2 patients (1 each from the 10 mg and 100 mg dose groups on day 9) were excluded due to incomplete data or low exposure]. The mean t 1/2 after a single dose ranged from 10.8 (25 mg dose) to 13.6 h (100 mg dose) and was similar at steady state (day 9; range 10.3–18.8 h) (Table 6). Consistent with the drug’s half-life, up to 23% accumulation of empagliflozin was observed at steady state. The fe ranged from 12.2% to 18.7% at steady state. On day 1, renal clearance of empagliflozin over 48 h (CLR, 0–48h) was 15.0–29.1 mL/min and at steady state on day 9 (CLR,τ,ss) was 23.5–34.4 mL/min (Table 6). Overall, steady state parameters were similar to single dose parameters, suggesting linear pharmacokinetics with respect to time (Table 6).

Mean plasma concentration–time profiles of empagliflozin after multiple oral doses of 2.5–100 mg (semi-log scale). Data from the pharmacokinetic analysis set (n = 36)
Pharmacodynamics
At all doses, oral administration of empagliflozin increased UGE above the level of placebo, with both the amount and rate of glucose excretion increasing with dose. After a single dose under OGTT conditions (day 1; Fig. 2), the amount of glucose excreted in the urine over 24 h (Ae0–24) was 46.3 g with 2.5 mg empagliflozin compared with 5.84 g with placebo. There was more than a twofold increase in UGE with the fourfold increase in dose from 2.5 to 10 mg, in contrast with about a 1.2-fold increase in UGE with the fourfold dose increase in dose from 25 to 100 mg. Glucose excretion seemed to plateau after the 10 mg dose, with total cumulative amounts excreted ranging from 77.9 mg to 89.8 g with 10 to 100 mg empagliflozin after a single dose. Similar results were observed after multiple doses with or without OGTT (days 9 and 8, respectively; Fig. 2). Empagliflozin inhibited reabsorption of 39%, 46%, 58%, and 64% of filtered glucose with 2.5, 10, 25, and 100 mg once-daily doses, respectively (Fig. 3). The mean decrease in FPG from baseline (day −1) to day 9 (based on least squares means) ranged from 17.2% to 25.8% with empagliflozin, compared with 12.7% with placebo. The overall decline in FPG was 4.6–13.2% greater with empagliflozin than with placebo (Fig. 4). The decrease in FPG from baseline was significantly different compared to placebo with the 10 mg dose (Fig. 4). A respective decrease in plasma glucose levels was also observed following OGTT immediately after the first dose. The mean decrease in MDG from baseline (based on least squares means) was 24.1–37.0% with empagliflozin, compared with 13.5% for placebo [comparison of day 8 (without OGTT) with baseline (day −2)] (Table 5). The decrease in MDG from baseline was significantly different compared to placebo with the 2.5 and 10 mg doses (Table 7). After OGTT, reductions in plasma glucose levels in empagliflozin groups were greater than in the placebo group. Mean AUEC1–5 values decreased approximately 22.9–27.5% from baseline with empagliflozin, compared with 14.2% with placebo (Table 8). Serum insulin levels did not change to a relevant degree (data not shown).

Mean (SD) cumulative urinary glucose excretion (UGE) over 24 h (Ae0–24) after single (day 1) and multiple (day 8 and 9) oral doses of empagliflozin or placebo. Data from the treated set (n = 48)

Mean (SD) percent inhibition of glucose reabsorption on day 8 following multiple oral administrations of empagliflozin (2.5–100 mg) once daily. Data from the treated set (n = 48)

Mean (SD) changes from baseline (day −1) in fasting plasma glucose (FPG) after treatment with multiple doses of empagliflozin or placebo (day 9). Data from the treated set (n = 48)
Discussion
This randomized, double-blind, placebo-controlled trial examined the safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple rising doses of empagliflozin (2.5, 10, 25, and 100 mg) administered to adult patients with T2DM. Analysis of safety data showed that empagliflozin was well tolerated in patients with T2DM. There were no serious AEs associated with drug treatment and the frequency of AEs with empagliflozin treatment was similar to placebo. Importantly, plasma glucose was lowered in the absence of non-OGTT-related episodes of hypoglycemia. Furthermore, there were no safety findings of concern, with no clinically significant changes observed in creatinine clearance, urine volume or micturition frequency following empagliflozin administration under the controlled fluid intake conditions of the trial, indicating that increased UGE is unlikely to affect renal function as assessed by these measures. No urogenital infections were observed in this short-term study. More long-term studies in patients with T2DM will provide more information on the safety and tolerability of empagliflozin.
Pharmacokinetic data demonstrated that oral doses of empagliflozin were rapidly absorbed, reaching steady state by day 5 with moderate accumulation. There was a rapid distribution phase and a slower elimination phase, with dose-proportional increases in exposure and a terminal elimination half-life of 10–19 h. These results are consistent with those from a single rising dose study conducted in healthy volunteers [21], which demonstrated approximately dose-proportional increases in exposure and a terminal elimination half-life of 8.6–13 h. These pharmacokinetic features demonstrate the suitability of empagliflozin for once-daily dosing in patients with T2DM.
All doses of empagliflozin had significant pharmacodynamic effects indicated by an increased rate and total amount of UGE relative to placebo. Cumulative amounts of glucose excreted in urine over 24 h after administration of a single dose of empagliflozin ranged from 46 to 90 g/day in this study, which is similar to the findings of the single rising dose study in healthy volunteers, in which cumulative amounts of glucose excreted in urine in the 24 h after administration of empagliflozin (2.5–100 mg) ranged from 30.6 to 78.6 g/day [21]. Reports of UGE following administration of the SGLT2 inhibitor dapagliflozin were 60 g/day in a study of 2.5–100 mg dapagliflozin doses administered to healthy subjects [22] and 52–85 g/day with 2.5–50 mg doses of dapagliflozin administered to patients with T2DM [23]. Canagliflozin treatment resulted in maximal UGE of 70 g/day with doses of 200–800 mg qd in healthy subjects [24]. Ipragliflozin dose dependently increased UGE up to a maximum of approximately 59 g/day following multiple doses of 5–600 mg in healthy volunteers and 90 g/day following multiple doses of 300 mg in patients with T2DM [25, 26]. However, cross-study pharmacodynamic comparisons are difficult, as the efficacy of SGLT2 inhibitors may depend on overall glycemic status [27].
In this study, empagliflozin inhibited reabsorption of renally filtered glucose by approximately 39–64% under steady state conditions. Again, these data are comparable with the results of the single rising dose study [21], reporting levels of inhibition of approximately 12–56% with single empagliflozin doses administered to healthy volunteers over the same dose range.
The increased UGE resulted in a substantial decrease in both FPG and PPG and therefore also in MDG versus placebo at all doses tested. Whereas UGE was similar after single and multiple dosing, the plasma glucose lowering effect of empagliflozin occurred immediately after the first dose and increased with treatment duration. Importantly, empagliflozin exhibited a sustained blood glucose lowering effect on day 10 (i.e., 1 day after the intake of the last dose). The FPG levels in the empagliflozin groups were at least 14% lower than on day 1, which was twice as high as the reduction observed with placebo. This supports the suitability of once-daily dosing with empagliflozin.
Conclusion
This first trial investigating the SGLT2 inhibitor empagliflozin in patients with T2DM demonstrated that multiple oral doses of empagliflozin (2.5–100 mg) increased UGE, reduced plasma glucose and were well tolerated in patients with T2DM. Empagliflozin treatment resulted in significant and clinically meaningful blood glucose lowering effects that were apparent on the first day of treatment, were enhanced over the course of this (short-term) trial and lasted beyond 24 h after intake of the last dose. The pharmacokinetic and pharmacodynamic properties of empagliflozin were consistent with once-daily dosing. These results warrant further long-term investigation of this compound as a potential treatment for T2DM.
References
Cefalu WT, Richards RJ, Melendez-Ramirez LY. Redefining treatment success in type 2 diabetes mellitus: comprehensive targeting of core defects. Cleve Clin J Med. 2009;76:S39–47.
Inzucchi SE, Bergenstal RM, Buse JB, et al. American Diabetes Association (ADA); European Association for the Study of Diabetes (EASD). Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35:1364–79.
Cheung BM, Ong KL, Cherny SS, Sham PC, Tso AW, Lam KS. Diabetes prevalence and therapeutic target achievement in the United States, 1999 to 2006. Am J Med. 2009;122:443–53.
Gerstein HC, Swedberg K, Carlsson J, CHARM Program Investigators, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–59.
Duckworth W, Abraira C, Moritz T, VADT Investigators, et al. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med. 2009;360:129–39.
Patel A, MacMahon S, Chalmers J, ADVANCE Collaborative Group, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560–72.
DeSouza C, Fonseca V. Therapeutic targets to reduce cardiovascular disease in type 2 diabetes. Nat Rev Drug Discov. 2009;8:361–7.
Boussageon R, Bejan-Angoulvant T, Saadatian-Elahi M, et al. Effect of intensive glucose lowering treatment on all cause mortality, cardiovascular death, and microvascular events in type 2 diabetes: meta-analysis of randomised controlled trials. BMJ. 2001;343:d4169.
Schernthaner G, Barnett AH, Betteridge DJ, et al. Is the ADA/EASD algorithm for the management of type 2 diabetes (January 2009) based on evidence or opinion? A critical analysis. Diabetologia. 2010;53:1258–69.
Preiss D, Ray KK. Intensive glucose lowering treatment in type 2 diabetes. BMJ. 2011;343:d4243.
Gerich JE. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabet Med. 2010;27:136–42.
Ferrannini E. Learning from glycosuria. Diabetes. 2011;60:695–6.
Mather A, Pollock C. Renal glucose transporters: novel targets for hyperglycemia management. Nat Rev Nephrol. 2011;6:307–11.
Bailey CJ, Gross JL, Pieters A, Bastien A, List JF. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:2223–33.
Ferrannini E, Ramos SJ, Salsali A, Tang W, List JF. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care. 2010;33:2217–24.
Zhang L, Feng Y, List J, Kasichayanula S, Pfister M. Dapagliflozin treatment in patients with different stages of type 2 diabetes mellitus: effects on glycaemic control and body weight. Diabetes Obes Metab. 2010;12:510–6.
Wilding JP, Norwood P, T’joen C, Bastien A, List JF, Fiedorek FT. A study of dapagliflozin in patients with type 2 diabetes receiving high doses of insulin plus insulin sensitizers: applicability of a novel insulin-independent treatment. Diabetes Care. 2009;32:1656–62.
Rosenstock J, Arbit D, Usiskin K, Capuano G, Canovatchel W. Canagliflozin, an inhibitor of sodium glucose co-transporter 2 (SGLT2), improves glycemic control and lowers body weight in subjects with type 2 diabetes (T2D) on metformin. Diabetes. 2010;59:0077-OR.
Kashiwagi A, Takinami Y, Kazuta K, Yoshida S, Utsuno A, Nagase I. Ipragliflozin improved glycaemic control with additional benefits of reductions of body weight and blood pressure in Japanese patients with type 2 diabetes mellitus: BRIGHTEN Study. Diabetologia. 2011;54:S68.
Grempler R, Thomas L, Eckhardt M, et al. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterization and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012;14:83–90.
Seman L, Macha S, Nehmiz G, et al. Empagliflozin (BI 10773), a potent and selective SGLT-2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharm Drug Dev. 2013;2:152–61.
Komoroski B, Vachharajani N, Boulton D, et al. Dapagliflozin, a novel SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Ther. 2009;85:520–6.
List JF, Woo V, Morales E, Tang W, Fiedorek FT. Sodium-glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care. 2009;32:650–7.
Sha S, Devineni D, Ghosh A, et al. Canagliflozin, a novel inhibitor of sodium glucose co-transporter 2, dose-dependently reduces calculated renal threshold for glucose excretion and increases urinary glucose excretion in healthy subjects. Diabetes Obes Metab. 2011;13:669–72.
Schwartz SL, Akinlade B, Klasen S, Kowalski D, Zhang W, Wilpshaar W. Safety, pharmacokinetic, and pharmacodynamic profiles of ipragliflozin (ASP1941), a novel and selective inhibitor of sodium-dependent glucose co-transporter 2, in patients with type 2 diabetes mellitus. Diabetes Technol Ther. 2011;13:1219–27.
Veltkamp SA, Kadokura T, Krauwinkel WJ, Smulders RA. Effect of Ipragliflozin (ASP1941), a novel selective sodium-dependent glucose co-transporter 2 inhibitor, on urinary glucose excretion in healthy subjects. Clin Drug Investig. 2011;31:839–51.
List JF, Whaley JM. Glucose dynamics and mechanistic implications of SGLT2 inhibitors in animals and humans. Kidney Int Suppl. 2011;120:S20–7.
Acknowledgments
The authors acknowledge Andreas Port for his contributions related to the planning and conduct of the study, and his support in preparing the clinical trial report and manuscript, and Bailuo Ren for contributions related to bioanalysis of plasma and urine pharmacokinetic samples. In addition, the authors thank John Gerich M.D., University of Rochester School of Medicine, Rochester, New York, USA, for helpful comments on the manuscript. Sponsorship and article processing charges for this study was funded by Boehringer Ingelheim. Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Lindsay Napier Ph.D. and Isobel Lever Ph.D. of Fleishman-Hillard Group Ltd during the preparation of this manuscript.
Conflict of interest
Tim Heise is a shareholder of a private research institute (Profil Institut für Stoffwechselforschung GmbH). Within the last year, this institute received research grants from the following pharmaceutical companies: Astellas, Becton–Dickinson, Biocon, Boehringer Ingelheim, Dance Pharmaceuticals, Evolva, Hoffmann LaRoche, Johnson & Johnson, Eli Lilly, Lundbeck, Novo Nordisk, Noxxon, Sanofi and Skye Pharma. In addition, he has received speaker honoraria and travel grants from Boehringer Ingelheim and Novo Nordisk and has been member of advisory panels for these companies. Peter Jones is a consultant statistician, and has worked with Boehringer Ingelheim and a number of other pharmaceutical and device companies. Leo Seman is an employee of Boehringer Ingelheim. Sreeraj Macha is an employee of Boehringer Ingelheim. Alexandra Marquart is an employee of Boehringer Ingelheim. Sabine Pinnetti is an employee of Boehringer Ingelheim. Hans. J. Woerle is an employee of Boehringer Ingelheim. Klaus Dugi is an employee of Boehringer Ingelheim.
Compliance with ethics guidelines
The Clinical Trial Protocol was submitted and approved by the local Independent Ethics Committee. The trial was carried out in compliance with the Helsinki Declaration of 1975, as revised in 2000, and the ICH Harmonised Tripartite Guideline for Good Clinical Practice. All patients gave signed and dated informed consent prior to inclusion.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development and have approved the final version. Tim Heise, Leo Seman, Sreeraj Macha and Sabine Pinnetti contributed to the research design. Sreeraj Macha and Peter Jones performed data analysis. An abstract of these data were presented at the 70th American Diabetes Association Annual Meeting, June 25–29, 2010, Orlando, Florida; Diabetes 2010: 59 [suppl 1]: A156 [571-P]. Tim Heise is the guarantor for this article, and takes responsibility for the integrity of the work as a whole.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Heise, T., Seman, L., Macha, S. et al. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Multiple Rising Doses of Empagliflozin in Patients with Type 2 Diabetes Mellitus. Diabetes Ther 4, 331–345 (2013). https://doi.org/10.1007/s13300-013-0030-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-013-0030-2