
Abstract
The object of this review is to provide the definitions, frequency, risk factors, pathophysiology, diagnostic considerations, and management recommendations for diabetic ketoacidosis (DKA) in children and adolescents, and to convey current knowledge of the causes of permanent disability or mortality from complications of DKA or its management, particularly the most common complication, cerebral edema (CE). DKA frequency at the time of diagnosis of pediatric diabetes is 10%–70%, varying with the availability of healthcare and the incidence of type 1 diabetes (T1D) in the community. Recurrent DKA rates are also dependent on medical services and socioeconomic circumstances. Management should be in centers with experience and where vital signs, neurologic status, and biochemistry can be monitored with sufficient frequency to prevent complications or, in the case of CE, to intervene rapidly with mannitol or hypertonic saline infusion. Fluid infusion should precede insulin administration (0.1 U/kg/h) by 1–2 hours; an initial bolus of 10–20 mL/kg 0.9% saline is followed by 0.45% saline calculated to supply maintenance and replace 5%–10% dehydration. Potassium (K) must be replaced early and sufficiently. Bicarbonate administration is contraindicated. The prevention of DKA at onset of diabetes requires an informed community and high index of suspicion; prevention of recurrent DKA, which is almost always due to insulin omission, necessitates a committed team effort.
Similar content being viewed by others

Introduction
Definition of Diabetic Ketoacidosis
Diabetic ketoacidosis (DKA) is biochemically defined as a venous pH <7.3 or serum bicarbonate concentration <15 mmol/L, serum glucose concentration >200 mg/dL (11 mmol/L) together with ketonemia, glucosuria, and ketonuria.1,2 Rarely, DKA may occur with normal circulating glucose concentrations if there has been partial treatment or with pregnancy.3,4 The severity of DKA is determined by the degree of acidosis:2
-
Mild: venous pH >7.2 and <7.3, bicarbonate <15 mmol/L
-
Moderate: venous pH >7.1 and <7.2, bicarbonate <10 mmol/L
-
Severe: venous pH <7.1, bicarbonate <5 mmol/L.
Pathophysiology
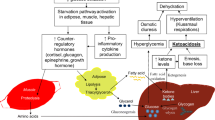
DKA is the result of a critical relative or absolute deficit of insulin, resulting in intracellular starvation of insulin-dependent tissues (muscle, liver, adipose), stimulating the release of the counter-regulatory hormones glucagon, catecholamines, cortisol, and growth hormone. The counter-regulatory hormonal responses may also be the result of stress-induced proinflammatory cytokines.5 They stimulate lipolysis and proteolysis, hepatic and renal glucose production, and hepatic oxidation of fatty acid to ketone bodies.6 Unlike during physiologic fasting, absence of citric acid cycle processing of glucose impedes the processing of these ketones for energy (Figure 1).

Pathophysiology of diabetic ketoacidosis. Absolute or relative insulin deficiency decreases glucose utilization in insulin-sensitive tissues and promotes lipolysis. Energy deficiency in these tissues is a stress, stimulating counter-regulatory hormones (glucagon, catecholamines, growth hormone, cortisol) and proinflammatory cytokines, which also stimulate these hormones. The counter-regulatory hormones in turn enhance lipolysis and proteolysis, providing a substrate for hepatic ketogenesis and hepatic and renal gluconeogenesis, all of which results in ketoacidosis, osmotic diuresis, dehydration, and tissue hypoperfusion, adding lactic acidosis to the metabolic acidosis from accumulated ketones and loss of base in the urine
Frequency and Risk Factors
DKA with New-Onset Diabetes
The frequency of new-onset diabetes presenting as DKA varies widely by geographic region, and correlates inversely with the regional incidence, and therefore the level of awareness in the community of pediatric diabetes. In Europe this frequency varies from 11% to 67%.7 In Australia the frequency between 1985 and 2000 was 26%,8 and in New Zealand 63% in 1988/89 and 42% in 1995/96.9 Children <5 years of age are more likely to have DKA at diagnosis as are those who are in social or economic situations that do not permit ready access to medical care.2,7–14 In patients from Colorado, DKA was seen at onset in 28.4%; the odds ratio for uninsured patients compared with insured patients was 6.2, with significantly greater severity in the uninsured group.12 Long-term reductions in frequency of DKA at onset have been reported following intensive education of the medical and lay community.8–10 DKA at the time of diagnosis has been estimated to occur in as many as 25% of children with type 2 diabetes (T2D).2
It is not uncommon for previously undiagnosed patients in DKA to have been seen in physicians’ offices or emergency rooms without adequate history and laboratory study that could have made the diagnosis before they became critically ill. A high index of suspicion is particularly warranted for infants and young children whose symptoms may be nonspecific.
Recurrent DKA
Among 1243 patients in Colorado, the risk of recurrent DKA was eight episodes per 100 patient years; 20% of the patients accounted for 80% of the episodes. The risk factors for recurrent DKA were poor metabolic control or previous episodes of DKA, female gender (peripubertal or adolescent), psychiatric disorders including eating disorders, difficult or unstable family circumstances, limited access to medical services, and insulin pump therapy.12 Only rapid- or short-acting insulin is used in pumps, so that interruption of insulin delivery for any reason rapidly leads to insulin deficiency. In the 1970s and 1980s, the establishment of treatment teams with intensive education of families on sick day management and 24-hour availability demonstrated a profound reduction in recurrent DKA, which is almost invariably due to intentional omission of insulin administration.15,16
Diagnosis and Initial Evaluation
Hyperglycemic hyperosmolar state (HHS) defined as serum glucose >600 mg/mL (33 mmol/L), serum osmolality >320 mOsm/L, and minimal ketonemia/ketonuria, is being seen with increasing frequency as the presenting indication of T2D and may be associated with mild-tomoderate acidosis from severe dehydration, leading to confusion with DKA.17 Conversely, patients with type 1 diabetes (T1D) may have features of HHS, especially if they have been satisfying their polydipsia with fluids containing a high concentration of glucose.18 HHS has a substantial mortality rate and requires aggressive reconstitution of the circulatory volume.17–20
Body weight should be determined for calculation purposes. Dehydration can be estimated as 5% if there is reduced skin elasticity, dry mucous membranes, tachycardia, and deep breathing, and up to 10% with capillary refill time greater than 3 seconds and sunken eyes (Table 1). Calculations of fluid deficit are commonly based on 10% dehydration, which in most cases is a modest overestimate that does not appear to have clinical significance.21–22 The level of consciousness should be recorded using the Glasgow Coma Scale (Table 2).23 An initial venous blood sample should be tested for glucose; electrolytes; pH; urea nitrogen; creatinine; osmolality; ketones or beta-hydroxybutyrate; hemoglobin and hematocrit or complete blood count, while keeping in mind that DKA is associated with leukocytosis.
Osmolality can be measured or calculated as: 2× (sodium [Na] + K) + glucose in millimoles per liter (or glucose in mg/dL/18). The urine should be checked for ketones. If there is any possibility of delay in obtaining a serum K result, electrocardiographic monitoring should be done for K status.24,25 Severely ill patients should have continuous electrocardiographic monitoring using standard lead II or if the initial K level is ≤3 mEq/L or ≥6 mEq/L (Figure 2).26

Electrocardiographic patterns for hypo- and hyperkalemia
Glycohemoglobin (HbA1c) concentration will provide information about the severity and duration of hyperglycemia in newly diagnosed patients and can be compared with the previous determination as an indicator of noncompliance leading to the DKA in an established patient. Measurement of serum-free insulin concentration before the administration of insulin can confirm an impression of failure to administer insulin as the cause of recurrent DKA.27
The severely obtunded child will require continuous nasogastric suction for the prevention of pulmonary aspiration and may require oxygen. Bladder catheterization should be performed only in individuals who are unable to void on demand; older patients may have an atonic bladder with urinary retention requiring initial catheterization, but not indwelling.28
Management
Children with established diabetes, who are not vomiting or severely ill, and whose caregiver has been trained in sick day management, can be managed at home or at an outpatient facility, with appropriate supervision and follow-up.2,29–32 Children who are vomiting and not severely ill may be managed with intravenous (IV) fluids in an emergency department and discharged to home if able to take fluids orally following rehydration, if venous pH is above 7.25, and telephone follow-up can be assured. Children requiring IV rehydration over an extended period need to be admitted to a unit in which neurological status and vital signs can be monitored frequently and blood glucose levels measured hourly.2 Nursing staff trained in monitoring and management of DKA should be available and written guidelines should be provided.2,32 With severe DKA (long duration of symptoms, compromised circulation, depressed level of consciousness) or if there are other factors increasing the risk for cerebral edema (CE; age under 5 years, low partial pressure of carbon dioxide [pCO2], high serum urea nitrogen), an intensive care unit, preferably pediatric, or an equivalent facility is appropriate.2
Fluid and Insulin Replacement
Goals of Treatment with Fluid and Insulin
-
Restore perfusion, which will increase glucose uptake in the periphery, increase glomerular filtration, and reverse the progressive acidosis.
-
Arrest ketogenesis with insulin administration, which reverses proteolysis and lipolysis while stimulating glucose uptake and processing, thereby normalizing blood glucose concentration.
-
Replace electrolyte losses.
-
Intervene rapidly when complications, especially CE, occur.
Fluid Therapy
-
Dehydration can be assumed to be 5%–10% (50–100 mL/kg). As noted above, the degree of dehydration is usually overestimated. The severity of extracellular fluid (ECF) contraction may be indicated by serum urea and hematocrit concentrations.11 Serum Na concentration is not reliable for determining ECF deficit because of the osmotic effect of hyperglycemia-induced dilutional hyponatremia33,34 and the low Na content of the elevated lipid fraction of the serum in DKA. Corrected Na, ie for normal glucose levels, can be estimated by adding 1.6 mEq to the measured value for each 100 mg/dL blood glucose above normal.33
-
During the first 1–2 hours, 10–20mL/kg 0.9% sodium chloride (NaCl) should be provided to restore peripheral perfusion.
-
Maintenance can be calculated as 1000 mL for the first 10 kg body weight + 500 mL for the next 10 kg + 20 mL/kg over 20 kg or 1500 mL/m2 body surface area.
-
The remainder of replacement after the loading dose, based on 5%–10% dehydration, and maintenance, can be distributed over the subsequent 22–23 hours. While many guidelines call for calculating replacement over 48 hours, there is no evidence basis for this being safer or more efficacious. Fluids that have recently been administered orally at home (if not vomited) and parenteral fluids provided in the emergency room or referring institution need to be incorporated into the calculation.
-
Except for severely ill and very young individuals, oral intake should begin before 24 hours.
-
While urinary output should be carefully documented, urinary losses should not be added to fluid requirements, except in the presence of HHS.2,17,20
-
After initial 0.9% NaCl bolus, rehydration/maintenance should be continued with 0.45% NaCl. The measured Na can increase to the level of the corrected Na during rehydration as glycemia declines and then decline to normal levels if the corrected level was elevated.
-
Continued use of 0.9% saline after the initial resuscitation may result in hyperchloremic metabolic acidosis.35,36
-
To prevent unduly rapid decline in plasma glucose concentration and hypoglycemia, 5% glucose should be added to the IV fluid when the plasma glucose falls to 300 mg/dL (17 mmol/L). An efficient method of providing glucose as needed without long delays entailed by the changing of IV solutions is to have two IV fluid bags connected, one containing 10% dextrose and the appropriate Na and K concentrations and the other with the same salt concentrations but no dextrose, the so-called “two bag system”.37
-
K (40 mEq/L or up to 80 mEq/L as needed) can be provided as half potassium chloride (KCl), half potassium phosphate (KPO4; to replenish low phosphate levels and to decrease the risk of hyperchloremia) or as half KPO4 and half K acetate (which, like lactate, is converted to bicarbonate to help correct acidosis) after serum K is <6 mmol/L or urine flow is established. Serum K concentration increases by approximately 0.6 mEq/L for every 0.1 decrease in pH, so that the serum K level does not reflect the large deficit from diuresis and vomiting, ∼5 mEq/kg body weight. Both K and phosphate shift markedly from intracellular to extracellular compartments with acidosis and re-enter cells rapidly with insulin-induced glucose uptake and rehydration.26,38
-
Bicarbonate administration is not indicated in pediatric DKA. There is no evidence that bicarbonate facilitates metabolic recovery. Restoring circulatory volume will improve tissue perfusion and renal function, increasing organic acid excretion and reversing lactic acidosis. The administration of insulin stops further synthesis of ketoacids and reactivates the Krebs cycle, permitting metabolism of ketoacids, and regeneration of bicarbonate. Bicarbonate therapy may cause paradoxical central nervous system (CNS) acidosis, rapid correction of acidosis can cause hypokalemia, the additional Na can add to hyperosmolality, and alkali therapy can increase hepatic ketone production, potentially slowing recovery.2
Insulin
-
Insulin should be started after initial fluid expansion. This provides a more realistic starting glucose level.
-
0.1 U/kg/hour is given as a continuous infusion, using a pump. Fifty units of regular insulin are diluted in 50 mL normal saline to provide 1 unit/mL.
-
A bolus dose of insulin is not indicated and may increase the risk of CE.39
-
In some settings it may be necessary to administer insulin subcutaneously. Studies in adults have indicated no significant difference in recovery time whether insulin was administered intravenously, intramuscularly, or subcutaneously after the first couple of hours of treatment.40,43 A study of subcutaneous insulin in children with DKA using a rapid-acting insulin analog (lispro) provided a dose of 0.15 units/kg every 2 hours; there were no significant differences from children randomized to receive 0.1 unit/kg per hour intravenously.44 The administration of 0.1 unit/kg subcutaneously every hour may be preferable and can be adjusted to maintain blood glucose concentrations at ∼180–200 mg/dL (10–11 mmol/L).
-
Fluid expansion alone will have a dilutional effect, lowering high blood glucose levels by as much as 180–270 mg/dL (10–15 mmol/L). With insulin infusion the rate of glucose decline should be 50–150 mg/dL (2.8–8.3 mmol/L/hour), but not >200 mg/dL (11 mmol/L/hour). If serum glucose values are not dropping adequately, the insulin dose should be increased; this is rarely necessary.
-
If the blood glucose concentration falls below 150 mg/dL (8.3 mmol/L) 10% dextrose solution should be given and the insulin dose reduced to 0.05 U/kg/hour if glucose concentration is not sustained by the 10% dextrose solution.
-
Insulin should not be stopped; a continuous supply of insulin is needed to prevent ketosis and permit continued anabolism. If the patient demonstrates marked sensitivity to insulin, the dose may be decreased to 0.05 units/kg/hour, or less, provided that metabolic acidosis continues to resolve.
-
Persistent acidosis, defined as bicarbonate value <10 mmol/L after 8–10 hours of treatment, is usually caused by inadequate insulin effect. Insulin dilution and rate of administration should be checked, and a fresh preparation made. Too dilute a solution may enhance insulin adherence to the tubing. If insulin is being given by subcutaneous injection, inadequate absorption may be occurring. Rare causes of persistent acidosis include lactic acidosis due to an episode of hypotension or apnea or inadequate renal handling of hydrogen ion as a result of an episode of renal hypoperfusion.28
Monitoring
Successful management and early intervention for complications requires close monitoring. A flow chart should be maintained to document all relevant incidents regarding the patient’s condition.28 Minimal monitoring frequency recommendations include vital signs and neurologic checks hourly; blood glucose hourly; venous blood gases every 2 hours for 6 hours, then every 4 hours; Na, K, and ionized calcium every 2 hours for 6 hours, then every 4 hours; magnesium and phosphorus every 4 hours; basic metabolic profile at admission and then every morning. These minimum requirements should be adapted to the situation; for example, more frequent (hourly) K measurements may be indicated, along with electrocardiogram (ECG) monitoring depending on the initial K value,24,25 or more frequent neurologic and vital sign checks (20–30 minutes) if there is a concern about the patient’s mental status.28
Ketonuria should not be used as a measure of improvement. The dip sticks used to measure ketones react with acetoacetate. Concentrations of beta-hydroxybutyrate are much higher than those of acetoacetate and will be converted to acetoacetate during successful treatment of the DKA, resulting in sustenance or increase of urinary ketone concentration. Laboratory measurement or the use of a bedside fingerstick sample monitor for beta-hydroxybutyrate will avoid this problem.
Transition
-
IV fluids can be stopped 1–2 hours after substantial consumption of oral fluids without vomiting.
-
Subcutaneous insulin injection can be started when IV fluids are no longer needed. Presupper or prebreakfast time is most convenient for starting or restarting intermediate- or long-acting insulin. Before then, rapid-acting or regular insulin 0.25 U/kg subcutaneously can be given every ∼6 hours, and the insulin infusion stopped 60–120 minutes after the first subcutaneous dose of regular insulin or 60 minutes after a rapid-acting insulin analog.
-
Patients should not be kept in the hospital simply to adjust insulin dosage because food, activity, and psychosocial environment are not normal in the hospital setting. Therefore, insulin requirements will not be particularly informative for home management.
-
Established patients with DKA can resume their usual home dose of insulin.
Complications of DKA or its treatment
Most of the diabetes-related morbidity and mortality in childhood T1D can be attributed to complications of DKA.2 Mortality risks for each episode of DKA have been reported from the United States (0.15%),45 Canada (0.18%–0.25%),46,47 and the United Kingdom (0.31%).48 CE accounts for 60%–100% of this mortality.45,50 Other causes of death or disability with DKA include hypokalemia, hypophosphatemia, hypoglycemia, other intracerebral complications, peripheral venous thrombosis, mucormycosis, rhabdomyolysis, acute pancreatitis, acute renal failure, sepsis, aspiration pneumonia, and other pulmonary complications.2 Prevention of hypokalemia was noted in the section of fluid therapy. Hypophosphatemia can cause progressive muscle weakness and death due to cardiorespiratory arrest days after metabolic recovery from DKA and can be prevented by administration of half of the K as KPO4; administration of all the K as KPO4 can result in hypocalcemia.51,52
Renal Failure
Renal failure is a common complication and cause of mortality with HHS, necessitating early consideration of dialysis,17 but is extremely rare with pediatric T1D.
Peripheral Venous Thrombosis
Central venous catheter (CVC) placement was associated with deep venous thrombosis (DVT) in four of eight children with DKA,53 and in three of six pediatric intensive care unit (PICU) patients with DKA who required femoral CVC (out of 113 DKA/PICU patients).54 The PICU patients were under 18 months of age and required long-term heparin therapy for persistent leg swelling; in contrast, seven comparably aged patients with shock who had femoral CVC for longer periods of time than the DKA patients had no episodes of DVT. This suggested that DKA is associated with a thrombotic diathesis beyond that which is attributable to dehydration. In the absence of DKA, coagulation factor abnormalities have not been demonstrated in children with diabetes,55 but von Willebrand factor activity remains elevated at 120 hours following admission for DKA.56 Thus, DKA and its treatment may promote a prothrombotic state and activation of vascular endothelium, predisposing to thrombosis.
More than 50% of children who have venous thromboses will carry a genetic thrombophilic defect, and two-thirds of such defects will be a mutation in the factor V gene referred to as factor V Leiden.57 Massive arterial thrombosis resulting in unilateral below-the-knee amputation has been reported in a 12-year-old female patient with a heterozygous factor V Leiden mutation and a 2-year history of poorly controlled T1D who was not in DKA.58
Pancreatitis
Acute pancreatitis is a recognized complication of DKA in adults and in new-onset T2D with HHS17 but is unusual in childhood T1D. Serum levels of amylase and lipase were commonly elevated in 50 children with DKA at onset of T1D.59 The amylase elevation is of salivary origin. The serum lipase level was directly associated with the degree of acidosis, and acute pancreatitis was demonstrated by abdominal computed tomography (CT) in one of the patients who had persistent abdominal pain.59 Acute pancreatitis must be considered with abdominal pain that does not resolve with correction of acidosis.
Rhabdomyolysis
Rhabdomyolysis is a common complication of HHS and is associated with renal failure.17 It occurs with DKA, but in the absence of HHS is not associated with mortality. Risk factors include severe hyperglycemia, high osmolality, and hypophosphatemia. A 15-month-old was added to the few reported pediatric cases in 2003.60 The occurrence of rhabdomyolysis in a 27-month-old patient 25 years earlier led to a chart review of 133 children admitted with new-onset diabetes, of whom 12 had orthotolidine reactions, which detect myoglobin in the urine.61
Mucormycosis
This acute, rapidly progressing, and often fatal facultative fungal infection occurs in young patients with diabetes who have chronically poor glycemic control and ketoacidosis. The principal locus may be rhinocerebral, pulmonary, cutaneous, gastrointestinal, or in the CNS, or it may be disseminated. In a series of five patients, the sole survivor had severe neurologic disability; risk factors were African American race and history of poor compliance (poor clinic attendance, risk-taking behaviors, and high rate of hospital admission).62
Cerebral Edema
In 1967, two teenage patients were reported with fatal CE and only three others were identified from the literature with a similar clinical history.63 In the subsequent 13 years, only 13 more cases were reported.64 In the next 10 years the total reported cases reached 40 and in 1990 an additional 29 were added in one report and 11 in another.49,65 Subsequently, another ∼160 cases have been added. These reports have provided incidence data and have emphasized the idiosyncratic nature of the occurrence of CE and the absence of iatrogenic causation.66,67
Frequency
A 3-year United Kingdom population-based study yielded an incidence of 0.7% overall, with a marked discrepancy between the newly diagnosed, 1.2%, and established patients, 0.4%.68 A higher incidence of 4.3% in newly diagnosed, 1.3% in established patients, and 2% overall was reported from Australia.8 The North American multicenter study reported an incidence of 0.9% for CE without differences between new-onset and established patients.50 In a prospective surveillance study in Canada of patients <16 years of age with DKA, there were 13 verified cases of CE, for a rate of 0.51% of episodes of DKA.47 Variations in case definition and referral patterns likely explain some of these differences in the reported frequencies of CE.
Morbidity and Mortality
Earlier reports may reflect biased fatality and residual morbidity data and later recognition than more recent population-based observations. Appropriate intervention was also less frequent in these earlier-reported and litigated cases. The first extensive series, in 1990, analyzed 69 cases, including 40 that were previously published, and found a case fatality rate of 64% overall. Survival without disability was noted in 14%, with disability that did not preclude independence in 9%, and severe disability or vegetative state in 13%. Among those who were treated before respiratory arrest, even if this was at the gasping stage, case fatality was only 30%, with 30% surviving without disability, 26% with disability not precluding independence, and 13% with severe disability or vegetative state.49 Although the intervention in these cases was often inadequate and after severe neurologic compromise, the outcome with treatment is similar to those in contemporary population-based studies. There have been a few case reports of CE developing before initiation of treatment of DKA and a report from Canada indicated that 19% of cases of CE were present before treatment was undertaken.47,49,50,68
Risk Factors
Young children, especially <5 years of age, are at increased risk for the development of CE. This may reflect the more rapid deterioration in this age group and greater delay in diagnosis because of nonspecificity of symptoms. The younger brain might also be more susceptible to metabolic and vascular changes associated with DKA. Severity of acidosis, degree of hypocapnia, and elevated serum urea nitrogen, indicators of severity of ketoacidosis and dehydration, have been noted to be risk factors.2 Although Na bicarbonate administration was significantly associated with the development of CE in one population-based case-control study,50 this association may simply reflect the greater severity of ketoacidosis that was not adequately controlled for in the case-control design, rather than an effect of the bicarbonate.69
CE associated with DKA is rarely seen beyond the pediatric age group (0–21 years)67 or in adult patients with HHS, despite rapid rehydration and restoration of normal glycemia.70 In several studies increased risk of CE has been associated with a failure of serum Na concentration to increase appropriately during treatment for DKA. This may be because those who are in the early, presymptomatic stages of CE experience changes in the brain that result in dysregulation of antidiuretic hormone secretion.50,71 The absence of evidence of associations between volume or tonicity of fluids or the rate of change in serum glucose and risk of CE indicates that this might be the result of altered renal Na handling due to cerebral injury, rather than the type or rate of fluid administration.2,47,49,65,71
In the 61 children with CE from a North American multicenter study, worse outcomes occurred in those with greater neurologic depression at the time of diagnosis of CE and with higher initial serum urea nitrogen concentrations.50,72 Intubation and hyperventilation to a pCO2 of <22 mmHg also was associated with worse outcome, dictating the need for caution in hyperventilating patients.2,72
Mechanisms
CE refers to an increase of cerebral tissue water causing an increase of tissue volume.73 The edema may be vasogenic, due to breakdown of the blood-brain barrier, such as around a tumor or with trauma; cytotoxic, from poisoning or metabolic derangement; or osmotic, as occurs with hyponatremia. Neither the cause nor the location of the fluid in the swollen brains of children with DKA is known. The mechanism is likely to be complex, and it may not be the same in all affected individuals, as reflected by the time of onset and the brain imaging findings. The time of onset is distributed in a bimodal fashion, with approximately two-thirds of patients developing signs and symptoms in the first 6–7 hours and the rest from 10–24 hours after the start of treatment, with the early-onset individuals tending to be younger.50,71,72
Initial brain CT undertaken 2–44 hours after the diagnosis of CE found 39% with no acute abnormalities visible, and this did not differ significantly between early and late-onset subjects. Twenty-six percent had diffuse edema, which was also similar between early and late onset. Three of the eight with diffuse edema also had hemorrhages, and 17% of the entire group had only subarachnoid or intraventricular hemorrhage. Five subjects had focal brain injury in the mesial basal ganglia and thalamus, the periaqueductal gray matter, and the dorsal pontine nuclei (22%). These localized injuries were only in the early-onset patients. They were not an artifact of the time that the studies were performed, and thus, truly reflect what appear to be widely varying pathology in the brain leading to the syndrome that is referred to as “idiopathic CE”.71 The observation that approximately 40% of initial brain imaging studies in children who have CE are normal emphasizes that CE is a clinical diagnosis requiring the initiation of treatment before imaging studies are undertaken.
Monitoring
Because treatment modification has not prevented CE and with the strong evidence that early administration of mannitol prevents brain damage and death from this complication, Muir et al. developed a model for early detection.71 Diagnostic criteria were abnormal motor or verbal response to pain; decorticate or decerebrate posture; cranial nerve palsy (especially III, IV, VI); and abnormal neurogenic respiratory pattern (eg, grunting, tachypnea, Cheyne-Stokes, apneustic). Major criteria were altered mentation/fluctuating level of consciousness; sustained heart rate deceleration (decline more than 20 per minute) not attributable to improved intravascular volume or sleep state; and age-inappropriate incontinence. Minor criteria were vomiting following initial treatment and its cessation, if present at admission; headache (recurrent and more severe than on admission); lethargy or not easily aroused from sleep; diastolic blood pressure greater than 90 mmHg; and age <5 years. The system allowed 92% sensitivity and 96% specificity for the recognition of CE early enough for intervention, using one diagnostic criterion, two major criteria, or one major plus two minor criteria. This means that five youngsters will be treated for CE to prevent brain damage or death in one patient, a reasonable proposition considering the alternative of waiting for more stringent criteria to be met.
Treatment
Improvement has been consistently observed with administration of IV mannitol in a dosage of 1.0 g/kg over 20 minutes with repeat as necessary in 1–2 hours and associated measures as soon as CE is suspected, especially before respiratory arrest.2,49,50,65 Intervention includes reduction in the rate of fluid administration and elevation of the head of the bed. Although early intervention with mannitol treatment has improved outcome, it is difficult to determine to what degree, because increased recognition of the problem has undoubtedly led to less stringent case definition. The application of criteria developed by Muir et al., when applied to a series of 69 consecutive cases thought to be uncomplicated and experiencing full recovery, yielded three (4.3%) who would have been appropriately treated with mannitol according to these criteria.71 This is remarkably close to the percentage of DKA patients treated as CE in a recent report of 18 years experience.76
Mannitol lowers the hematocrit and blood viscosity, improving cerebral blood flow (CBF) and oxygenation, red cell deformability, and vasoconstriction in areas of the brain with intact autoregulation. It may also improve autoregulation from the effects on CBF and oxygenation, and has direct osmotic effects with reduction in extracellular free water. Intensivists frequently express concern about the use of mannitol because of their experience with the risk of rebound edema and renal failure when mannitol is used over an extended period. However, there are no reports of complications of mannitol use in this acute situation.
Hypertonic saline (HS) has become the standard for treating acute intracranial hypertension in head injury and following surgical procedures in the supratentorial region and in these circumstances has been considered at least as effective as mannitol.75–79 HS for treatment of CE in DKA was initially described in a 13-year-old female patient who developed a severe headache 20 minutes after the start of treatment and who had CT evidence of diffuse CE with transtentorial herniation. She continued to deteriorate despite mannitol treatment and was given 5 mL/kg of 3% saline rapidly; she awoke with recovered neurologic function within 5 minutes.80 The use of 5–10 mL/kg 3% saline in patients who have not responded adequately to mannitol infusion of a dose of 1 g/kg appears justified.
Intubation should be reserved for those with respiratory compromise, but should not be undertaken simply because of the diagnosis of CE. Aggressive hyperventilation was a significant risk factor for poor outcome in the study of Marcin et al.,72 similar to detrimental effects reported in head trauma and high-altitude exposure.81,82
Intracerebral Complications other than CE
Approximately 10% of all instances of neurologic collapse during ketoacidosis can be attributed to intracerebral complications, with or without associated edema, but by definition not idiopathic CE.2 The causes include subarachnoid hemorrhage,49 basilar artery thrombosis,49 cerebral venous thrombosis,83,85 meningoencephalitis,86 and disseminated intravascular coagulation.87,88
Prevention of DKA
Prevention of DKA at onset is most dramatically demonstrated when early diagnosis is made through genetic and immunologic screening of high-risk children.7,15 For the general population, an example has been provided by the Italian School and Physician Awareness Program directed at 6–14-year-olds, which reduced the rates of new-onset DKA from 78% to nearly 0% over 6 years10 Materials used in this effort are available online.89
In the 1970s, a comprehensive approach involving outreach clinics, frequent routine and emergency telephone contact, and a camping program supported by state funding for children with special healthcare needs dramatically reduced recurrent DKA episodes. Private patients in the program had a reduction in hospital admission days from preintervention of 2.8/patient/year to 0.3 and in the second year to 0. The children sponsored by the state program had a reduction from 4.9/patient/year to 1.8 and in the second year to 0.9.15
Patients with compliance problems account for a disproportionate number of recurrent DKA episodes. In the UK surveillance study, 4.8% of patients accounted for 22.5% of all episodes68 and as noted above, 20% of patients in Colorado accounted for 80% of recurrent DKA episodes.14 The principal immediate reason for the recurrent DKA in children and adolescents is insulin omission, reflected in low or absent levels of free insulin.27 The necessity for assuring administration of insulin by responsible adults is critical.
Conclusion
DKA is the result of absolute or relative deficiency of insulin in combination with exuberant secretion of counter-regulatory hormones (glucagon, catecholamines, cortisol, growth hormone) resulting in blockade of glucose utilization in insulin-sensitive tissues (liver, fat, muscle) and a cascade of derangement progressing to ketoacidosis and dehydration. Much new-onset DKA can be prevented by increased awareness of early signs and symptoms of diabetes and, in principle, almost all recurrent DKA is preventable by informed sick day management, recognition of psychosocial problems leading to insulin omission, and careful monitoring of insulin pump function.
Management of DKA should occur in centers with treatment experience and monitoring capability. Fluid replacement begins with 0.9% saline to restore circulation and subsequent 0.45% saline for maintenance and replacement of 5%–10% dehydration, according to severity indicators. Whether levels of serum K are normal or elevated, there will be a total K deficit that must be dealt with early and sufficiently. Bicarbonate administration is neither necessary nor safe. Mannitol or hypertonic saline should be at the bedside, for rapid intervention as indicated for CE, the most common serious complication of DKA. Other complications include hypokalemia, hypophosphatemia, hypoglycemia, intracerebral complications other than CE, peripheral venous thrombosis, mucormycosis, aspiration pneumonia, rhabdomyolysis, acute pancreatitis, and acute renal failure. Among questions that need to be addressed regarding DKA in children and adolescents are whether feasible and cost-effective methods can reduce the frequency of new-onset and recurrent DKA, what characterizes optimal fluid replacement therapy, how effective proposed monitoring and intervention criteria are in the reduction of CE morbidity and mortality, and whether hypertonic saline is as or more effective than mannitol in the treatment of CE.
Key points
-
Treatment of DKA requires first and foremost fluid resuscitation with 0.9% saline, followed by replacement for 5%–10% dehydration, depending on severity, and maintenance with 0.45% saline, and early and adequate K replacement.
-
Insulin should not be given as a bolus, but infused after the initial fluid resuscitation in a dose of 0.1 U/kg of body weight/hour.
-
Bicarbonate infusion is contraindicated in pediatric DKA.
-
CE is the most common serious complication, requiring careful neurologic and vital sign monitoring and early intervention with mannitol or hypertonic saline infusion.
References
American Diabetes Association. Position statement: hyperglycemic crises in patients with diabetes mellitus. Diabetes Care. 2001;24:S83–S90.
Wolfsdorf J, Craig ME, Daneman D, et al. Diabetic ketoacidosis in children and adolescents with diabetes. Pediatr Diabetes. 2009;10(Suppl. 12):118–133.
Burge MR, Hardy KJ, Schade DS. Short-term fasting is a mechanism for the development of euglycemic ketoacidosis during periods of insulin deficiency. J Clin Endocrinol Metab. 1993;76:1192–1198.
Pinkey JH, Bingley PJ, Sawtell PA, Dunger DB, Gale EA. Presentation and progress of childhood diabetes mellitus: a prospective population-based study. The Bart’s-Oxford Study Group. Diabetologia. 1994;37:70–74.
Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN. Hyperglycemic crises in adult patients with diabetes. Diabetes Care. 2009;32:1335–1343.
Foster DW, McGarry JD. The metabolic derangements and treatment of diabetic ketoacidosis. N Engl J Med. 1983;309:159–169.
Lévy-Marchal C, Patterson CC, Green A, on behalf of the EURODIAB ACE study group. Geographical variation of presentation at diagnosis of type 1 diabetes in children: the EURODIAB Study. Diabetologia. 2001;44(Suppl. 3):B75–B80.
Bui TP, Werther GA, Cameron EJ. Trends in diabetic ketoacidosis in childhood and adolescence: a 15-yr experience. Pediatr Diabetes. 2002;3:82–88.
Jackson W, Hoffman PL, Robinson EM, Elliot RB, Pilcher CC, Cutfield WS. The changing presentation of children with newly diagnosed type 1 diabetes mellitus. Pediatr Diabetes. 2001;2:154–159.
Vanelli M, Chiari G, Ghizzoni L, Costi G, Giacalone T, Chiarelli F. Effectiveness of a prevention program for diabetic ketoacidosis in children. An 8-year study in schools and private practices. Diabetes Care. 1999;22:7–9.
Mallare JT, Cordice CC, Ryan BA, Carey DE, Kreitzer PM, Frank GR. Identifying risk factors for the development of diabetic ketoacidosis in new onset type 1 diabetes mellitus. Clin Pediatr. 2003;42:591–597.
Maniatis AK, Goehrig SH, Gao D, Rewers A, Walravens P, Klingensmith G. Increased incidence and severity of diabetic ketoacidosis among uninsured children with newly diagnosed type 1 diabetes mellitus. Pediatr Diabetes. 2005;6:79–83.
Neu A, Willasch A, Ehehalt S, Hub R, Ranke MB, on behalf of the DIARY group Baden-Wuerttemberg. Ketoacidosis at onset of type 1 diabetes mellitus in children — frequency and clinical presentation. Pediatr Diabetes. 2003;4:77–80.
Roche EF, Menon A, Gill D, Hoey H. Clinical presentation of type 1 diabetes. Pediatr Diabetes. 2005;6:75–78.
Giordano B, Rosenbloom AL, Heller DR, Weber FT, Gonzales R, Grgic A. Regional services for children and youth with diabetes. Pediatrics. 1977;60:492–498.
Golden MP, Herrold AJ, Orr DP. An approach to prevention of recurrent diabetic ketoacidosis in the pediatric population. J Pediatr. 1985;107:195–200.
Rosenbloom AL. Hyperglycemic hyperosmolar state: an emerging pediatric problem. J Pediatr. 2010;156:180–184.
McDonnell CM, Pedreira CC, Vadamalayan B, Cameron FJ, Werther GA. Diabetic ketoacidosis, hyperosmolarity and hypernatremia: are high-carbohydrate drinks worsening initial presentation? Pediatr Diabetes. 2005;6:90–94.
Morales A, Rosenbloom AL. Death caused by hyperglycemic hyperosmolar state at the onset of type 2 diabetes. J Pediatr. 2002;144:270–273.
Canarie MF, Bogue CW, Banasiak KJ, Weinzimer SA, Tamborlane WV. Decompensated hyperglycemic hyperosmolarity without significant ketoacidosis in the adolescent and young adult population. J Pediatr Endocrinol Metab. 2007;20:1115–1124.
Koves IH, Neutze J, Donath S, et al. The accuracy of clinical assessment of dehydration during diabetic ketoacidosis in childhood. Diabetes Care. 2004;27:2485–2487.
Fagan MJ, Avner J, Khine H. Initial fluid resuscitation for patients with diabetic ketoacidosis: how dry are they? Clin Pediatr. 2008;47:851–855.
Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2:81–84.
Malone JI, Brodsky SJ. The value of electrocardiogram monitoring in diabetic ketoacidosis. Diabetes Care. 1980;3:543–547.
Soler NG, Bennett MA, Fitzgerald MG, Malins JM. Electrocardiogram as a guide to potassium replacement in diabetic ketoacidosis. Diabetes. 1974;23:610–615.
Schatz DA, Rosenbloom AL. Diabetic ketoacidosis: management tactics in young patients. J Crit Illness. 1988;3:29–41.
Malone JI, Root AW. Plasma free insulin concentrations: keystone to effective management of diabetes mellitus in children. J Pediatr. 1981;99:862–867.
Rosenbloom AL, Hanas R. Diabetic ketoacidosis (DKA): treatment guidelines. Clin Pediatr. 1996;35:261–266.
Bonadio WA, Gutzeit MF, Losek JD, Smith DS. Outpatient management of diabetic ketoacidosis. Am J Dis Child. 1988;142:448–450.
Linares MY, Schunk JE, Lindsay R. Laboratory presentation in diabetic ketoacidosis and duration of therapy. Pediatr Emerg Care. 1996;12:147–151.
Chase HP, Garg SK, Jelley DH. Diabetic ketoacidosis in children and the role of outpatient management. Pediatr Rev. 1990;11:297–304.
Rosenbloom AL, Schatz DA. Diabetic ketoacidosis in childhood. Pediatr Ann. 1994;23:284–288.
Katz MA. Hyperglycemia-induced hyponatremia — calculation of expected serum sodium depression. N Engl J Med. 1973;289:843–844.
Hillier TA, Abbott RD, Barrett EJ. Hyponatremia: evaluating the correction factor for hyperglycemia. Am J Med. 1999;106:399–403.
Adrogue HJ, Eknoyan G, Suki WK. Diabetic ketoacidosis: role of the kidney in the acid-base homeostasis re-evaluated. Kidney Int. 1984;25:591–598.
Oh MS, Carroll HJ, Uribarri J. Mechanism of normochloremic and hyperchloremic acidosis in diabetic ketoacidosis. Nephron. 1990;54:1–6.
Poirier MP, Greer D, Satin-Smith M. A prospective study of the “two bag system” in diabetic ketoacidosis management. Clin Pediatr. 2004;43:809–813.
Rosenbloom AL, Ongley JP. Serum calcium phosphorus and magnesium decrement during oral glucose tolerance testing: alteration in pre clinical and overt diabetes mellitus in childhood. In: Catin M, Selig M, eds. Magnesium in Health and Disease. New York: Spectrum Publishers Inc.; 1980:297–304.
Edge J, Jakes R, Roy Y, et al. The UK case-control study of cerebral oedema complicating diabetic ketoacidosis in children. Diabetologia. 2006;49:2002–2009.
Fisher J, Shahshahani M, Kitabch A. Diabetic ketoacidosis: low dose insulin therapy by various routes. N Engl J Med. 1977;297:238–243.
Sacks HS, Shahshahani M, Kitabchi, AE, Fisher JN, Young RT. Similar responsiveness of diabetic ketoacidosis to low-dose insulin by intramuscular injection and albumin-free infusion. Ann Intern Med. 1979;90:36–42.
Umpierrez GE, Latif K, Stoever J, et al. Efficacy of subcutaneous insulin lispro versus continuous IV regular insulin for the treatment of patients with diabetic ketoacidosis. Am J Med. 2004;117:291–296.
Umpierrez GE, Cuervo R, Karabell A, Latif K, Freire AX, Kitabchi AE. Treatment of diabetic ketoacidosis with subcutaneous insulin aspart. Diabetes Care. 2004;27:1873–1878.
Della Manna T, Steinmetz L, Campos PR, et al. subcutaneous use of a fast-acting insulin analog: an alternative treatment for pediatric patients with diabetic ketoacidosis. Diabetes Care. 2005;28:1856–1861.
Levitsky L, Ekwo E, Goselink CA, Solomon EL, Aceto T. Death from diabetes (DM) in hospitalized children (1970–1998). Pediatr Res. 1991;29: Abstract A195.
Curtis JR, To T, Muirhead S, Cummings E, Daneman D. Recent trends in hospitalization for diabetic ketoacidosis in Ontario children. Diabetes Care. 2002;25:1591–1596.
Lawrence SE, Cummings EA, Gaboury I, Daneman D. Population-based study of incidence and risk factors for cerebral edema in pediatric diabetic ketoacidosis. J Pediatr. 2005;146:688–692.
Edge JA, Ford-Adams ME, Dunger DB. Causes of death in children with insulin-dependent diabetes 1990–96. Arch Dis Child. 1999;81:318–323.
Rosenbloom AL. Intracerebral crises during treatment of diabetic ketoacidosis. Diabetes Care. 1990;13:22–33.
Glaser N, Barnett P, McCaslin I, et al. Risk factors for cerebral edema in children with diabetic ketoacidosis. N Engl J Med. 2001;344:264–269.
Zipf WB, Bacon GE, Spencer ML, Kelch RP, Hopwood NJ, Hawker CD. Hypocalcemia, hypomagnesemia, and transient hypoparathyroidism during therapy with potassium phosphate in diabetic ketoacidosis. Diabetes Care. 1979;2:265–268.
Winter RJ, Harris CJ, Phillips LS, Green OC. Diabetic ketoacidosis. Induction of hypocalcemia and hypomagnesemia by phosphate therapy. Am J Med. 1979;67:897–900.
Gutierrez JA, Bagatell R, Samson MP, Theodorou AA, Berg RA. Femoral central venous catheter-associated deep venous thrombosis in children with diabetic ketoacidosis. Crit Care Med. 2003;31:80–83.
Worly JM, Fortenberry JD, Hansen I, Chambliss CR, Stockwell J. Deep venous thrombosis in children with diabetic ketoacidosis and femoral central venous catheters. Pediatrics. 2004;113:57–60.
Zeitler P, Thiede A, Muller HL. Prospective study on plasma clotting parameters in diabetic children — no evidence for specific changes in coagulation system. Exp Clin Endorinol Diabetes. 2001;109:146–152.
Carl CF, Hoffman WH, Passmore GG, et al. Diabetic ketoacidosis promotes a prothrombotic state. Endo Res. 2003;29:73–82.
De Stefano V, Rossi E, Paciaroni K, Leone G. Screening for inherited thrombophilia: indications and therapeutic implications. Haematologica. 2002;87:1095–1108.
Haller MJ, Valladares A, Rosenbloom AL. Arterial thrombosis resulting in amputation in a child with poorly controlled type 1 diabetes and heterozygous factor V Leiden mutation. Pediatr Diabetes. 2006;7:229–231.
Haddad NG, Croffie JM, Rugster EA. Pancreatic enzyme elevations in children with diabetic ketoacidosis. J Pediatr. 2004;145:122–124.
Casteels, K, Beckers D, Wouters C, Van Geet C. Rhabdomyolysis in diabetic ketoacidosis. Pediatr Diabetes. 2003;4:29–31.
Buckingham BA, Roe TF, Yoon J-W. Rhabdomyolysis in diabetic ketoacidosis. Am J Dis Child. 1981;135:352–354.
Moye J, Rosenbloom AL, Silverstein J. Clinical predictors of mucormycosis in type 1 diabetes in children. J Ped Endocrinol Metab. 2002;15:1001–1004.
Young E, Bradley RF. Cerebral edema with irreversible coma in severe diabetic ketoacidosis. N Engl J Med. 1967;276:665–669.
Rosenbloom AL, Riley WJ, Weber FT, Malone JI, Donnelly WH. Cerebral edema complicating diabetic ketoacidosis in childhood. J Pediatr. 1980;96;357–361.
Bello FA, Sotos JF. Cerebral oedema in diabetic ketoacidosis in children. Lancet. 1990;336;64.
Brown TB. Cerebral oedema in childhood diabetic ketoacidosis: is treatment a factor? Emerg Med J. 2004;21:141–144.
Azzopardi J, Gatt A, Zammit A, Albert G. Lack of evidence of cerebral edema in adults treated for diabetic ketoacidosis with fluids of different tonicity. Diabetes Res Clin Practice. 2002;57:87–92.
Edge JA, Hawkins MM, Winter DL, Dunger DB. The risk and outcome of cerebral oedema developing during diabetic ketoacidosis. Arch Dis Child. 2001;85:16–22.
Dunger DB, Edge JA. Predicting cerebral edema during diabetic ketoacidosis. N Engl J Med. 2001;344:302–303.
Carroll P, Matz R. Uncontrolled diabetes mellitus in adults: experience in treating diabetic ketoacidosis and hyperosmolar nonketotic coma with low dose insulin and a uniform treatment regimen. Diabetes Care. 1983;6:579–585.
Muir AB, Quisling RG, Yang MCK, Rosenbloom AL. Cerebral edema in childhood diabetic ketoacidosis. Natural history, radiographic findings, and early identification. Diabetes Care. 2004;27:1541–1546.
Marcin JP, Glaser N, Barnett P, et al. Factors associated with adverse outcomes in children with diabetic ketoacidosis-related cerebral edema. J Pediatr. 2002;141:793–797.
Pappius HM. Fundamental aspects of brain edema. In: Vinkin PJ, Bruyn GW, eds. Handbook of Clinical Neurology. Volume 16. Part 1. Tumors of the Brain and Skull. Amsterdam: North Holland Publishing Co.; 1974:167–185.
Fiordalisi I, Novotny WE, Holbert D, Finberg L, Harris, GD. Critical Care Management Group. An 18-yr prospective study of pediatric diabetic ketoacidosis: an approach to minimizing the risk of brain herniation during treatment. Pediatr Diabetes. 2007;8:142–149.
Freshman SP, Battistella FD, Matteucci M, Wisner DH. Hypertonic saline (7.5%) versus mannitol: a comparison for treatment of acute head injuries. J Trauma. 1993;35:344–348.
Berger S, Schurer L, Hartl R, Messmer K, Baethmann A. Reduction of post-traumatic intracranial hypertension by hypertonic/hyperoncotic saline/dextran and hypertonic mannitol. Neurosurgery. 1995:37:98–107.
Gemma M, Cozzi S, Tommasino C, et al. 7.5% hypertonic saline versus 20% mannitol during elective neurosurgical supratentorial procedures. J Neurosurg Anesthesiol. 1997:9:329–334.
Worthley LI, Cooper DJ, Jones N. Treatment of resistant intracranial hypertension with hypertonic saline. Report of two cases. J Neurosurg. 1988:68:478–481.
Qureshi AL, Suarez JI. Use of hypertonic saline solutions in treatment of cerebral edema and intracranial hypertension. Crit Care Med. 2000:28:3301–3313.
Curtis JR, Bohn D, Daneman D. Use of hypertonic saline in the treatment of cerebral edema in diabetic ketoacidosis (DKA). Pediatr Diabetes. 2001;2:191–194.
Shapiro HM, Drummond JC. Neurosurgical anesthesia. In: Miller RD (ed). Anesthesia. 3rd edition. New York: Churchill Livingstone; 1990:1913–1914.
Muizelaar JP, Marmarou A, Ward JD, et al. Adverse effects of prolonged hyperventilation in patients with severe head injury: a randomized clinical trial. J Neurosurg. 1991;75:731–739.
Roberts MD, Slover RH, Chase HP. Diabetic ketoacidosis with intracerebral complications. Pediatr Diabetes. 2001;2:109–114.
Keane S, Gallagher A, Aykroyd S, McShane MA, Edge JA. Cerebral venous thrombosis during diabetic ketoacidosis. Arch Dis Child. 2002;86:204–206.
Rosenbloom AL. Fatal cerebral infarctions in diabetic ketoacidosis in a child with previously unknown heterozygosity for factor V Leiden deficiency. J Pediatr. 2004;145:561–562.
Yoon J-W, Austin M, Onodera T, Notkins AL. Virus-induced diabetes mellitus. N Engl J Med. 1979;300:1173–1179.
Cooper RM. Turner RA, Hutaff L, Prichard R. Diabetic keto-acidosis complicated by disseminated intravascular coagulation. South Med J. 1973;66:653–657.
Bonfanti R, Bognetti E, Meschi F, Medaglini S, D’Angelo A, Chiumello G. Disseminated intravascular coagulation and severe peripheral neuropathy complicating ketoacidosis in a newly diagnosed diabetic child. Acta Diabetol. 1994;31:173–174.
World Diabetes Day. DKA Awareness. Available at: http://www.worlddiabetesday.org/DKA. Accessed December 1, 2010.
Acknowledgments
The author has no conflicts of interest to declare. A.L.R. is the guarantor for this article, and takes responsibility for the integrity of the work as a whole.
Open Access. This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is published with open access at Springerlink.com
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Rosenbloom, A.L. The management of diabetic ketoacidosis in children. Diab Ther 1, 103–120 (2010). https://doi.org/10.1007/s13300-010-0008-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-010-0008-2