
Abstract
Introduction
Dapagliflozin is an orally administered selective sodium-glucose cotransporter 2 (SGLT2) inhibitor under development for the treatment of type 2 diabetes mellitus (T2DM). Dapagliflozin lowers blood glucose through a reduction in renal glucose reabsorption. This study was performed to assess the effect of the oral antidiabetic agent voglibose [0.2 mg thrice daily (t.i.d.)] at steady-state, on the pharmacokinetics, safety and tolerability of dapagliflozin administered as a single oral dose (10 mg) to Japanese patients with T2DM.
Methods
This was an open-label, multi-center, drug–drug interaction study. A single oral dose of dapagliflozin (10 mg) was administered to 22 Japanese patients with T2DM in the presence and absence of voglibose (0.2 mg t.i.d.). Serial blood samples were collected before and at regular prespecified intervals after each dapagliflozin dose to determine dapagliflozin plasma concentrations and to evaluate pharmacokinetic parameters. Based on a mixed effect analysis of variance model, including the dosing condition as a fixed effect and patients as a random effect, the ratios of geometric means of area under curve from time 0 to infinity (AUC0-inf) and maximum observed plasma concentration (C max) with and without voglibose were estimated along with two-sided 90% confidence intervals (CIs).
Results
In Japanese patients with T2DM, the exposure to dapagliflozin following a single oral dose of dapagliflozin 10 mg was not influenced by the concomitant administration of voglibose (0.2 mg t.i.d.). The geometric ratio (90% CI) for dapagliflozin AUC0-inf with/without voglibose was 1.009 (0.954, 1.067), and for C max 1.040 (0.899, 1.204). The median time to C max (t max) and plasma clearance of dapagliflozin were also similar between treatments. The mean half-life (t ½) for dapagliflozin was slightly higher when administered in combination with voglibose. Dapagliflozin 10 mg was well tolerated when administered alone and in combination with voglibose in Japanese patients with T2DM.
Conclusion
The results presented here support the co-administration of dapagliflozin and voglibose without dose adjustment of either agent.
Similar content being viewed by others

Introduction
The number of people diagnosed with diabetes worldwide has more than doubled since 1980 to nearly 350 million [1]. The rise in prevalence of diabetes has been particularly rapid in Japan and is likely to have been fueled by changes in lifestyle [2]. In Japanese medical practice there is an increasing use of combination therapy with multiple antidiabetic drugs in the treatment of type 2 diabetes mellitus (T2DM) [3]. Frequently an alpha-glucosidase inhibitor is a part of this treatment regimen. Voglibose is an oral antihyperglycemic agent of the alpha-glucosidase inhibitor class that is widely used in the management of T2DM in Japan [4]. Voglibose undergoes minimal systemic absorption and primarily functions within the gastrointestinal tract to decrease and delay the intestinal absorption of carbohydrate [5–7].
Dapagliflozin is a first-in-class selective sodium-glucose cotransporter 2 (SGLT2) inhibitor under development for the treatment of T2DM that improves glycemic control by reducing the reabsorption of glucose in the kidney and enhancing the urinary excretion of excess glucose [8, 9]. As with voglibose, the mechanism of action of dapagliflozin is independent of insulin secretion or action and efficacy, and its safety has been evaluated in a broad range of patients with T2DM as a monotherapy [10, 11] and as an add-on to metformin, glimepiride, pioglitazone, sitagliptin, and insulin [11–17]. Both dapagliflozin and voglibose effectively control postprandial blood glucose excursions in patients with T2DM [18, 19]. Thus, the co-administration of dapagliflozin with voglibose could potentially provide a complementary approach to glycemic control.
Predictable dose-proportional pharmacokinetic and pharmacodynamic parameters with dapagliflozin have been demonstrated in both Japanese and non-Japanese subjects [20–22]. Dapagliflozin is metabolized via the uridine diphosphate-glucuronosyltransferase 1A9 pathway to an inactive metabolite, dapagliflozin 3-O-glucuronide [23]. No clinically significant drug–drug interaction has been observed in patients when dapagliflozin was administered in combination with some commonly employed antihyperglycemic agents (metformin, pioglitazone, glimepride, or sitagliptin) [24] or other agents that are commonly used in patients with T2DM (warfarin, simvastatin, valsartan, and digoxin) [25].
Due to the differing pharmacokinetics of each treatment no drug–drug interaction was anticipated between dapagliflozin and voglibose. However, due to the possible future co-administration of dapagliflozin with voglibose in Japan, the aim of the study reported here was to assess the potential for drug–drug interaction between dapagliflozin and voglibose in Japanese patients with T2DM. As voglibose is not orally absorbed, only the effect of voglibose on the plasma pharmacokinetics of dapagliflozin was studied.
Materials and Methods
Subjects
An open-label, multi-center, drug–drug interaction study was conducted to assess the effect of voglibose [0.2 mg thrice daily (t.i.d.)] on the pharmacokinetics, safety and tolerability of dapagliflozin administered as a single oral dose (10 mg) to Japanese patients with T2DM. The study was performed in accordance with the Declaration of Helsinki and is consistent with ICH/Good Clinical Practice, applicable regulatory requirements, and the AstraZeneca policy on Bioethics and Human Biological Samples. All subjects provided written informed consent.
Male or female subjects ≥20 years of age were eligible for entry into the trial if they had a clinical diagnosis of T2DM, were already on voglibose treatment with a steady dosage for at least 8 weeks (one change in voglibose dose to 0.2 mg t.i.d. was permitted up to 4 weeks before study commencement), had a fasting C-peptide >1.0 ng/mL (0.33 nmol/L), fasting plasma glucose levels ≤180 mg/dL, and glycated hemoglobin (HbA1c) levels ≤8.5%. Women of child-bearing potential were required to have had a negative urine pregnancy test and to be using an adequate method of contraception throughout the study and for up to 4 weeks after the study.
Key exclusion criteria included a diagnosis of type 1 diabetes mellitus, a history of diabetic ketoacidosis, a calculated creatinine clearance <50 mL/min (calculated by Cockroft-Gault formula), urine albumin:creatinine ratio >1,800 mg/g (>203.4 mg/mmol), severe hepatic insufficiency and/or significant abnormal liver function defined as aspartate aminotransferase (AST) more than three-times the upper limit of normal (ULN) and/or alanine aminotransferase (ALT) more than three-times the ULN, total bilirubin >2.0 mg/dL (>34.2 μmol/L), congestive heart failure defined as New York Heart Association (NYHA) class III and IV, unstable or acute congestive heart failure, significant cardiovascular history within the past 6 months prior to the screening visit (defined as: myocardial infarction, unstable angina pectoris, transient ischemic attack, unstable, or previously undiagnosed arrhythmia), cardiac surgery, or revascularization (coronary angioplasty or bypass grafts), or cerebrovascular accident, systolic blood pressure (BP) ≥170 mmHg and/or diastolic BP ≥100 mmHg as an average value or creatine kinase more than three-times the ULN.
Study Design
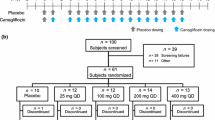
The study consisted of two pharmacokinetic sampling periods (Fig. 1). In period 1, each patient (already on a stable dose of 0.2 mg t.i.d. voglibose) took a single oral dose of 10 mg dapagliflozin on Day 1 together with voglibose (0.2 mg t.i.d.) and plasma samples (3 mL) were collected serially at predetermined time points for up to 72 h after the dose for pharmacokinetic assessment. Voglibose was administered just before each meal. This was followed by a dapagliflozin and voglibose washout period of 7 days. In period 2, a single oral dose of 10 mg dapagliflozin was administered without voglibose and no voglibose was taken until after the last blood sampling for pharmacokinetic assessment on study Day 14. Plasma samples were collected at regular intervals for up to 72 h following each dapagliflozin dose for pharmacokinetic assessment of dapagliflozin.

Flow chart of study design. PK pharmacokinetics
Pharmacokinetic and Pharmacodynamic Evaluation
Noncompartmental pharmacokinetic parameters for dapagliflozin were derived using WinNonlin® Version 5.2.1 (Pharsight Corporation, Mountain View, CA, USA) from plasma concentration versus time data and comprised area under the plasma concentration versus time curve from time 0 to infinity (AUC0-inf), maximum observed plasma concentration (C max), time to C max (t max) and half-life (t ½).
Blood Sampling and Drug Analyses
Samples (3 mL) for dapagliflozin analysis were collected in di-potassium ethylenediaminetetraacetic acid (K2EDTA) tubes and were then shipped on dry ice and placed in approximately −20 °C storage until analyzed (Atlanbio, Cedex, France). Dapagliflozin concentration in plasma was analyzed within the known period of stability by high-performance liquid chromatography tandem mass spectrometry (HPLC–MS) methodology after solid phase extraction (Atlanbio). The lower limit of quantification of dapagliflozin in plasma was 1.0 ng/mL and the upper limit of quantification was 500.0 ng/mL. Run-to-run variability as described by between-run and within-run coefficients of variation (% CV) were 6.5 and 7.5, respectively.
Safety Assessments
Laboratory variables, vital signs, and 12-lead electrocardiogram (ECG) were monitored throughout the study. Adverse events (AEs) were collected and summarized using the Medical Dictionary for Regulatory Activities (MedDRA version, version 14.0) vocabulary.
Statistical Analysis
All pharmacokinetic and safety data were summarized by dosing condition using descriptive statistics. The pharmacokinetic variables AUC and C max were log-transformed before analysis. They were analyzed using a mixed effect analysis of variance model, including the dosing condition (with voglibose or without voglibose) as a fixed effect and patients as a random effect. Estimates and confidence intervals (CIs) of the difference between the dosing conditions were first constructed in the log scale. Then the estimates and CIs of the ratio of geometric means between the dosing conditions in the original scale were obtained by taking anti-logarithms. The estimates and two-sided 90% CIs of the ratio of geometric means were presented for: AUC (with voglibose)/AUC (without voglibose) and C max (with voglibose)/C max (without voglibose). Lack of pharmacokinetic drug–drug interaction was concluded if the CI fell within 0.80–1.25 for the AUC and C max of dapagliflozin.
Sample Size Rationale
With 20 patients, the two-sided 90% CIs for ratios of AUC (with voglibose)/AUC (no voglibose) and C max (with voglibose)/C max (no voglibose) would fall within 0.8 and 1.25 with power over 99 and 90%, respectively, if voglibose had no true effect on AUC and C max of dapagliflozin and assuming that within-subject standard deviation of the log(AUC) and log(C max) was 0.095 and 0.2024, respectively. Two (10%) additional subjects were included to account for possible dropouts.
Results
Subject Disposition and Demographics
All 22 patients enrolled in the study were Japanese. Thirteen of the patients were male and nine were female. The median age of the patients was 55 years and the median body mass index (BMI) was 25.7 kg/m2 (Table 1).
Pharmacokinetics
The mean dapagliflozin plasma concentration–time curves following the single dose of dapagliflozin (10 mg) alone and in combination with voglibose (0.2 mg t.i.d.) are illustrated in Fig. 2. The mean plasma concentration profiles of dapagliflozin over time were similar when dapagliflozin was administered alone and in combination with voglibose. No meaningful effect (as defined by the Japanese Pharmaceutical and Medical Safety Bureau Notification No. 813, 2001 [26]) of voglibose on dapagliflozin exposure was observed (Table 2). The percent differences in geometric means of the AUC0-inf and C max with and without voglibose were less than 5%. Furthermore, the entirety of the two-sided 90% CIs for the ratio of geometric means of the AUC and C max with/without voglibose fell within the prespecified “no-effect” range of 0.80–1.25. t max and oral plasma clearance were also similar when dapagliflozin was given alone or in combination with voglibose. Dapagliflozin t ½ was slightly higher when dapagliflozin was administered together with voglibose.

Dapagliflozin plasma concentrations following treatment with or without voglibose. SD standard deviation
Three subjects (one subject during treatment with dapagliflozin and voglibose combination and two subjects during treatment with dapagliflozin alone) were excluded from the evaluation of the AUC, plasma clearance and t ½ due to the low reliability of the extrapolated portion of the AUC from the last measurable point to infinity i.e., the extrapolation ratio of AUC for the relevant subjects was >20%.
Safety
Dapagliflozin (10 mg) was found to be well tolerated by the subjects with T2DM in this study when administered alone or co-administered with voglibose. There were no serious adverse events, discontinuations of the investigational product due to adverse events, or any other significant adverse event in the study.
Five AEs (gingivitis, paresthesia oral, edema peripheral, bronchitis and pollakiuria) were reported in five patients (one patient in the pharmacokinetic sampling period 1: dapagliflozin with voglibose, one patient in the drug treatment washout period and three patients in the pharmacokinetic sampling period 2: dapagliflozin only). Three AEs were considered to be related to treatment (one during the washout phase and two during the dapagliflozin-only treatment phase). All observed AEs were mild or moderate in intensity. No treatment-related trends were observed in clinical laboratory results, vital sign measurements, physical examination findings, or 12-lead ECG evaluation in either study.
Discussion
Numerous factors can affect the bioavailability and intestinal absorption of orally administered drugs, and among them is an elevation of gastrointestinal carbohydrate levels [27]. Orally administered voglibose is poorly absorbed by the gastrointestinal tract [5, 6], and the persistence of carbohydrates in the gastrointestinal tract following the inhibition of α-glucosidases with voglibose may potentially affect the absorption of other orally administered drugs. Voglibose also increases gastrointestinal motility, which could conceivably affect the absorption of concomitantly administered oral agents due to a shortened intestinal transit time [28].
This study was performed to evaluate whether the administration of voglibose would affect the total exposure to dapagliflozin. Previously, a lack of clinically relevant interaction was observed between dapagliflozin and the oral antidiabetic agents metformin, pioglitazone, glimepiride and sitagliptin [24]. The 10 mg dose of dapagliflozin was chosen as the highest expected usual dose for T2DM patients based on the results of several phase three studies and the dose of voglibose administered in this study (0.2 mg t.i.d.) is a commonly utilized dose in clinical practice in Japan.
Results from this study indicate that there is no clinically meaningful pharmacokinetic drug–drug interaction between dapagliflozin and voglibose in Japanese patients with T2DM. The exposure to dapagliflozin (as measured by the AUC0-inf and C max) after a single oral dose of dapagliflozin 10 mg was not influenced by concomitant doses of voglibose (0.2 mg t.i.d.). The median t max and plasma clearance were similar between treatments. Dapagliflozin t ½ was slightly longer in the presence of voglibose, although this was not considered clinically relevant.
Mean changes in dapagliflozin exposure were less than 5% with voglibose co-administration, and dapagliflozin appeared to be safe and well tolerated by the subjects in this study when administered as a single oral dose of 10 mg alone or when co-administered with voglibose.
Thus, the absorption of dapagliflozin across the gastrointestinal epithelium appears unaffected by the enhanced gastrointestinal levels of carbohydrate and increased gastrointestinal motility observed in the presence of voglibose. A clinically meaningful drug–drug interaction between dapagliflozin and voglibose appears unlikely and these results support the co-administration of dapagliflozin and voglibose without dose adjustment of either agent.
A potential limitation of the present study is that it was performed solely in patients of Japanese ethnicity. However, the results are still likely to be applicable to patients of other ethnicities, because voglibose has minimal systemic absorption from the gastrointestinal tract regardless of ethnicity and dapagliflozin shows similar pharmacokinetics in Japanese and non-Japanese subjects [20–22].
It is interesting to speculate regarding the additional glycemic benefits that may be gained by reducing glucose absorption across the gastrointestinal tract with voglibose, while at the same time reducing renal glucose reabsorption and increasing the elimination of glucose in the urine with dapagliflozin. This could provide a complementary and insulin-independent approach to glycemic control, which is likely to be associated with a low risk of hypoglycemia. In the present study no episodes of hypoglycemia were noted.
Conclusion
Both voglibose and dapagliflozin effectively control the rapid postprandial excursions observed following high carbohydrate meals [18, 19] and together could potentially provide an effective approach to improving glucose spikes in patients with T2DM. To date, however, there are no clinical studies evaluating combination therapy with dapagliflozin and voglibose. Additional studies would be required to evaluate the effects of voglibose on the pharmacodynamics and efficacy of dapagliflozin.
References
Danaei G, Finucane MM, Lu Y, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet. 2011;378:31–40.
The Japan Diabetes Society. Introduction to the Japan Diabetes Society. http://www.jds.or.jp/e/about_jds/message.html. Accessed 25 Apr 2012.
Neville SE, Boye KS, Montgomery WS, Iwamoto K, Okamura M, Hayes RP. Diabetes in Japan: a review of disease burden and approaches to treatment. Diabetes Metab Res Rev. 2009;25:705–16.
Odaka H, Shino A, Ikeda H, Matsuo T. Antiobesity and antidiabetic actions of a new potent disaccharidase inhibitor in genetically obese-diabetic mice, KKA(y). J Nutr Sci Vitaminol (Tokyo). 1992;38:27–37.
Fuder H, Kleist P, Birkel M, et al. The alpha-glucosidase inhibitor voglibose (AO-128) does not change pharmacodynamics or pharmacokinetics of warfarin. Eur J Clin Pharmacol. 1997;53:153–7.
Goke B, Fuder H, Wieckhorst G, et al. Voglibose (AO-128) is an efficient alpha-glucosidase inhibitor and mobilizes the endogenous GLP-1 reserve. Digestion. 1995;56:493–501.
Horii S, Fukase H, Matsuo T, Kameda Y, Asano N, Matsui K. Synthesis and alpha-D-glucosidase inhibitory activity of N-substituted valiolamine derivatives as potential oral antidiabetic agents. J Med Chem. 1986;29:1038–46.
Chao EC, Henry RR. SGLT2 inhibition–a novel strategy for diabetes treatment. Nat Rev Drug Discov. 2010;9:551–9.
Meng W, Ellsworth BA, Nirschl AA, et al. Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J Med Chem. 2008;51:1145–9.
Ferrannini E, Ramos SJ, Salsali A, Tang W, List JF. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase III trial. Diabetes Care. 2010;33:2217–24.
Henry R, Murray A, Marmolejo MH, Hennicken D, Ptaszynska A, List JF. Dapagliflozin, metformin-XR, or both together as initial therapy for T2DM [abstract]. Diabetes. 2011;60:A84. Abstract 307-OR.
Bailey CJ, Gross JL, Pieters A, Bastien A, List JF. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:2223–33.
Bailey CJ, Gross JL, Yadav M, Iqbal N, Mansfield TA, List JF. Long-term efficacy of dapagliflozin as add-on to metformin (MET) in T2DM inadequately controlled with metformin alone [abstract]. Diabetes. 2011;60:A271. Abstract 988-P.
Jabbour S, Hardy E, Sugg J, Parikh S. Dapagliflozin as add-on therapy to sitagliptin with or without metformin: a randomized, double-blind, placebo-controlled study. Diabetes. 2012;61:A275. Abstract 1071-P.
Nauck MA, Del PS, Meier JJ, et al. Dapagliflozin versus glipizide as add-on therapy in patients with type 2 diabetes who have inadequate glycemic control with metformin: a randomized, 52-week, double-blind, active-controlled noninferiority trial. Diabetes Care. 2011;34:2015–22.
Rosenstock J, Vico M, Wei L, Salsali A, List JF. Effects of dapagliflozin, a sodium-glucose cotransporter-2 inhibitor, on hemoglobin A1c, body weight, and hypoglycemia risk in patients with type 2 diabetes inadequately controlled on pioglitazone monotherapy. Diabetes Care. 2012;35:1473–8.
Strojek K, Hruba V, Elze M, Langkilde A, Parikh S. Efficacy and safety of dapagliflozin in patients with type 2 diabetes mellitus and inadequate glycaemic control on glimepiride monotherapy [abstract]. Diabetologia. 2010;53:S347. Abstract 870.
Kageyama S, Nakamichi N, Sekino H, Nakano S. Comparison of the effects of acarbose and voglibose in healthy subjects. Clin Ther. 1997;19:720–9.
Salsali A, Hruba V, Ying L, et al. Reduction in postprandial glucose with dapagliflozin in type 2 diabetes [abstract]. Diabetes. 2011;60:A303. Abstract 1104-P.
Kasichayanula S, Chang M, Hasegawa M, et al. Pharmacokinetics and pharmacodynamics of dapagliflozin, a novel selective inhibitor of sodium-glucose co-transporter type 2, in Japanese subjects without and with type 2 diabetes mellitus. Diabetes Obes Metab. 2011;13:357–65.
Komoroski B, Vachharajani N, Feng Y, Li L, Kornhauser D, Pfister M. Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus. Clin Pharmacol Ther. 2009;85:513–9.
Komoroski B, Vachharajani N, Boulton D, et al. Dapagliflozin, a novel SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Ther. 2009;85:520–6.
Obermeier M, Yao M, Khanna A, et al. In vitro characterization and pharmacokinetics of dapagliflozin (BMS-512148), a potent sodium-glucose cotransporter type II inhibitor, in animals and humans. Drug Metab Dispos. 2010;38:405–14.
Kasichayanula S, Liu X, Shyu WC, et al. Lack of pharmacokinetic interaction between dapagliflozin, a novel SGLT2 inhibitor, and metformin, pioglitazone, glimepiride, or sitagliptin in healthy subjects. Diabetes Obes Metab. 2011;13:47–54.
Kasichayanula S, Chang M, Liu X, et al. Lack of pharmacokinetic interactions between dapagliflozin and simvastatin, valsartan, warfarin, or digoxin. Adv Ther. 2012;29:163–77.
Methods of Investigation for Drug Interactions. PMSB/ELD Notification, vol No 813. 2001.
Trovato A, Nuhlicek DN, Midtling JE. Drug-nutrient interactions. Am Fam Physician. 1991;44:1651–8.
Nakamura T, Takebe K, Kudoh K, et al. Effect of an alpha-glucosidase inhibitor on intestinal fermentation and faecal lipids in diabetic patients. J Int Med Res. 1993;21:257–67.
Acknowledgments
Medical writing assistance was provided by Karen Pemberton, PhD, of PAREXEL and was funded by AstraZeneca and Bristol-Myers Squibb. The study was sponsored by AstraZeneca and Bristol-Myers Squibb. Dr. Ueda is the guarantor for this article, and takes responsibility for the integrity of the work as a whole.
Conflict of interest
SU and NH were employees of AstraZeneca K.K. and YI was an employee of Bristol-Myers Squibb K.K. at the time the studies were performed. AI and MK have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
ClinicalTrials.gov: #NCT01055652.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Imamura, A., Kusunoki, M., Ueda, S. et al. Impact of Voglibose on the Pharmacokinetics of Dapagliflozin in Japanese Patients with Type 2 Diabetes. Diabetes Ther 4, 41–49 (2013). https://doi.org/10.1007/s13300-012-0016-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-012-0016-5