
Abstract
Modern taxonomy has developed towards the establishment of global authoritative lists of species that assume the standardized principles of species recognition, at least in a given taxonomic group. However, in fungi, species delimitation is frequently subjective because it depends on the choice of a species concept and the criteria selected by a taxonomist. Contrary to it, identification of fungal species is expected to be accurate and precise because it should predict the properties that are required for applications or that are relevant in pathology. The industrial and plant-beneficial fungi from the genus Trichoderma (Hypocreales) offer a suitable model to address this collision between species delimitation and species identification. A few decades ago, Trichoderma diversity was limited to a few dozen species. The introduction of molecular evolutionary methods resulted in the exponential expansion of Trichoderma taxonomy, with up to 50 new species recognized per year. Here, we have reviewed the genus-wide taxonomy of Trichoderma and compiled a complete inventory of all Trichoderma species and DNA barcoding material deposited in public databases (the inventory is available at the website of the International Subcommission on Taxonomy of Trichoderma www.trichoderma.info). Among the 375 species with valid names as of July 2020, 361 (96%) have been cultivated in vitro and DNA barcoded. Thus, we have developed a protocol for molecular identification of Trichoderma that requires analysis of the three DNA barcodes (ITS, tef1, and rpb2), and it is supported by online tools that are available on www.trichokey.info. We then used all the whole-genome sequenced (WGS) Trichoderma strains that are available in public databases to provide versatile practical examples of molecular identification, reveal shortcomings, and discuss possible ambiguities. Based on the Trichoderma example, this study shows why the identification of a fungal species is an intricate and laborious task that requires a background in mycology, molecular biological skills, training in molecular evolutionary analysis, and knowledge of taxonomic literature. We provide an in-depth discussion of species concepts that are applied in Trichoderma taxonomy, and conclude that these fungi are particularly suitable for the implementation of a polyphasic approach that was first introduced in Trichoderma taxonomy by John Bissett (1948–2020), whose work inspired the current study. We also propose a regulatory and unifying role of international commissions on the taxonomy of particular fungal groups. An important outcome of this work is the demonstration of an urgent need for cooperation between Trichoderma researchers to get prepared to the efficient use of the upcoming wave of Trichoderma genomic data.
Similar content being viewed by others


Introduction into the predicament of Trichoderma identification
Fungi are ubiquitous. They penetrate their environment and impact multiple facets of human life, ranging from biotechnology, phytopathology, and medicine to biodiversity conservation (Hyde et al. 2019). Precise identification of fungi is required for all mycological investigations and applications. It allows us to predict beneficial or pathogenic properties of individual fungal strains, monitor their distribution, and establish safety measures. The recent introduction of DNA Barcoding in fungal identification has significantly improved species identification and reduced the associated labor (Schoch et al. 2012; Vu et al. 2019). However, the precision of fungal identification is frequently impeded by development of the underlying taxonomy (Lücking et al. 2020).
Taxonomy, which is naming, classifying, and describing living organisms based on the similarity of their characteristics and evolutionary history, is not an exact science (Garnett et al. 2020; Lücking et al. 2020; Schoch et al. 2020). Different groups of organisms are classified based on their specific characteristics and their role in the ecosystem (see below). These differences can apply even to related organisms that have unique lifestyles (such as obligate biotrophs or saprotrophs) that are considered in species delimitation. Fungal species can be frequently delimitated by expert taxonomists, other fungal researchers, and amateurs. Although they all will provide sufficient material for the formal taxonomic descriptions, the taxonomic approaches will not be the same (Fontaine et al. 2012; Garnett et al. 2020). Expert taxonomists can represent different schools and generations, and thus, they will use unequal approaches and methodologies. Therefore, no nomenclatural codes can specify the criteria that were used to recognize taxa. Zoologists have recently proposed the establishment of global species lists that should be based on universal principles of science, transparency, and political compliance (Garnett et al. 2020). They specified the key role of taxonomic communities in consolidation of such a list and taxa approval/rejection. The implementation of such high-level taxonomic regulations supported by stakeholders (taxonomy users) can consolidate expert groups.
In fungi, which comprise one of the most diverse group of eukaryotes with the predicted diversity of several million species (Choi and Kim 2017; Hawksworth and Lücking 2017), the unification of taxonomic criteria is impeded by the scarcity of fossils, irregular lifecycles, and relative morphological simplicity. Species delimitation is hindered by the difficulties of defining boundaries of individual fungal organisms or populations, diminutive bodies that develop inside of a substrate, and exceptional metabolic and ecological plasticity for which observation may be hampered. Therefore, DNA-based techniques allowed a virtual restart of fungal taxonomy based on the new level of precision (Lücking et al. 2020), and unprecedented success with unification and standardization was achieved (Taylor 2011; May et al. 2019). Molecular techniques also led to discovery of the hidden fungal diversity and fueled the ongoing debate on the classification and naming rules for the fungal “dark taxa” that are only known from their DNA sequences and have attracted great attention of fungal taxonomists (Nilsson et al. 2019). The main consequence of the new methodology is probably not the taxonomic criteria unification but the sharp increase in the number of taxa (of all ranks) among known fungal groups (Taylor et al. 2000; Hawksworth and Lücking 2017). Numerous genera of common and industrially or agriculturally important fungi such as Penicillium and Aspergillus (Houbraken and Samson 2011; Sklenar et al. 2017; Steenwyk et al. 2019; Houbraken et al. 2020) have been recently taxonomically revised, and ample species combinations were proposed within previous species complexes or clades. Recognition of more species is considered to be a useful practice because it leads to the accurate and precise diagnosis of potential pathogens, prediction of beneficial properties, and an improved overall understanding of fungal diversity and ecology (Hyde et al. 2019; Bajpai et al. 2019). However, because the identifiability of new taxa (Box 1) is not always evaluated, even well-studied groups of fungi can rapidly move from the rear of fungal taxonomy to its frontline.
Trichoderma as a suitable model for integrative fungal taxonomy
Ubiquitous mycotrophic and phytosaprotrophic fungi from the genus Trichoderma (syn. Hypocrea, Hypocreales) have been known to mycologists from the beginning of the formal taxonomic records for fungi from the late 18th century (see Persoon 1794). For 200 years, investigation of Trichoderma (and Hypocrea) developed with the pace of all mycology, and it was mainly based on investigation of its teleomorphic stage Hypocrea [the name is not in use, (Taylor 2011; Rossman et al. 2013)] that is tractable in the scientific literature (reviewed elsewhere, for example in Rossman et al. 2013; Jaklitsch and Voglmayr 2013). In the mid-20th century, only a few species (or “species aggregates”) of Trichoderma were proposed (Rifai 1969). However, similar to other common fungi, the last two decades sharply transformed Trichoderma to the species-rich genus (Druzhinina et al. 2006; Kubicek et al. 2008; Jaklitsch 2009, 2011; Atanasova et al. 2013; Bissett et al. 2015) that made it comparable to such fungi as Fusarium (Hypocreales), Aspergillus, or Penicillium (Eurotiales) and left all sister hypocrealean or even the model genus for fungal biology Neurospora (Sordariales) far behind (Fig. 1). The increase in the total number of Trichoderma species was not strongly influenced by the general mycological movement “One fungus—one name” (Taylor 2011), as the connection with the single Hypocrea teleomorph (with only a few exceptions) has been established earlier and considered in the first species counts (Druzhinina et al. 2006; Atanasova et al. 2013). In addition to the unprecedented effort of Trichoderma taxonomists (see below), the drastic increase in Trichoderma species number has several explanations that are related to the technologies and applications. The first reason is the emerging importance of Trichoderma for humankind. Approximately 50 years ago, T. reesei was recognized as a highly efficient producer of plant biomass-degrading enzymes for biofuel and other industries. A couple of decades later, several other species (T. atroviride, T. virens, T. harzianum, and others) were proposed as potent bioeffectors for plant protection (biofungicides) and plant growth promotion (biofertilizers) (reviewed by Harman et al. 2004, Druzhinina et al. 2011 and others), and they are now widely used for biological control of fungal pests in sustainable agriculture (biocontrol). Trichoderma was also documented as the causative agent of the green mold disease on mushroom farms (Komoń-Zelazowska et al. 2007) and as an opportunistic pathogen in humans (Sandoval-Denis et al. 2014). This resulted in the rapid increase of scientific publications based on Trichoderma species (Fig. 1). The second reason that ultimately contributed to the sudden increase in the species number is the use of either phylogenetic (PSR, Box 1) or the genealogical concordance phylogenetic species recognition (GCPSR, Box 1) concepts and DNA Barcoding techniques in Trichoderma taxonomy and the subsequent modification of the criteria for species delimitation. Before the introduction of DNA Barcoding, Trichoderma species were recognized based on their morphology and growth characteristics. However, the introduction of molecular methods and, in particular, the extensive use of GCPSR (Box 1) resulted in the recognition of several hundred Trichoderma species (reviewed in Druzhinina et al. 2006; Atanasova et al. 2013) many of which were delimitated within previously existing species complexes or clades. Although the applications are still restricted to a few species, the growth of species richness positively influences the Trichoderma science development as the number of Trichoderma-based publications grows proportionally to it (Fig. 1).

Research interest to Trichoderma spp. as of July 2020. a The number of records in PubMed Central for the key word “Trichoderma” compared to other fungi with noticeable importance for humankind such as plant pathogens, industrial producers, and research model organisms. b Trends in research interest over last 100 years for Trichoderma compared to Neurospora, Aspergillus, and Fusarium. c The number of records in IndexFugorum. d The relationship between the number of species described per year and the number of Trichoderma-based research articles recorded in PubMed Central. e Research interest for Trichoderma in different countries, which is estimated as the number of publications and affiliations (including joint studies)
Another striking property of Trichoderma that makes it a useful model of taxonomic studies is the evident lack of hidden diversity or “dark Trichoderma species” (Migheli et al. 2009; Friedl and Druzhinina 2012; Hagn et al. 2007; Meincke et al. 2010; López-Quintero et al. 2013; Röhrich et al. 2014; Jaklitsch 2009, 2011; Jaklitsch and Voglmayr 2015), meaning that most or all species can be successfully cultivated in vitro. Therefore, Trichoderma spp. can potentially be extensively phenotypically and physiologically characterized along with taxonomic or nomenclatural acts (Samuels et al. 2006, 2012; Druzhinina et al. 2010b; Chaverri et al. 2015; Bissett et al. 2015). The possibility of the extended ecophysiological profiling paves the way for the introduction of the integrative (polyphasic) taxonomy for species delimitation, i.e., the combination of genealogy (phylogeny), phenotype (including autecology), and reproductive biology (when feasible) (Lücking et al. 2020). The analysis of a relatively large number of whole-genome sequences (WGS) for Trichoderma spp. (see below) also provided insights into the evolutionary timeline of this genus (Druzhinina et al. 2018; Kubicek et al. 2019). Thus, Trichoderma can serve as a useful model for the observation of taxonomic development with an impact on the precision, accuracy, and ambiguity of species delimitation and subsequent identification.
The challenge and the aim: identification of Trichoderma species
To address the current state of Trichoderma identifiability at the species level, we invited researchers working with these fungi to perform an exercise on DNA Barcoding. The respondents were offered an anonymous online survey where they could insert their identification results along with the description of the identification procedure, their experience in the area, and comments. For this test, we picked two unpublished Trichoderma strains that had sequences of DNA barcoding loci that were similar but not identical to those that were available in public databases in May 2020. Each strain was represented by a set of the three sequences (ITS, partial sequences of tef1, and rpb2 genes, respectively, see Box 1 and below) and a brief description of the habitat. No information on biogeography, morphology, or physiology was provided. As shown below, one strain belongs to T. guizhouense (TUCIM 10063, nick-named a “mycoparasite” in the survey), which is a sister species to T. harzianum (Li et al. 2013; Chaverri et al. 2015). Another strain (TUCIM 5640, nick-named an “epiphyte”) represents a putative new Trichoderma species (T. sp. TUCIM 5640), which is awaiting its formal description if additional material will become available.
The survey was completed by 47 respondents (Fig. 2). Among them, 82% described themselves as experienced Trichoderma researchers, including 15% who were also experienced in advanced DNA Barcoding of fungi (putative taxonomists). Ten (21%) replies diagnosed both strains correctly (see below), while 23 respondents (49%) failed to identify both sequences. T. guizhouense was identified correctly by 20 respondents, and the second strain was assigned to a putative new species by 14 respondents (see below). The accuracy of identification did not correlate with the experience because nearly one-half of the correct answers were given by beginners, while ten highly experienced Trichoderma scientists failed to identify both strains (Fig. 2). Similarly, time had no effect on the identification because the average time spent for the correct and incorrect answers was similar to the total average (55 min; ANOVA, P > 0.05).

Molecular identification of Trichoderma strains by experts. a The online survey results on the identification of the two unknown Trichoderma isolates based on the combination of primary (ITS) and secondary (tef1 and rpb2) DNA barcodes. The survey was completed by 47 volunteers with experience in the area. The level of their expertise was provided by the respondents. b The correctness of species identification of 42 Trichoderma isolates, for which WGS are available in public databases in July 2020. “Uncertain” correspond to strains that were deposited as Trichoderma sp.
Identification of the WGS strains provided an alternative measurement of Trichoderma species identifiability by the experts because genomes are usually deposited by researchers who specialize in this fungus. Therefore, we have assessed the identification of Trichoderma strains for which the WGSs have been available in public databases (Table 1). Among the 42 strains, two strains were deposited without species names (as Trichoderma sp. IMV 00454 and Trichoderma sp. TW21990_1), while the original identification of 15 strains (35%) was not accurate (Fig. 2, and below).
Thus, these two tests demonstrate that the accurate molecular identification of Trichoderma species is a considerable challenge for experts who do research on this fungus. It is not easy even for specialists in fungal taxonomy. The difficulties related to identification are also reflected in the fact that more than 2000 Trichoderma records in the NCBI Taxonomy Browser were deposited as “Trichoderma sp.” Identification of these 44 (2 + 42) strains also challenged our skills and triggered the study on how to identify a Trichoderma species, which is presented below.
Thus, this work addresses the problem of molecular identification of Trichoderma at the species level. We have selected the “white paper” format to provide a review of Trichoderma taxonomy and prepare the authoritative guidelines for the accurate unambiguous molecular identification of Trichoderma diversity that is recognized by the year 2020. For this, we first provided a complete inventory and a cumulative summary of Trichoderma nomenclature, and reviewed the current state of its molecular taxonomy. Second, we developed and explained the protocol for molecular identification of currently valid Trichoderma species. The comparison of ITS sequences for Trichoderma spp. and its neighboring genera allowed us to set up a similarity threshold to estimate a query strain for its possibility of being a member of the genus. We also used the variability of the two DNA barcoding markers (rpb2 and tef1, Box 1) between the currently defined species and set the numerical standards of the similarity threshold at the level at which it is sufficient for species identification for most of the existing species. We then provided practical examples of DNA Barcoding showing how the identification results can be presented and gave examples on how a new species hypothesis can be proposed. Finally, we developed recommendations for Trichoderma taxonomy providers and taxonomy users on performing diversity studies. For this, we introduced the www.trichokey.com and the www.trichoderma.info web resources that dedicated to Trichoderma taxonomy and molecular identification. We concluded that the genus Trichoderma is highly suitable for the application of the integrative (polyphasic) taxonomy based on genealogy, ecophysiology, and biogeography, which was initially proposed by John Bissett for these and other fungi (Kubicek et al. 2003; Komoń-Zelazowska et al. 2007; Hoyos-Carvajal et al. 2009), and therefore, we dedicate this work to his memory. We also proposed a regulatory and unification role of International Commissions on Taxonomy of Trichoderma (ICTT) for the approval/rejection of new species proposals.
Assumptions made in this study
In this study, we assumed that the genus Trichoderma included species that were originally described as Trichoderma (basionym) or transferred to Trichoderma from other genera (combinatio nova; comb. nov.) such as Hypocrea, Protocrea, Aphysiostroma, or Sarawakus, according to Rossman et al. (2013). We also considered all Hypocrea and Protocrea records in the NCBI Taxonomy Browser that were transferred to Trichoderma because they were consistent with the aim of this study (molecular identification of Trichoderma). However, we did not consider all species names of Hypocrea that were deposited in the Index Fungorum and Mycobank that had not been formally transferred to Trichoderma because they may be members of other hypocrealean genera (e.g., Hypomyces, Hypocrella, Moelleriella, Protocreopsis, Clintoniella, Atkinsonella, Stilbocrea, Battarrina, Podocrea, Nectriopsis, Myriogenospora, Ophiocordyceps, Arachnocrea, Dialhypocrea, Selinia, Nectria, Epichloe, and others) or unrelated taxa (Broomella, Amphisphaeria, Thuemenella, Hypoxylon, Penzigia, or Amplistroma and Plowrightia).
Here, we focused on molecular identification using in silico methods and corresponding records in public databases. In some places, we indicated instances of incomplete reference material that were deposited into public databases or revealed identifications that could have increased accuracy, precision, and ambiguity. However, we assumed that the sequences and species descriptions were correct (i.e., we ignored incorrect sequences, not incorrect identifications).
We also assumed that all formally described species complied with the requirements of the Code (May et al. 2019; Box 1) irrespective of the species criteria applied, and that the material studied must be identifiable.
The importance of the Trichoderma taxonomic history, the scope of phenotypic assessments, morphology, biogeography, ecology, chemotaxonomy, reliability, and availability of reference specimens were highly appreciated but the detailed consideration of these aspects was beyond the scope of this survey.
For the sake of easier reading, we used the short taxonomic names, i.e. avoided listing authors’ name(s) and the publication year of species names. For all species, this information is available in tables and in the accessory websites www.trichoerma.info and www.trichokey.com. Exceptions made for the case where these parts of the formal species name are discussed.
The state of Trichoderma nomenclature, taxonomy, and DNA Barcoding by the year 2020
To estimate the state of Trichoderma taxonomy, we first collected all Trichoderma names and the former Hypocrea names transferred to Trichoderma according to Rossman et al. (2013) that have been deposited in the three major taxonomic databases, which are Index Fungorum (http://www.indexfungorum.org/), Mycobank (http://www.mycobank.org/), and the NCBI Taxonomy Browser (https://www.ncbi.nlm.nih.gov/taxonomy). The cumulative list is presented in Table 2 (see the digital sortable version at https://trichokey.com/index.php/trichoderma-taxonomy-2020 and a printable version at https://trichoderma.info/trichoderma-taxonomy-2020/). It summarizes the results in which we screened Trichoderma for the names that are currently in use, names that are not in use, orthographic variants, and other synonyms. Then, for each species, we collected the records for the reference strain (holotype or ex-type specified with the original species description or its valid substitute) and recorded the distribution of DNA Barcoding markers and the total number of DNA Barcoding sequences archived per each species. The assessment of the accuracy of individual sequence attribution to a given species name was beyond the scope of this research (see “Assumptions” above), but this issue is partially addressed below.
Trichoderma nomenclature
The inventory of Trichoderma nomenclature resulted in a complete list of 464 nonredundant species epithets (Table 2). Among them, 90 names are not currently in use (Bissett et al. 2015), including 22 grammatically incorrect names (orthographic variants) that have been replaced by their corrected versions (Table 2). Several names are considered to be invalid because their description did not follow the requirements of the Code (May et al. 2019) or the deposition to public databases was not performed or was made incompletely (refer to T. cyanodichotomous nom. inval. at NCBI Taxonomy Browser as an example). The contemporary valid nomenclature of Trichoderma spp. consists of 375 species names.
The Latin names of Trichoderma spp. most commonly reflect macromorphology of the teleomorph and the culture appearance in vitro (e.g., T. viride, T. citrinum, T. citrinoviride, T. pulvinatum) or the microscopic features of the species (e.g., T. helicum, T. spirale, T. crystalligenum, T. compactum, T. oblongisporum, T. brevicompactum, T. longibrachiatum). Some names indicate the species ecology (e.g., T. psychrophilum, T. aggressivum, T. endophyticum) or the substrates and hosts (e.g., T. arenarium, T. bannaense, T. alni, T. parepimyces, T. epimyces, T. pleuroti, T. taxi). The etymology of many Trichoderma species names corresponds to the names of continents or regions (e.g., T. caribbaeum, T. sinense, T. americanum, T. sinoaustrale, T. europaeum, T. mediterraneum), famous geographic hallmarks such as mountains or river basins (e.g., T. shennongjianum, T. changbaiense, T. amazonicum, T. alpinum), or they reflect political or historical–geographical names of the sampling locations (e.g., T. aethiopicum, T. linzhiense, T. austriacum, T. britannicum, T. britdaniae, T. camerunense, T. costaricense, T. danicum, T. estonicum, T. guizhouense, T. hainanense, T. henanense, T. hispanicum, T. hongkongensis, T. hubeiense, T. istrianum, T. italicum, T. koreanum, T. moravicum, T. novae-zelandiae, T. sulawesense, T. taiwanense, T. thailandicum, T. tibetense, T. yunnanense). Naming after colleagues that contributed to Trichoderma research or the development of Trichoderma-based applications appears to be increasingly popular and appreciated, such as T. beinartii, T. bissettii, T. chetii, T. christiani, T. dingleyae, T. eijii, T. evansii, T. gamsii, T. harzianum, T. lieckfeldtiae, T. parmastoi, T. petersenii, T. priscilae, T. reesei, T. rifaii, T. rogersonii, T. samuelsii, T. simmonsii, T. voglmayrii, and others.
For cryptic species that are morphologically identical to previously described taxa, authors frequently compose Latin names by adding Greek or Latin affixes “neo-” (new) (e.g., T. neocrassum, T. neokoningii, T. neorufoides, T. neorufum, T. neosinense, T. neotropicale), “pseudo-” (false) (e.g., T. pseudobritdaniae, T. pseudocandidum, T. pseudodensum, T. pseudogelatinosum, T. pseudokoningii, T. pseudolacteum, T. pseudonigrovirens, T. pseudostramineum,), “para-” (near) (e.g., T. parareesei, T. pararogersonii, T. paratroviride, T. paraviridescens), or “-oides” (likeness) (e.g., T. asprelloides). Prefixes such as “eu-” (true), “sub-” (under), “mega-”, “megalo-” (big), “proto” (first), and “zelo” (zeal) are also used (e.g., T. eucorticioides, T. euskadiense, T. subviride, T. subeffusum, T. megalocitrinum, T. melanomagnum, T. zeloharzianum). There are no preferences for one naming strategy for Trichoderma. The etymology of each name is usually justified and explained along with the species description.
The common issue of Trichoderma nomenclature that is difficult to correct is the use of grammatically wrong formal scientific names (Table 2) (May et al. 2019). We calculated that nearly 80 Trichoderma spp. were first described using incorrect grammar. Even when corrected, such orthographic variants remain recorded in public databases as synonyms. This ultimately affects the identifiability of the species and confuses the taxonomy users. For example, in MycoBank, the orthographic variant “T. pleurotum Yu & Park (2006)” [MB#504755] is recorded as synonym of grammatically correct T. pleuroti Yu & Park (2006) [MB#546965]. Although the details on the name status appear on the page with the detailed profile of the MycoBank record, the main page for the orthographic variant MB#504755 has no indications that the name should not be used (http://www.mycobank.org/BioloMICSDetails.aspx?Rec=440470). The NCBI Taxonomy Browser does not have the incorrect orthographic variant listed, but incorrect sequence information appeared largely in the NCBI Entrez search system. For example, “Trichoderma pleurotum” was used in the definitions of at least 14 nucleotide sequences and will appear in the results of the sequence similarity search (BLAST). Although on GenBank pages such as https://www.ncbi.nlm.nih.gov/nuccore/EU279975.1, the “Source” and “Organism”, are correct (as Trichoderma pleuroti), unexperienced users may mistake the incorrect orthographic variant for species identification and, thus, unintentionally amplify the number of incorrect records. Therefore, it is highly recommended to carefully consider the grammar of the Latin language and ask the experts for the grammatic verification of a new name proposal. The San Juan Chapter F of the Code (Box 1) introduces the correctability for incorrectly cited identifiers of names and typifications (May et al. 2019).
Timeline of Trichoderma taxonomy
The current taxonomy of Trichoderma was provided by 179 mycologists who researched the diversity of this genus for the last 236 years starting from the first proposed and still-valid species name T. viride Pers. (1832) (Fig. 3) (see below). Before introducing molecular methods in fungal taxonomy, the most significant contributions were made by C. H. Persoon (11 species, http://www.indexfungorum.org/) and M. A. Rifai (six species) (Rifai 1969). J. Bissett with colleagues, worked on the edge of DNA Barcoding times (the 90's of the 20th century) and recognized 24 species, including some that have been DNA barcoded (Table 2). The most substantial contribution to Trichoderma taxonomy of all time was made by the groups of W. M. Jaklitsch [> 120 species, (for example, Atanasova et al. 2010; Jaklitsch 2009, 2011; Jaklitsch et al. 2005, 2006, 2008a, b, 2012, 2013, 2014; Jaklitsch and Voglmayr 2012, 2013, 2015)] and G. J. Samuels [> 70 species, (for example, Samuels et al. 2002, 2006, 2010, 2012; Chaverri et al. 2015; Chaverri and Samuels 2003; Lu et al. 2004; Chaverri et al. 2011)] that worked alone or collaborated with each other and such researchers as C. P. Kubicek, E. Lieckfeldt, H. Voglmayr, and P. Chaverri (Fig. 2). Most of the above-listed taxonomists except P. Chaverri have completed their research in Trichoderma diversity. Current active taxonomy providers for Trichoderma are W. Y. Zhuang and her colleagues who have named > 85 species in the last five years (for example, Chen and Zhuang 2016; Qin and Zhuang 2016a, c; Chen and Zhuang 2017a, b, c, d; Qin and Zhuang 2016b, 2017). However, the most recent species that appeared in 2020 were also described by scientists who are new to Trichoderma taxonomy (Tomah et al. 2020; Ding et al. 2020) (Table 2). Contact details for the current experts in Trichoderma taxonomy are available on the International Committee on Taxonomy of Trichoderma (ICTT) website (www.trichoderma.info), which replaces the currently unsupported www.isth.info (see below).

Development of Trichoderma taxonomy over the last 236 years. a Groups of the most significant providers of Trichoderma taxonomy. b The number of Trichoderma species introduced to IndexFungorum per year. c The total number of Trichoderma species recorded in IndexFungorum
Beginning in the late 18th century and for the first 200 years, cumulative taxonomy for Trichoderma and Hypocrea developed at a steady rate, accumulating one or two new species every two years (Fig. 2). This mainly includes the teleomorphic species that were originally described as Hypocrea spp. and were recently transferred to Trichoderma according to the contribution of Rossman et al. (2013). In the 1990s, shortly before introducing DNA Barcoding in fungal diversity research, there were almost 100 Hypocrea/Trichoderma names deposited for this genus. However, with the introduction of DNA-based techniques, molecular phylogeny, and the GCPSR concept, the number of Trichoderma basionyms started to increase exponentially, resulting in a “hockey stick” shape of the plot showing the species number against time (Fig. 3). In 2006, the first 100 species were characterized using molecular data (Druzhinina et al. 2006), which was predicted to be accounted for one-half of the total diversity at that time, while in 2013, the number of DNA barcoded species doubled to about 200 (Atanasova et al. 2013). The maximum productivity of taxonomy providers was reached between 2014 and 2017 when > 50 molecularly characterized Trichoderma species were added per year (Fig. 3). The recent growth of molecular diversity in this genus has slightly declined, but it still leads to the addition of roughly a dozen new molecularly defined Trichoderma basionyms each year.
By the year 2020, most Trichoderma species have been characterized using DNA-based techniques. We have counted only 14 currently valid names that have not been characterized molecularly because DNA extraction from old specimens failed (e.g., T. latizonatum, T. sino-australe, and T. viridiflavum) or samples were not available for this analysis (Bissett et al. 2015; Zhu and Zhuang 2014). The following eight unsequenced members of Trichoderma were transferred to this genus from Sarawakus (Hypocreales): T. hexasporum, T. fragile, T. izawae, T. rosellum, T. sordidum, T. subtrachycarpum, T. succisum, and T. trachycarpum by Jaklitsch et al. (2014) and T. brevipes from Cordyceps (Hypocreales) (Bissett et al. 2015) (Table 2). None of these species are available for DNA barcoding.
The remaining 361 Trichoderma species (96%) have been sequenced for at least one DNA Barcoding locus. In the following section, we provide an overview of the taxonomy and molecular identifiability of these species.
Reference materials that are available for molecular identification of Trichoderma
We first reviewed the Trichoderma species names that were deposited into the three main mycological taxonomic databases by May 2020. The largest number (all/valid) were recorded in Mycobank (436/361) and Index Fungorum (422/359) (See “Assumptions” above). The NCBI Taxonomy browser contained 336 names, among which 12 are not in use (Table 2), as follows: T. album, T. glaucum, H. pachybasioides, T. luteffusum, T. fomitopsis, T. subsulphureum, T. undatipile, T. cyanodichotomus, T. subalni, T. rugosum, T. acremonioides, and T. subiculoides. The four currently abandoned names—T. album, T. glaucum, T. fomitopsis, and T. subsulphureum,—were retrievable as valid from all three databases.
Although all three depositories are powered with an option to distinguish between currently legitimate names, synonyms, and names that are not in use, these records showed frequent disagreements. In Mycobank, T. album is correctly synonymized with T. polysporum, while in IndexFungorum, T. citrinum is listed as the currently correct name. The NCBI Taxonomy browser has no notes on the current status of T. album while 17 DNA sequences are attributed to this outdated taxonomic name, which appears in similarity search results (BLAST). Thus, none of the three depositories contain all 375 taxonomically valid names of Trichoderma spp. Therefore, none of the databases can be considered to be the only sufficient reference for currently valid Trichoderma nomenclature. Only 309 (82%) currently accepted names were deposited into all three databases (Table 2).
The description of the new fungal species requires deposition of the name into MycoBank (Seifert and Rossman 2010; May et al. 2019). Upon acceptance of the publication, a taxonomy provider (the author of the species name) is expected to manually release the name in this database for consideration by the curators. The name will be automatically copied to Index Fungorum without any manual update (Redhead and Norvell 2012), and therefore, these two databases will have concordant records. However, at least for Trichoderma, the validity of all names should still be cautiously considered, irrespective of the entry date.
The deposition of the name into the NCBI Taxonomy Browser is only possible along with the submission of DNA barcode sequences. Thus, this database does not contain currently used taxonomic names of the species for which DNA barcode sequences are not available.
Alternatively, deposition into the NCBI GenBank (and the Taxonomy Browser, respectively) without the deposition into Mycobank/Index Fungorum leads to an invalid species description (May et al. 2019). Some names have been abandoned by Mycobank/Index Fungorum because of the application of the “One fungus–one name” concept (Taylor 2011), which is specified for the order Hypocreales in Rossman et al. (2013), but it is still being recorded in the NCBI Taxonomy Browser. In Trichoderma, it refers to the name of the teleomorphic stage Hypocrea, or species of such sister genera as Protocrea (Jaklitsch 2009) or Sarawakus (Jaklitsch et al. 2014), which have been transferred to Trichoderma. For example, the NCBI Taxonomy Browser links the currently unused name “Hypocrea pachybasioides Doi 1972” to the correct name T. polysporum, but the “Definition” of the numerous individual sequences of T. polysporum remains “Hypocrea pachybasioides”. This disagreement should be considered when the results of the sequence similarity search (BLAST) against the NCBI GenBank are evaluated (see below). Five recently introduced species names were present in NCBI Taxonomy Browser but not deposited in Mycobank/Index Fungorum (Table 2). We assigned them as invalid for now based on the Code (May et al. 2019). Among them, T. cyanodichotomus is noted in the NCBI Taxonomy Browser as “Trichoderma cyanodichotomus J.S. Li & K. Chen, 2018, nom. inval.” with the note “Nom. inval. (i.e., nomen invalidum, or invalid name) refers to a name that is not published in accordance with rules that were enumerated in the ICN”, while T. subalni, T. rugosum, T. acremonioides, and T. subiculoides are not noted as such. T. dorothopsis (Tomah et al. 2020) has been deposited into MycoBank but not yet released. Therefore, we consider this species name to be valid. Thus, the status of each species name should be verified using multiple sources. Table 2 is designed to aid this search.
The name of the generic type species (Trichoderma viride) is presented differently in the three databases. The NCBI Taxonomy Browser contains T. viride Pers. 1832, while MycoBank and Index Fungorum refers to T. viride Pers. 1794, which is absent in the NCBI Taxonomy Browser. Jaklitsch et al. (2006) outlined the history of this species description in the 18th to 19th centuries, which allowed them to conclude that the correct taxonomic name should refer to both publications and be presented as Trichoderma viride Pers., Neues Mag. Bot. ([Roemer’s] 1: 92. 1794: Fries, Syst. Mycol. 3: 215. 1832) (Jaklitsch et al. 2006). However, none of the databases accepts the double records for the authors, publications, and years, and only one of them should be chosen (Table 2).
To review the material that is available for molecular identification of Trichoderma species, we manually recorded the distribution of DNA barcodes that were deposited in the NCBI GenBank per each Trichoderma species that were recorded in NCBI Taxonomy Browser (Table 2). This analysis aimed to reveal gaps in the deposition of DNA barcoding markers, but could not allow verification of the correctness of available materials (see “Assumptions”). It showed that 224 (66%) Trichoderma species were characterized by four or more loci, 80 (22%) species were characterized by three loci, and 35 (10%) remain characterized by one or two loci. The most commonly deposited DNA barcode loci were tef1 (322) and rpb2 (310), followed by ITS (293). For 270 species (76% from the molecularly characterized and 72% from all taxa), these three DNA barcodes were available, and tef1 and rpb2 were available for 307 species (85% and 82%, respectively). ITS was missing for 73 (20%), rpb2 was missing for 56 (16%), and tef1 was missing for 43 (12%) species. The other phylogenetic markers were deposited for considerably fewer species, as follows: acl1 for 140 (39%), cal1 for 113 (32%), act for 103 (29%), and chi18-5 for 87 (24%). Genes encoding LSU and SSU rRNA loci were sequenced for the small number of species (Table 2).
This analysis shows that the providers of molecular taxonomy of Trichoderma agreed on the use of the three DNA barcode loci (ITS, rpb2, and tef1) and deposited them for most of the molecularly characterized species. Consequently, independent of their properties and suitability for the purpose, only ITS, rpb2, and tef1 can be used for molecular identification of contemporary diversity of Trichoderma. The community of Trichoderma taxonomy providers currently has no agreement on the suitability of other loci. Therefore, all other markers have incomparably smaller collections of reference sequences and cannot be considered for the comparison unless reference strains are available for sequencing. Below, we will also show that this lack of agreement and the resulting incomplete databases for phylogenetic loci and their distribution along the infrageneric clades considerably and adversely influenced the process of species delimitation by the taxonomists.
Properties of ITS, rpb2, and tef1 DNA barcoding markers for Trichoderma spp.
In this study, we aimed to expand upon the protocol for accurate and unambiguous molecular identification of existing Trichoderma spp. based on the available DNA barcodes. In the following section, we estimate the genus-wide differences and similarities between the three DNA barcoding loci that are available for most molecularly defined species.
ITS is required to identify the genus Trichoderma
The theory suggests that accurate and precise molecular identification of such common and large fungal genera as Trichoderma, Fusarium, Aspergillus, and the others relies of the combined use of primary and secondary DNA barcodes (Stielow et al. 2015; Bissett et al. 2015; O’Donnell et al. 2015; Sklenar et al. 2017). The complete ITS region or more precisely, the internal transcribed spacers 1 and 2 of the rRNA gene cluster (See Box 1 and the discussion on the structure of ITS DNA barcoding locus below, Fig. 9), has been assigned as the primary DNA barcode marker for all fungi (Schoch et al. 2012). Although this locus can have insufficient polymorphism at a species level and numerous fungal sister species cannot be distinguished by the comparison of ITS sequences (e.g., Atanasova et al. 2013; Stielow et al. 2015; O’Donnell et al. 2015; Sklenar et al. 2017), it has the advantages of easy amplification and of the largest reference database (Nilsson et al. 2019; Schoch et al. 2020). The latter makes it more suitable for metabarcoding of fungal communities (Tedersoo et al. 2014; Abdelfattah et al. 2015) and thereby leads to the rapid growth of the number of records on the environmental ITS sequences (usually either ITS1 or ITS2) that are deposited in public databases [e.g., UNITE (Nilsson et al. 2019)].
ITS was the first locus that was introduced in DNA Barcoding of Trichoderma in late 1990s (Kuhls et al. 1996), while in 2005, we used it to develop the on-line oligonucleotide DNA Barcoding tool to identify all 88 Trichoderma species that have been molecularly characterized at that time (Druzhinina et al. 2005). Although most species were reliably identified by the unique combinations of oligonucleotide ITS hallmarks, sister species such as T. longibrachiatum - T. orientale, T. koningii - T. ovalisporum, and others could not be distinguished at that time. Since then, and particularly along with the recent boom of Trichoderma taxonomy in 2014–2017, ITS was repeatedly criticized for the high number of homoplasious sites that evolve due to the high mutation rate and saturation (Samuels et al. 2006; Druzhinina et al. 2005; Chaverri et al. 2015) and for its insufficient resolution at the species level (Atanasova et al. 2010; Druzhinina et al. 2012; Sandoval-Denis et al. 2014; Samuels et al. 2006). Therefore, this locus has even been abandoned in some large surveys of Trichoderma diversity (Jaklitsch 2009, 2011; Jaklitsch and Voglmayr 2015), resulting in the description of at least 73 species that were not characterized by ITS (Table 2). This essentially compromised the status of ITS as a primary DNA barcode locus, at least for Trichoderma spp. identification.
In this study, we analyzed the pairwise similarities between the full-length reference ITS sequences (including the 5.8S rRNA gene, see the exact length in the Supplementary Datasets) representing all infrageneric groups of Trichoderma and compared it to sequences of Protocrea, Hypomyces, Escovopsis, Sepedonium, Cladobotryum, Sphaerostilbella, Hypocreopsis, Mycogone, and Beauveria (all from Hypocreales). The polymorphism reached 300 mutations from the total length of 760 base pairs in the alignment (63% similarity) (Fig. 4). However, we noticed that the ITS sequences in Trichoderma were significantly more similar to each other compared to the related genera (Fig. 4). The heat map and the principal component analysis showed that the infrageneric similarity of ITS in Trichoderma spp. is between 71 and 100% while the similarity between Trichoderma spp. and the currently recognized neighboring genera is almost 76%, which indicates that if a query ITS sequence shares a similarity ≥ 76% to at least one of the known Trichoderma spp., it most likely belongs to Trichoderma genus, and vice versa. This calculation allowed us to compose an ITS56 Dataset that contains representative ITS sequences from the genus Trichoderma. The dataset can be used for the identification of a query sequence on the generic level if its similarity is ≥ 76% to at least one of the records in the dataset (Supplementary Datasets). We then verified the above assumption by particularly checking the sequences of “basal” species from the genus Trichoderma such as T. albolutescens (Jaklitsch 2011), T. undulatum (du Plessis et al. 2018), and T. alcalifuscescens (Overton et al. 2006; Jaklitsch and Voglmayr 2013) that were characterized by the relatively long genetic distance to the core species of the genus (Jaklitsch and Voglmayr 2013). Moreover, this threshold was not contradicted by the results that were generated from other loci (see below).

Sequence pairwise similarities of the three main DNA barcoding loci of Trichoderma. a Heatmap of ITS pairwise similarity between Trichoderma and other Hypocreales and within Trichoderma genus. Representative ITS sequences from 56 type strains belonging Trichoderma spp. (see ITS56 Dataset in Supplementary Datasets) and 22 other Hypocreales were respectively collected. b Principal component analysis (PCA) of the ITS pairwise similarity matrix. c Heatmap of rpb2 pairwise similarities within Trichoderma genus (355 species). d Heatmap of tef1 pairwise similarity within Trichoderma genus (200 species that produced significant alignment)
Similar to previous studies, we also revealed that many closely related Trichoderma species shared the same ITS phylotypes [Fig. 4, (Samuels et al. 2006; Druzhinina et al. 2006, 2012)]. Thus, this locus cannot be used for the identification at the species level. We also showed that although ITS sequences are highly conserved between some infrageneric groups of Trichoderma (Section Trichoderma or Viride Clade, Fig. 4), it is not suitable for the identification of currently proposed infrageneric groups, which is likely due to the high level of homoplasious sites (Druzhinina et al. 2005; Sandoval-Denis et al. 2014).
We conclude that because ITS is highly diagnostic at the genus level and provides essential information for the molecular identification of Trichoderma spp., it remains the primary locus that is required for DNA Barcoding.
Trichoderma species can be identified based on ≥ 99% and ≥ 97% pairwise similarities of rpb2 and tef1, respectively
We then analyzed pairwise interspecific similarity values for the two other DNA barcoding loci that are available for Trichoderma—the partial sequences of rpb2 and tef1 (Fig. 4) genes. The exact length of the used fragments is given in the Supplementary Datasets and discussed below, Fig. 9). For this reason, we collected reference strains for all DNA barcoded species (Table 2) and used NCBI Entrez to retrieve the respective sequences. The lists of accession numbers for DNA sequences in public databases are highly prone to errors and become rapidly outdated because of taxonomic revisions of individual fungal groups. Therefore, we provided the list of suggested reference strains. We would like to recommend that taxonomy users address the literature and retrieve the reference strains for species of interest and then search the databases for the corresponding DNA barcode sequences. In this study, the correctness of each sequence was verified using taxonomic literature and records in Index Fungorum, MycoBank, and/or NCBI Taxonomy Browser. The sequences were trimmed to the standard length of a phylogenetic marker that was established for Trichoderma [see below, Kopchinskiy et al. (2005) and “Materials and Methods”].
The results indicated that the genetic border of the genus was not apparent on rpb2 or tef1 similarity plots (data not shown). Therefore, these two loci cannot be used for identification at the generic level.
The sequences of tef1 (Box 1) were highly polymorphic (Fig. 4) and showed > 50% of mismatches between individual fragments, and therefore, they frequently did not produce a statistically significant alignment for most of their length. Consequently, most individual species can be distinguished by the tef1 DNA barcode (Fig. 4). The high level of tef1 polymorphism has the drawback of a high level of infraspecific variability that can lead to ambiguity and false-positive species hypotheses. Thus, a single 28 bp indel in the tef1 sequence was used to recognize a cryptic species T. bissettii within the common putative agamospecies T. longibrachiatum (Sandoval-Denis et al. 2014). However, the polyphasic approach, i.e. the application of the GCPSR concept integrated with the detailed ecophysiological profiling and analysis of biogeography did not support the existence of T. bissettii as a single taxon because no other differences were detected (Hatvani et al. 2019).
Reference strains of several currently valid species shared highly similar (> 99.5%) phylotypes of tef1 (for example, T. afarasin and T. endophyticum). Moreover, the history of tef1 application for DNA Barcoding consists of several periods when researchers used different fragments of this large gene for phylogenetic reconstructions (Druzhinina and Kubicek 2005). Thus, in the early 2000s, we used the short fifth intron of this gene, and J. Bissett’s group then tested the applicability of the first two introns at the 5ʹ end of the gene, while P. Chaverri and G. J. Samuels et al. proposed the large portion of the last (sixth) exon (Chaverri and Samuels 2003). Most resolution is provided by the fragment spanning over the fourth intron, fifth exon, and fifth intron (Kopchinskiy et al. 2005). Consequently, the NCBI GenBank contains all these frequently non-overlapping fragments of the tef1 gene, which complicates its use and in particular affects the results of the sequence similarity search. Together, these findings make the tef1 locus insufficient to be used as the only DNA barcode marker for Trichoderma identification at the species level as it was also proposed by Rahimi et al. (2020) for the identifiction of T. reesei. The limitations outlined above also reveal that the application of tef1 together with ITS will not allow unambiguous identification of Trichoderma species.
The sequences of rpb2 (Box 1) were most conserved because many Trichoderma spp. shared highly similar phylotypes. Figure 4c shows large clusters of highly similar species and even clades indicating that the single use of this DNA barcode was also not suitable for species identification.
Thus, currently none of the three DNA barcode loci can be used as a sole sufficient marker for the identification of the 361 Trichoderma species.
In this study, we aim to determine how to distinguish currently valid Trichoderma species using the DNA barcode sequences that have been provided. To assess the sequence similarity threshold in a manner that is sufficient to identify species, we screened the subclades of species that exhibited highly similar rpb2 and tef1 sequences (Fig. 5). In such groups, we ignored rare species that were available from a low number of isolates, and focused on the well-established and common species with recorded values for humankind. As a reference example, we selected (1: reesei) the main industrial cellulase producer T. reesei (e.g., Druzhinina et al. 2016) and two of its sibling species T. parareesei (Atanasova et al. 2010) and T. thermophilum (Qin and Zhuang 2016a). (2: harzianum) The most common environmental opportunistic species with high suitability for biocontrol, plant growth promotion, and enzyme production are as follows: T. harzianum (Chaverri et al. 2015), and the two sibling species, T. afroharzianum (Chaverri et al. 2015) and T. guizhouense (Li et al. 2013; Grujic et al. 2019); and (3: asperellum) another common species with multiple applications in agriculture, T. asperellum (Rivera-Méndez et al. 2020) and the two recently recognized sibling species, T. asperelloides (Samuels et al. 2010) and T. yunnanense (Yu et al. 2007).

Sequence pairwise similarities of each DNA barcoding locus between sets of selected model species. The three closely related sibling species. T. reesei, T. parareesei, and T. thermophium represent the Longibrachiatum Clade; T. harzianum, T. afroharzianum, and T. guizhouense represent the Harzianum Clade; and T. asperellum, T. asperelloides, and T. yunnanense represent the Section Trichoderma. Sequences were collected from the type strains and consistently trimmed as described in the Materials and Methods and in Fig. 9
ITS was polymorphic in the 2: harzianum group, but T. reesei–T. parareesei (the 1: reesei group) and T. asperelloides–T. yunnanense (the 3: asperellum group) shared the same ITS phylotypes. In all three groups, the rpb2 sequences were different, with similarities that were 98.15–98.77% for the 1: reesei group, 94.93–95.82% for the 2: harzianum cluster, and 98.65–99.14% for 3: asperellum. Thus, if none of these species hypotheses to be rejected based on rpb2, Trichoderma species should be only by 1% different. It corresponds to the maximum level of infraspecific polymorphism of eight mutations (substitutions or indels) if the total length of the alignment is fixed to the diagnostic region of 820 base pairs (see Fig. 9 below and “Materials and Methods” for the details). Thus, assignment to an existing species is possible if the similarity of rpb2 is ≥ 99%. However, in this case, the uniqueness of T. yunnanense rpb2 appears to be compromised (Fig. 5, Table 2).
Similar consideration of the tef1 polymorphism resulted in 82.63–96.10% similarities between the 1: reesei group, 80.29–86.85% for the 2: harzianum cluster, and 89.29–95.39% for the 3: asperellum group. Thus, these species can be distinguished based on tef1 similarity < 97% or identified based on ≥ 97%. This assumes that different strains of the same species can have up to 27 mutations in the diagnostic area of the tef1 DNA barcode, which agrees well with the species where large populations were studied (Druzhinina et al. 2012; Hatvani et al. 2019).
We, therefore, conclude that a query strain can be assigned to the existing Trichoderma species if it is ≥ 99% similar for rpb2 and has ≥ 97% tef1 similarities to that of the reference strains. The molecular identification can only be achieved if both loci point to the same result species.
The high level of infrageneric conservation of rpb2 (Atanasova et al. 2013; Jaklitsch 2009, 2011; Jaklitsch and Voglmayr 2015) has the advantage that allows construction of the most complete phylogram for the genus Trichoderma (Fig. 6) and, thus, reveal the “phylogenetic order” (“PhyloOrder”) of the species that is provided in Table 2. To achieve this for all DNA barcoded 361 species, the approximate position of the species for which rpb2 is not available or for which it is available but not attributed to the species in the NCBI Taxonomy Browser was determined based on the similarities of other loci and respective taxonomic literature (Fig. 6). The phylogenetic analysis of the alignment of 356 rpb2 sequences revealed at least eight statistically supported rpb2-based infrageneric clades that largely correspond to those presented in previous reviews of Trichoderma taxonomy (Atanasova et al. 2013). To avoid further confusion and discrepancies, we skipped naming the clades, but we numbered them and highlighted the most prominent species within each clade (Fig. 6, Table 2).

The list of all DNA barcoded Trichoderma spp. (361) sorted based on the phylogenetic position (PhyloOrder in Table 2). The core topology of the phylogram is based on the maximum-likelihood (ML) phylogeny of the currently rpb2-barcoded Trichoderma species. Eight main clades were collapsed and numerically named (see “Clade” in Table 2). Species names are sorted alphabetically within each clade. Well-known species are highlighted in red font for convenience purpose. The attribution of species that have no rpb2 sequence available was approximately determined based on the other available loci. The nucleotide substitution model of TIM3 + F + R6 was chosen based on the Bayesian Information Criterion (BIC). Circles at the nodes indicate ultrafast bootstrap values > 80 given by IQ-TREE. The sequences of rpb2 from Arachnocrea stipata, Hypomyces austrlasiaticus, and Sphaerostilbella aureonitens were used as the outgroups. The inset (top left) shows the complete topology of the rpb2 phylogram
Sorting all molecularly defined Trichoderma species according to their approximate phylogenetic position in Table 2 (“PhyloOrder”) revealed the distribution of other phylogenetic markers (chi18-5 = ech42, cal1, act, acl1,18S rRNA = SSU, 28S rRNA = LSU) along the genus genealogy. This demonstrates that the usability of such loci is limited because none of Trichoderma clades have a complete reference dataset for any of them. Therefore, they can only be used if the providers of Trichoderma taxonomy will complement missing sequences or if all Trichoderma reference strains will become available for the research community (see “Discussions and suggestions” below). Consequently, molecular identification of Trichoderma spp. is only possible based on ITS, tef1 and rpb2 that are available in public databases.
Accuracy, precision, and ambiguity in DNA Barcoding of Trichoderma
With all the molecularly defined Trichoderma spp. ordered based on their approximate phylogenetic relation, we can estimate the potential identifiability of individual species and list warnings that should be considered by the users of Trichoderma taxonomy (Table 2).
Our analysis suggests that for at least 216 Trichoderma species (60%), molecular characteristics are sufficient for accurate and precise species identification based on three DNA barcodes (ITS, tef1, and rpb2) assuming that the deposited data are correct (Table 2) (See “Assumptions”). This group includes the most common species such as T. harzianum (= T. harzianum sensu stricto), T. virens, T. gamsii, T. atroviride, T. koningiopsis, T. hamatum, and T. citrinoviride, T. reesei, and around 100 rare species that are only known from a few or even one isolate (Table 2). Although these species have mostly complete records in all databases, some minor deviations should be considered. For example, T. longipile is deposited in IndexFungorum as T. longipilis (orthographic variant). T. undatipile Chen & Zhuang 2017 was molecularly characterized and deposited in MycoBank under its correct name, but it was deposited in IndexFungorum as T. undatipilosum. Four species, T. pinicola, T. guizhouense, T. kunigamense, and T. tsugarense are absent in MycoBank, which jeopardizes the validity of these taxa (Table 2).
Molecular identifiability of 141 Trichoderma species (40%) is compromised either by the lack of DNA barcodes or by the high similarity of tef1 and/or rpb2 sequences to their sister species. Among 73 species that lack ITS, 34 have tef1 and rpb2 and, therefore, can be potentially identified if their attribution to the genus is not in question. This group includes the very common or even dominant European species T. europaeum and T. mediterraneum, while many others are rare or very rare. Ten species, including Hypocrea subcitrina, T. cornu-damae, H. dichromospora, T. aestuarinum, T. cerebriforme, T. poronioideum, T. densum, H. ampulliformis, T. surrotundum, and T. patellotropicum, have ITS but lack either tef1 or rpb2 sequences and, therefore, cannot be accurately identified. It also suggests that these species were described without considering the GCPSR concept (see “Discussions and suggestions” below). H. mikurajimensis is only characterized using 28S rRNA sequence, and therefore, its molecular identification is not possible.
The following 37 species has been molecularly and phylogenetically characterized, but their taxonomic status was not updated in the NCBI Taxonomy Browser, and they are not available for sequence similarity search (Table 2): T. limonium, T. grande, T. pruinosum, T. dimorphum, T. angustum, T. gregarium, T. bomiense, T. viridulum, T. pollinicola, T. tenue, T. purpureum, T. perviride, T. globoides, T. confertum, T. changbaiense, T. viridicollare, T. adaptatum, T. beijingense, T. panacis, T. tardum, T. bifurcatum, T. vulgatum, T. mangshanicum, T. shaoguanicum, T. citrinella, T. asterineum, T. pseudobritdaniae, T. henanense, T. odoratum, T. thermophilum, T. xanthum, T. centrosinicum, T. virgineum, T. fruticola, T. medogense, T. palidulum, and T. alboviride. The reference cultures for these species were mainly deposited into the Fungarium (also as HMAS, Herbarium Mycologicum Academiae Sinicae) at the Institute of Microbiology, Chinese Academy of Sciences, and therefore, they are mainly available for researchers in China. The insertion of these species into the NCBI Taxonomy Browser and the attribution of respective undefined isolates (which are currently deposited as “Trichoderma sp.”) will allow molecular identification of other strains that belong to these species if all three DNA barcodes are provided.
For 49 Trichoderma spp., the rpb2 sequences of reference strains showed high similarity to neighboring species (Fig. 7). Each of these species is marked by a respective warning in Table 2. Most of these species have rpb2 similarity > 99% with only one other species, but T. viridescens, T. viridarium, T. paraviridescens, T. trixiae, T. appalachiense, T. rossicum, T. sichuanense, T. verticillatum, T. alpinum, T. concentricum, T. alni, and T. pseudodensum have from three to eight species that each shares a highly similar rpb2 phylotype (> 99%). T. cremeoides also has no deposited ITS sequence, and thus, its molecular identification can only be putative. Our analysis also shows that tef1 of T. cremeoides is > 97% similar to T. sinuosum and T. brevicrassum and accurate molecular identification of these three species is also not possible. The type strain of T. asperellum shares highly similar phylotypes of rpb2 with T. yunnanense and T. kunmingense (Table 2, Fig. 4). Warnings related to the identification of all DNA barcoded Trichoderma spp. that are available to date are listed in Table 2.

Distribution of the pairwise similarities of rpb2 between the 352 Trichoderma species and of each respective most closely-related species. The bars represent the number of observations at a certain similarity range. The delimitation of rpb2 is set at 99%. Values < 99% are shown in grey and values ≥ 99% are in red
Thus, accurate DNA Barcoding of a large portion (40%) of Trichoderma species is not possible based on the provided molecular characters, and further sampling and an integrated analysis of molecular, ecophysiological, and biogeographic features are required.
Validation of DNA barcoding results
Although DNA Barcoding is presented as a tool that provides the final level of precision in microbial identification (Valentini et al. 2009), studies on other fungi (Lücking et al. 2020) and this work indicate that verification is required. It appears to be reasonable to conclude that in silico analysis may result in a putative identification or a formulation of the species hypothesis (including the new species hypothesis), while final identification can be achieved after the verification step. Following the principle of scientific falsification, verification should consist of critical considerations of the putative identification result. Verifying of the molecular identification should include the consideration of biological features such as concordant phenotypes, growth profile, lifecycle, and habitat. However, before this, the correctness of the molecular identification can also be considered critically (i.e., it has been validated) because it depends on the correctness of the deposited reference materials.
The correctness of reference materials that are used to formulate the species hypothesis should be critically assessed. The curators of public sequence databases (NCBI GenBank, EMBL, and DDBJ) take multiple measures to verify the quality of submitted materials (Lücking et al. 2020; Schoch et al. 2020). However, verification of species identification along with sequence submission is not a realistic task. Consequently, public databases contain a high proportion of sequences with incorrect species assignments. More than a decade ago, we estimated that 40% of such sequences were deposited into the NCBI GenBank for Trichoderma (Druzhinina et al. 2006; Atanasova et al. 2013). Molecular identification became essentially more complicated due to the rapid growth of species number, and we envision that the proportion of inaccurately identified sequence depositions will increase dramatically. Another source of incorrect species assignment for DNA barcode sequences is the common practice of taxonomic reclassifications that intends to improve the taxonomy of the group. However, such actions are not always reflect in the sequence annotations in public databases (see also above). Thus, hundreds of sequences that are available in the NCBI GenBank remain deposited under currently non-used “Hypocrea lixii”, which has been maintained since the time when this combination was used for Trichoderma harzianum sensu lato (Chaverri and Samuels 2003; Druzhinina et al. 2010b). The latter species has been divided into a dozen sibling species including a rare T. lixii, which is known from a single isolate from Thailand (Chaverri et al. 2015). Thus, most sequences named “Hypocrea lixii” in the NCBI Taxonomy Browser should be considered to be inaccurately identified. Even T. harzianum name that has been assigned to the sequences of the most frequently deposited species is doubtful (irrespective of the DNA barcoding locus) because it may refer to the species concept that existed before the work of Chaverri et al. (2015), in which T. harzianum sensu lato was divided into several newly defined species form this complex including T. harzianum sensu stricto.
To show a quantitative example, we collected the 100 best hits from the sequence similarity search of the DNA barcode sequences for one of the strains (TUCIM 10063, T. guizhouense) that was used for the online survey earlier in this study (Supplementary Table S1). For ITS, at least 15 hits were incorrectly labeled as unrelated T. atroviride and T. aureoviride or as “Hypocrea lixii”, and 31 were not identified. The tef1 gene sequence can be submitted as it is (Supplementary Table S1, see “Materials and methods”) or it can be trimmed for the length of the diagnostic fragment [see Kopchinskiy et al. (2005) or Fig. 9below]. The respective lists of the best hits for untrimmed and trimmed tef1 sequences contained at least 13 and 27 incorrect species names, and seven and 20 were not identified, respectively. We also detected Trichoderma sequences that were deposited as Dothideomycetes fungi such as Neofusicoccum spp. (KY024676.1 & KY024614.1) and Lasiodiploidia sp. (KY024673.1). It is likely that in these studies, Trichoderma parasitized these fungi [refer to the work of Druzhinina et al. (2018)], and its DNA was amplified instead of its hosts. These sequences were deposited under wrong names. Similarly, at least 27 rpb2 sequences were also incorrectly named and six were not identified. This analysis revealed only the minimum number of incorrect records in the NCBI GenBank, but because the species borders in this group are difficult to establish (Druzhinina et al. 2010b), the actual number of incorrect records is likely to be higher.
The manually curated databases of sequences have fewer incorrect records, but they are usually outdated. The first multiloci database of reference Trichoderma sequences was powered by several on-line identification tools that were available at www.isth.info (Druzhinina et al. 2005; Kopchinskiy et al. 2005), and it is no longer supported (however we offer some updated tools below). The new tool, Multiloci Identification System for Trichoderma (MIST) is available at http://mmit.china-cctc.org/ (Dou et al. 2020), and it is based on the sequential sequence similarity search of ITS, rpb2, or tef1 DNA barcode loci for a query strain against a MIST databases of reference and non-reference sequences. Although it provided correct identification of the query sequence in this case (T. guizhouense), for many other species it also exports numerous false-positive results (many species assigned at the identification step). When it was released in July 2020, it contained a database of tef1 and rpb2 sequences for 349 species (out of the current 361). Its usability will depend on the frequency of updates. If new species are not regularly added to the MIST database, it will lose its identification function but remain a useful support for searching for the approximate position of a query strain.
The use of the largest fungal database for sequence identification, UNITE https://unite.ut.ee/index.php#panel3, is not suitable for Trichoderma species identification because it is only based on partial ITS (see above). Analysis of the test strain of T. guizhouense TUCIM 10063 in UNITE resulted in four species hypotheses, none of which were correct (T. harzianum, T. tawa, T. lixii, and T. virens). However, all these species are closely related to T. guizhouense, and therefore, this tool provides identification at the level of the Harzianum and Virens Clades. Trichoderma spp. are not yet included in the collection of MycoBank Polyphasic Identifications Databases (http://www.mycobank.org/DefaultInfo.aspx?Page=polyphasicID).
Thus, the molecular identification is solely dependent on sequences that are deposited into public databases (curated and non-curated). The current diversity of Trichoderma requires manual analysis of sequence similarities and phylogenetic analyses, but accurate automated identification of Trichoderma species is not available. However, several Trichoderma-dedicated tools provide useful supporting material (www.trichokey.com, www.trichoderma.info, and MIST http://mmit.china-cctc.org/).
The solution: molecular identification guideline for Trichoderma spp.
Synopsis of molecular taxonomic inventory for the genus Trichoderma
-
The introduction of molecular evolutionary analyses resulted in exponential growth in the number of Trichoderma species, up to 50 new species that were described per each year.
-
Among the 375 species with valid names as of July 2020, 361 (96%) are DNA barcoded.
-
IndexFungorum and Mycobank do not contain complete lists of Trichoderma species. The NCBI Taxonomy Browser includes 90% of the species. Numerous species names that are not currently in use or not legitimate are listed in IndexFungorum and Mycobank. The NCBI Taxonomy Browser contains the fewest such names.
-
As for July 2020, identification (DNA Barcoding) and evolutionary analyses of Trichoderma spp. are possible only based on three phylogenetic markers: ITS, tef1, and rpb2. Other DNA barcodes (chi18-5 = ech42, cal1, act, acl1,18S rRNA = SSU, and 28S rRNA = LSU) are sequenced for less than one-half of the species, and therefore, they have limited or no suitability for molecular identification.
-
Trichoderma spp. cannot be identified by phylogenetic analysis without considering the sequence similarity values.
-
ITS can be used to identify Trichoderma at the generic level.
-
For the accurate and precise molecular identification of Trichoderma isolates at the species level, sequencing of the three DNA barcodes (ITS, tef1, and rpb2) is required.
-
Most closely related species of Trichoderma differ by 1% (approximately eight mutations) of rpb2 and/or 3% (approximately 27 mutations including indels) of tef1 sequences (if the specified region of each phylogenetic marker is considered, see Fig. 9 below). Some species and infrageneric groups share phylotypes of individual markers (ITS, tef1, or rpb2).
-
Molecular identification can be achieved based on the analysis of sequence similarities between the query strain and the reference strains that are analyzed for tef1 (≥ 97%) and rpb2 (≥ 99%). If this condition is not met, the identification can be made based on sequence similarities and phylogenetic concordance, i.e., analysis of single loci tree topologies for tef1 and rpb2.
-
Molecular identification must be validated by the critical evaluation of non-biological aspects (quality and completeness of the reference taxonomic materials) and verified based on biological criteria (morphology, ecophysiology, biogeography, habitat, and occurrence).
-
The inventory of DNA barcoding materials that were deposited in public databases revealed that only 60% of molecularly characterized Trichoderma species can potentially be unambiguously identified based on the reference sequences that were deposited by taxonomy providers.
-
Identifiability of 40% of species is compromised by any of the following factors or their combinations: incomplete DNA barcoding, incomplete deposition of reference cultures or reference sequences, or insufficient polymorphism of one or several diagnostic sequences.
-
Trichoderma spp. cannot be identified by the automated sequence similarity search (such as BLAST) irrespective of the reference database or DNA barcodes that are used as such results require in silico validation and biological verification.
-
On-line tools for Trichoderma identification can provide a useful estimation of the taxonomic (phylogenetic) surroundings for a given strain. However, the tools that are currently available do not offer precise identification at the species level.
-
Identification of Trichoderma species is an intricate and laborious task that requires a background in mycology, molecular biological skills, training in molecular evolution, and in-depth knowledge of taxonomic literature. For ambiguous cases, a consultation with Trichoderma taxonomy experts is recommended.
Molecular identification protocol for a single Trichoderma isolate
The following molecular identification protocol enables a user to do the following: (1) identify the genus Trichoderma, i.e., to exclude fungi other than Trichoderma; (2) identify Trichoderma species; and (3) verify the ambiguity of the identification. The protocol allows recognition of a putative new species as a particular case of species identification.
All steps proposed below refer to the taxonomic limitations that constrain the molecular diversity of the genus Trichoderma and recognized species that existed as of July 2020.
A Trichoderma species can be identified if its ITS sequence reaches at least one similarity value ≥ 76% compared to the sequences in the dataset that is attached to the protocol and the two other DNA barcoding markers are highly similar to the corresponding sequences of the reference strain from one species, with rpb2 ≥ 99% and tef1 ≥ 97%. These conditions can be shortened as shown in the following sequence similarity standard:
Trichoderma [ITS 76 ] ~ sp∃!( rpb2 99 ≅ tef1 97 ),
where “Trichoderma” means the genus Trichoderma, “sp” means a species, “~” indicates an agreement between ITS and other loci, “≅” refers to the concordance between “rpb2” and “tef1”, and “∃!” indicates the uniqueness of the condition (only one species can be identified). Subscripts show the similarity per locus that is sufficient for the identification based on the assumptions of the protocol below. A flowchart of the protocol is presented in Fig. 8.

The flowchart of the molecular identification protocol of Trichoderma based on three DNA barcode sequences. A species of Trichoderma can be identified if its ITS sequence reaches a similarity value ≥ 76% (ITS76) compared to the sequences in the dataset that is attached to the protocol and the two other DNA barcoding markers are highly similar to the corresponding sequences of the reference strain of one species as rpb2 ≥ 99% and tef1 ≥ 97% (rpb299≅ tef197); “≅” refers to the concordance between rpb2 and tef1
The result of molecular identification requires biological verification (Lücking et al. 2020) and consideration of the original taxonomic literature. The morphology and growth profile of the query strain should not contradict the published records for the identified species. It is recommended to compare the biogeography and occurrence records for the identified species with metadata for the query strain. The observed lifecycle, ecology (habitat and interactions with other organisms), and ecophysiology of the query strain should be in agreement with the description of the identified species. For ambiguous cases, it is useful to consult taxonomy experts.
The check-list for materials, tools, and preparation steps.
-
Isolate a single spore (asco- or conidiospore) culture from the putative Trichoderma sp. strain.
Note: Although the fast growth on rich nutritional media, mycoparasitism, resistance to xenobiotics, and greenish conidiation are characteristic features for most of the Trichoderma cultures, some species have hyaline conidia or do not produce them in vitro (they appear white in culture), some are sensitive to fungicides, and some do not parasitize other fungi and/or have slow growth in vitro. Refer to the diversity of Trichoderma spp. morphotypes in monographs by Jaklitsch (2009, 2011) or elsewhere.
-
Use PCR to amplify and sequence the three DNA barcode loci as follows: the complete fragment of ITS1 and 2 (including the 5.8S rRNA) of the rRNA gene clusters, and partial sequences of rpb2 and tef1 genes.
Note: PCR protocols including the corresponding primer pairs are provided in Table 3, and the structure of the loci is shown in Fig. 9.
Table 3 PCR conditions for the amplification of the three Trichoderma DNA barcodes Fig. 9 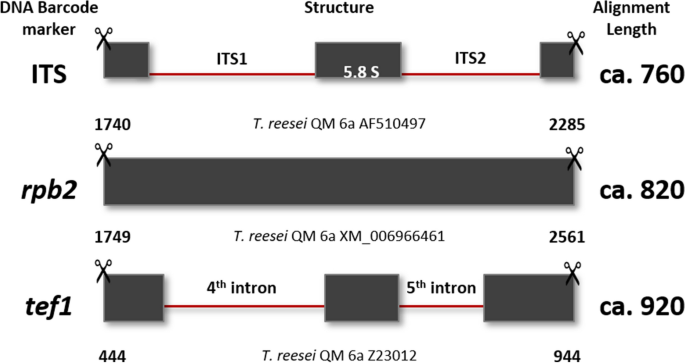
Structure of DNA barcoding loci trimmed for molecular identification. Numbers below each locus show the 5ʹ and 3ʹ positions on the trimmed fragment that were suitable for molecular identification using reference loci from T. reesei QM 6a (Druzhinina et al. 2010a; Druzhinina et al. 2005; Chenthamara et al. 2020) as an example
Note: The quality of obtained sequences is crucially important for this protocol. No ambiguity in sequencing reaction is accepted. Ideally, sequences should be verified by sequencing from the 3ʹ and 5ʹ ends.
-
Connect to the Internet.
-
Trim the sequences. Use TrichoMARK 2020, which is available at www.trichokey.com, or use the reference datasets (Supplementary Datasets and www.trichoderma.info) and trim the length of the query sequences such that they correspond to the length of the reference DNA barcode loci, as shown in Fig. 9.
Note: this step is required for the molecular identification protocol. If online tools are not available, the sequences can be trimmed manually using Aliview (Larsson 2014) or other sequence editors. The technical requirement to trim the sequences is also explained in Kopchinskiy et al. (2005).
-
Use a text editor (e.g., Notepad) and put your trimmed query DNA barcode sequences into FASTA format and save the input files separately.
-
Install Aliview, IQ-TREE (Nguyen et al. 2015b), and FigTree (http://tree.bio.ed.ac.uk/software/figtree/), or softwares with similar functions.

Step 1: ITS
Estimate the pairwise similarity between the ITS sequence of the query strain and the sequences that are given in the ITS56 dataset as described in Comment 1 at the end of the protocol.
If the maximum similarity is ≥ 76%,
the query strain belongs to the genus Trichoderma spp. Continue to Step 2.
If the maximum similarity is < 76%,
the query strain belongs to a genus other than Trichoderma. Identification of Trichoderma spp. is not possible.
Step 2: rpb2 and tef1
For each locus (rpb2 and tef1), estimate the pairwise similarities between the query strain and the sequences of closely related reference strains, as described in Comment 2.
If the condition ∃!(rpb299 ≅ tef197) is met,
Record the identified species and continue to Step 4.
If the condition ∃!(rpb299 ≅ tef197) is not met,
Continue to Step 3.
Step 3: phylogenetic analyses of rpb2 and tef1
-
Open rpb2 and tef1 (separately) alignments that are produced in Step 2 (see Comment 2).
-
Record cases of short or missing sequences for reference strains (if any).
-
Run phylogenetic analyses separately for rpb2 and tef1 sequences. Given that the correct parameters were selected, the maximum likelihood or Bayesian methods are recommended; however maximal parsimony is also suitable. See details in the “Materials and methods”.
-
Visualize the tree files in Figtree and (optionally) export the data to a graphics software.
-
On each tree, locate the query sequence and the most similar reference sequences; mark the pairwise similarities that were estimated in Step 2 (examples are shown on Figs. 10–13).
Fig. 10 
Molecular identification of genome-sequenced strains from the Harzianum and Virens clades using maximum-likelihood (ML) phylogeny and pairwise sequence similarity calculation. The ML phylograms of rpb2 and tef1 were constructed in IQ-TREE 1.6.12 (bootstrap replicates = 1000) using the nucleotide substitution models of TNe + R3 and HKY + F+G4. Circles at the nodes indicate ultrafast bootstrap values > 80 given by IQ-TREE. Genome sequenced strains were shaded in green. The reference strains were provided with the GenBank accessions and the strain name, among which, strains with uncompleted reference information were shaded in orange. Results of the pairwise sequence similarity were illustrated on the dashed lines between the query strain and its closely related species (arrows point to the reference strains). The pairwise sequence similarity calculation was performed using the online tool of ClustalOMEGA (https://www.ebi.ac.uk/Tools/msa/clustalo/)
-
Interpret the concordance of rpb2 and tef1 phylograms considering the similarity values that were estimated in Step 2.

Note: Consideration of single-loci phylograms for tef1 and rpb2 is required. The concatenated phylogram of the two loci is optional in addition to analysis of single-locus trees.
Note: For the interpretation of phylogenetic trees, refer to Comment 3 and practical examples below.
Step 4: Validation of molecular identification
For the validation of the molecular identification and assignment of ambiguity status, the literature on Trichoderma taxonomy should be studied. Table 2 of this study provides supplementary information.
In some cases, results of phylogenetic analysis (Step 3) can be used to validate the identification results (Comment 3).
Validation of species identification
If all of the following criteria are met:
-
The identified species is represented by the complete set of reference DNA barcodes (Table 2, taxonomic literature).
-
The identifiability of the species is not compromised by insufficient polymorphism of tef1 and rpb2, or other parameters (i.e., none of the warnings from Table 2 are present).
-
The identified species was recognized based on the GCPSR concept using a polyphasic approach.
The identification is unambiguous, precise, and accurate.
If any of the following criteria are met:
-
The identified species is represented by the incomplete set of reference DNA barcodes (see warnings in Table 2).
-
The identifiability of the species is compromised by low tef1 and rpb2 polymorphism, or the quality of the reference sequences is not sufficient (usually, too short) (see warnings in Table 2).
-
The identified species is recognized based on insufficient reference material or ambiguous species criteria.
The identification is ambiguous; the species name can be assigned as “confer” or “cf.” (i.e., compared to ) or as “affinis” or “aff.” (i.e., related to ) the most closely related species.
Note: In this case, the most closely related species can be revealed based on the results of phylogenetic analyses of tef1 and rpb2 (Step 3, Comment 3). Note: Precise and accurate identification will usually require either taxonomic revision of reference materials, additional DNA sequencing, or/and sampling.
Note: If phylogenetic analyses of both loci point to a single sister species but it can’t be identified because of incomplete reference materials, “aff.” can be used to specify the related taxon: T. aff. [related species name]. If several sister species are proposed, the use of “cf.” is more appropriate: T. cf. [one of the related species]. Here, it is suggested to point to the related species that is best studied or has similar features.
Validation of the new species hypothesis
If all of the following criteria are met:
-
The query strain belongs to the genus Trichoderma (meets Trichoderma[ITS76] standard).
-
The query strain has unique sequences of rpb2 or tef1 (does not meet the sp∃!(rpb299 ≅ tef197) standard for known species).
-
The existing closely related species have complete sets of reference DNA barcodes.
-
The new species hypothesis is supported by the topology of both phylograms (rpb2 and tef1) and is not contradicted by other markers (GCPSR concept).
The new species hypothesis is unambiguous, precise, and accurate. Record the results as “ T . sp. strain ID” before the formal name is given.
Note: the formal taxonomic description of a new fungal species requires the guidelines of Seifert and Rossman (2010) to be followed, including naming (see The Code), registration of the type (May et al. 2019), deposition of the reference materials into public databases, microbiological investigation, and imaging of microscopic features. It comprises the molecular evolutionary analysis (Comment 3) and comparison of morphological, eco-physiological, and biogeographical characteristics between the query strain(s) and closely related taxa.
If any of the following criteria met:
-
Attribution of the query strain to the genus Trichoderma is ambiguous (does not meet the Trichoderma[ITS76] standard, in particular if the similarity is < 70%)
-
Closely related species have incomplete sets of DNA barcodes, the quality of the reference sequences is not satisfactory, or related species were recognized based on insufficient DNA barcoding material.
-
The position of a new species is not supported by the topology of both phylograms (rpb2 and tef1) or is contradicted by other markers (GCPSR concept is not applicable).
The hypothesis of a new species remains ambiguous.
Note: In this case, the species name can be assigned as T. sp. with the addition of either “affinis” or “aff.” [i.e., related to] (if there is only one sister species) or “confer” or “cf.” [i.e., compared to] (if there is a group of related species) the most closely related species that can be revealed based on the results of phylogenetic analysis (Step 3, Comment 3). Precise and accurate identification of a new species will usually require either taxonomic revision of reference materials, additional sequencing, or/and sampling.
Step 5: Presentation of the identification result and data archiving
Record the identification results. An example is given in Table 4.
Archive your non-trimmed query DNA barcode sequences along with their identification (FASTA format is suggested).
Comments:
Comment 1. Calculation of pairwise similarities between the query and reference sequences using ITS:
-
Download the sequence ITS56 dataset from Supplementary Datasets from this study or www.trichokey.com and open in the text editor. Add the query ITS sequence to the dataset.
-
Insert the sequences in Aliview and use “Realign everything” option in “Align” menu.
-
Check whether the length of the query sequence fits the ITS56 dataset. If not, the identification result will be ambiguous.
-
Export the alignment as a .fasta file and save it.
-
Upload the exported .fasta file or paste the sequences into the input box of the online ClustalOMEGA tool for pairwise similarity calculation (https://www.ebi.ac.uk/Tools/msa/clustalo/) or use other tools for pairwise sequence similarity calculation.
-
Select the option of “DNA”, setup your parameters (“ClustalW” is recommended), and click the “submit” button.
-
Download the .pim file, which contains the results of the pairwise similarity calculation, from the “results summary” page.
-
(Optional) A “guide tree” can also be obtained from the “results summary” page and visualized in FigTree for your interest.
-
Open the .pim file using Microsoft Excel or a text editor, search for the maximum similarity value(s) between your query sequence and the references. Make sure you have excluded the value showing the similarity to the query sequence (100%).
Note: The ITS56 dataset contains 56 selected reference ITS sequences that represent intrageneric polymorphism of the Trichoderma genus.
Comment 2. Manual calculation of pairwise similarities between the query and reference sequences using tef1 or rpb2:
-
Submit the trimmed rpb2 sequence to TrichoBLAST (www.trichokey.com) and detect the most closely related species.
-
Use the most updated data in Table 2 (i.e., the latest updated version is on www.trichokey.com) and taxonomic literature that was published after the release of this manual, and compose lists of the most closely related species, 6 < N < 10.
-
Find the taxonomically confirmed reference strains (ex-type, type, vouchered; Table 2) for each species and retrieve rpb2 and tef1 sequences from public databases.
-
Align and trim the sequences, and calculate the pairwise sequence similarities as described in Comment 1.
Comment 3. Application of phylogenetic analysis in molecular identification and its use for the validation of identification results.
Phylogenetic analysis can contribute to unambiguous or ambiguous identification of either a known species or a putative new species, as described below.
-
If the sequence similarity standard (whether it is rpb2 and/or tef1) indicates several species (e.g., T. cf. endophyticum CFAM-422, Tables 1 and 4), phylogenetic analysis of both loci will reveal the closest species and allow accurate but imprecise (ambiguous) identification as Trichoderma cf. [closest species]. Thus, this analysis will usually indicate a need for the taxonomic revision of the reference group. In this case, phylogeny is used as an identification step.
-
If the two loci indicate different species (existing or putatively new), the phylogenetic analysis results can demonstrate that the loci are not concordant (e.g., T. sp. NJAU 4742, Tables 1 and 4). In this case, and considering that only two markers are currently available, phylogeny is used as a validation step. With the introduction of genomic techniques in fungal taxonomy, such cases may be resolved by the application of phylogenomic analyses (Galtier and Daubin 2008).
-
If the reference sequences are not complete, the results of phylogenetic analysis will reveal the closest species and allow accurate but imprecise (ambiguous) identification as Trichoderma aff. [closest species] or Trichoderma cf. [closest species] (e.g., T. cf. atrobrunneum ITEM 908, Tables 1 and 4). In this case, phylogeny is used as a validation step.
-
If a new species is found, phylogeny is a required as part of the new species recognition. In this case, the topologies of both phylograms are expected to be concordant and pairwise sequence similarities should support the unambiguous new species hypothesis.
Practical examples of Trichoderma identification
To verify the suitability of the molecular identification protocol and to demonstrate how the identification results can be presented, we list below the detailed identification diagnoses for the two strains that were used for the on-line survey (see above) and the 42 WGS Trichoderma strains that were available in public databases as of July 2020.
Note: Sequences of all phylogenetic markers were trimmed before the analysis using TrichoMARK 2020, which is available at www.trichokey.com or the reference datasets (Supplementary Datasets and www.trichoderma.info), so that they correspond to the length of the reference DNA barcode loci, as shown in Fig. 9.
Identification of strains that were used in the on-line survey
(1) TUCIM 10063 (called “mycoparasite” in the on-line survey)
Identification: The pairwise sequence similarity of ITS (MT792072) between strain TUCIM 10063 and the references that are given in the ITS56 dataset showed several values > 76%, which indicated that strain TUCIM 10063 belongs to the genus Trichoderma. The similarity of rpb2 (MT802437) between strain TUCIM 10063 and the most closely related species T. guizhouense (strain CBS 131803) that is found at this locus was 100.00% (Table 2), and the similarity of tef1 (MT802439) between strain TUCIM 10063 and the most closely related species T. guizhouense (strain CBS 131803) that is found at this locus was 100.00% (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete, and identification was precise, accurate, and unambiguous. Therefore, strain TUCIM 10063 can be identified as T. guizhouense.
(2) TUCIM 5640 (called “epiphyte” in the on-line survey)
Identification: The pairwise sequence similarity of ITS (MT792073) between strain TUCIM 5640 and the references that is given in the ITS56 dataset showed several values > 76%, which indicated that strain TUCIM 5640 belongs to the genus Trichoderma. The similarity of rpb2 (MT802438) between strain TUCIM 5640 and the most closely related species including T. compactum (strain CBS 121218) and T. aggregatum (strain HMAS 248863) that are found at this locus was 96.55% and 96.05% (Table 2), respectively, and the similarity of tef1 (MT802440) between strain TUCIM 5640 and the most closely related species including T. compactum (strain CBS 121218) and T. aggregatum (strain HMAS 248863) that are found at this locus was 95.84% and 91.51% (Table 2), respectively, (i.e., the condition ∃!(rpb299 ≅ tef197) was not met). This indicates that strain TUCIM 5640 can be recognized as a putative new species (Comment 3).

Molecular identification of genome-sequenced strains from the Section Longibrachiatum using maximum-likelihood (ML) phylogeny and pairwise sequence similarity calculation. The ML phylograms of rpb2 and tef1 were constructed in IQ-TREE 1.6.12 (bootstrap replicates = 1000) using the nucleotide substitution models of TN + F + I + G4 and TN + F + R2. Circles at the nodes indicate ultrafast bootstrap values > 80 given by IQ-TREE. Genome sequenced strains were shaded in green. The reference strains were provided with the GenBank accessions and the strain name, among which the strains with uncompleted reference information were shaded in orange. Results of the pairwise sequence similarity were illustrated on the dashed lines between the query strain and its closely related species (arrows point to the reference strains). The pairwise sequence similarity calculation was performed using the online tool Clustal OMEGA (https://www.ebi.ac.uk/Tools/msa/clustalo/)
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain TUCIM 5640 can be identified as T. sp. TUCIM 5640.
Identification of Trichoderma isolates for which WGSs have been deposited in public databases before July 2020
Corresponding sequences can be retrieved from public databases. Accession numbers, references for WGS, and the initial species identifications are listed in Table 1 and Fig. 2. The dataset includes several ex-type strains that do not require identification (i.e., they are reference strains). However, the sequence similarity analysis is also described for these strains.
We deliberately skipped the WGS mutants of T. reesei because the pedigree for the type strain QM 6a that leads to diverse industrial mutants is well documented in the literature (Druzhinina and Kubicek 2016). However, we included mutants of several other species that are used in agriculture and may be confused with the wild-type strains.
(3) NJAU 4742 (Tables 1 and 4; Fig. 10)
Identification: Pairwise sequence similarity of ITS between strain NJAU 4742 and the references that are given in the ITS56 dataset showed several values > 76%, which indicated that strain NJAU 4742 belongs to the genus Trichoderma. The similarity of rpb2 between strain NJAU 4742 and the most closely related species T. pyramidale (strain CBS 135574) that is found at this locus was 97.79% (Table 2; Fig. 10), while the similarity of tef1 between strain NJAU 4742 and the most closely related species T. guizhouense (strain CBS 131803) that is found at this locus was 100.00% (Table 2; Fig. 10) (i.e., the condition ∃!(rpb299 ≅ tef197) was not met). This indicates that strain NJAU 4742 can be recognized as a putative new species that has non-concordant phylogenies of rpb2 and tef1 (Comment 3).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain NJAU 4742 can be identified as a putative new species T. sp. NJAU 4742. Due to the value of this strain for the development of biofertilizers, we propose a provisional name to this species as T. shenii nom. prov. The formal taxonomic description will be presented elsewhere upon additional sampling.
(4) M10 (Tables 1 and 4; Fig. 10)
Identification: The pairwise sequence similarity of ITS between strain M10 and the references that are given in the ITS56 dataset showed several values > 76%, which indicated that strain M10 belongs to the genus Trichoderma. The similarity of rpb2 between strain M10 and the most closely related species T. bannaense (strain HMAS 248840) that is found at this locus was 97.79% (Table 2; Fig. 10), and the similarity of tef1 between strain M10 and the most closely related species that are found at this locus were all < 97% (Table 2; Fig. 10) (i.e., the condition ∃!(rpb299 ≅ tef197) was not met). This indicates that strain M10 can be recognized as a putative new species (Comment 3).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain M10 can be identified as T. sp. M10.
(5) T. harzianum CBS 226.95, type strain (Tables 1 and 4; Fig. 10)
Identification: not required for the type strain
The pairwise sequence similarity of ITS between strain CBS 226.95 and the references that are given in the ITS56 dataset showed several values > 76%, which indicated that strain CBS 226.95 belongs to the genus Trichoderma. Strain CBS 226.95 is the ex-type strain of species T. harzianum sensu stricto. The similarity of rpb2 and tef1 between strain CBS 226.95 and the most closely related species T. harzianum (itself) that is found at this locus was 100.00% (Table 2; Fig. 10), and the similarity of tef1 between strain CBS 226.95 and the most closely related species T. harzianum (itself) that is found at this locus was 100.00% (Table 2; Fig. 10) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete.
(6) B97 (Tables 1 and 4; Fig. 10)
Identification: The pairwise sequence similarity of ITS between strain B97 and the references that are given in the ITS56 dataset showed several values > 76%, which indicated that strain B97 belongs to the genus Trichoderma. The similarity of rpb2 between strain B97 and the most closely related species T. harzianum (strain CBS 226.95) that is found at this locus was 99.51% (Table 2; Fig. 10), and the similarity of tef1 between strain B97 and the most closely related species T. harzianum (strain CBS 226.95) that is found at this locus was 99.60% (Table 2; Fig. 10) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain B97 can be identified as T. harzianum.
(7) TR274 (Tables 1 and 4; Fig. 10)
Identification: The pairwise sequence similarity of ITS between strain TR274 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain TR274 belongs to the genus Trichoderma. The similarity of rpb2 between strain TR274 and the most closely related species T. harzianum (strain CBS 226.95) that is found at this locus was 99.51% (Table 2; Fig. 10), and the similarity of tef1 between strain TR274 and the most closely related species T. harzianum (strain CBS 226.95) that was found at this locus was 100.00% (Table 2; Fig. 10) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain TR274 can be identified as T. harzianum.
(8) T6776 (Tables 1 and 4; Fig. 10)
Identification: The pairwise sequence similarity of ITS between strain T6776 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain T6776 belongs to the genus Trichoderma. The similarity of rpb2 between strain T6776 and the most closely related species T. afroharzianum (strain CBS 124620) that is found at this locus was 99.88% (Table 2; Fig. 10), and the similarity of tef1 between strain T6776 and the most closely related species T. afroharzianum (strain CBS 124620) that is found at this locus was 99.61% (Table 2; Fig. 10) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain T6776 can be identified as T. afroharzianum. The same conclusion was obtained in Kubicek et al. (2019).
(9) T22 (Tables 1 and 4; Fig. 10)
Identification: The pairwise sequence similarity of ITS between strain T22 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain T22 belongs to the genus Trichoderma. The similarity of rpb2 between strain T22 and the most closely related species T. afroharzianum (strain CBS 124620) that is found at this locus was 100.00% (Table 2; Fig. 10), and the similarity of tef1 between strain T22 and the most closely related species T. afroharzianum (strain CBS 124620) that was found at this locus was 98.82% (Table 2; Fig. 10) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain T22 can be identified as T. afroharzianum. Similar conclusion was obtained in Chaverri et al. (2015).
Note: This is a laboratory strain that was obtained in vitro as a UV treated protoplast fusion hybrid of the benomyl-resistant strain T-95 (ATCC 60850) and T12m (ATCC 20737) (Stasz et al. 1988).
(10) IMV 00454 (Tables 1 and 4; Fig. 10)
Identification: The pairwise sequence similarity of ITS between strain IMV 00454 and the references that are given in the ITS56 dataset showed several values > 76%, which indicated that strain IMV 00454 belongs to the genus Trichoderma. The similarity of rpb2 between strain IMV 00454 and the most closely related species T. simmonsii (strain CBS 130431) that is found at this locus was 100.00% (Table 2; Fig. 10), and the similarity of tef1 between strain IMV 00454 and the most closely related species T. simmonsii (strain CBS 130431) that is found at this locus was 99.69% (Table 2; Fig. 10) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain IMV 00454 can be identified as T. simmonsii.
(11) CFAM-422 (Tables 1 and 4; Fig. 10)
Identification: The pairwise sequence similarity of ITS between strain CFAM-422 and the references that are given in the ITS56 dataset showed several values > 76%, which indicated that strain CFAM-422 belongs to the genus Trichoderma. The similarity of rpb2 between strain CFAM-422 and the most closely related species including T. afarasin (strain CBS 130742), T. lentiforme (strain DIS 253B), and T. endophyticum (strain CBS 130730) that are found at this locus was 99.75%, 99.75%, and 99.51%, respectively (Table 2; Fig. 10), while the similarity of tef1 between strain CFAM-422 and the most closely related species including T. afarasin (strain CBS 130742) and T. endophyticum (strain CBS 130730) that are found at this locus was 98.23% and 99.80%, respectively (Table 2; Fig. 10) (i.e., the condition ∃!(rpb299 ≅ tef197) was not met. This indicates that strain CFAM-422 can be recognized as T. afarasin or T. endophyticum (Comment 3).
Validation: The reference materials that were used in the molecular identification in this case were not complete due to the lack of sequences from the ex-type strains of several related species (the reference sequences used in this case were obtained from the published voucher materials, which may require taxonomic revision) (Comment 3). The identification was precise, but inaccurate, and ambiguous. Therefore, the strain CFAM-422 can be identified as T. cf. endophyticum.
(12) TPhu1 (Tables 1 and 4; Fig. 10)
Identification: The pairwise sequence similarity of ITS between strain TPhu1 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain TPhu1 belongs to the genus Trichoderma. The similarity of rpb2 between strain TPhu1 and the most closely related species T. amazonicum (strain CBS 126898) and T. pleuroti (strain CBS 124387) that are found at this locus were 98.89% and 98.87%, respectively (Table 2; Fig. 10), and the similarity of the tef1 between strain TPhu1 and the most closely related species T. pleuroti (strain CBS 124387) that was found at this locus was 98.10% (Table 2; Fig. 10) (i.e., the condition ∃!(rpb299 ≅ tef197) was not met), indicating strain TPhu1 can be recognized as a putative new species (Comment 3).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain TPhu1 can be identified as T. sp. TPhu1.
(13) Tr1 (Tables 1 and 4; Fig. 10)
Identification: The pairwise sequence similarity of ITS between strain Tr1 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain Tr1 belongs to the genus Trichoderma. The similarity of rpb2 between strain Tr1 and the most closely related species T. pleuroticola (strain CBS 124383) that is found at this locus was 99.02% (Table 2; Fig. 10), and the similarity of tef1 between strain Tr1 and the most closely related species T. pleuroticola (strain CBS 124383) that is found at this locus was 100.00% (Table 2; Fig. 10) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain Tr1 can be identified as T. pleuroticola.
(14) ITEM 908 (Tables 1 and 4; Fig. 10)
Identification: The pairwise sequence similarity of ITS between strain ITEM 908 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain ITEM 908 belongs to the genus Trichoderma. The similarity of rpb2 between strain ITEM 908 and the most closely related species including T. atrobrunneum (strain G.J.S. 98-183) that is found at this locus was 100.00% (Table 2; Fig. 11), while the similarity of tef1 between strain ITEM 908 and the most closely related species T. atrobrunneum (strain G.J.S. 98-183) that is found at this locus was 95.55% (Table 2; Fig. 11) (i.e., the condition ∃!(rpb299 ≅ tef197) was not met).
Validation: The reference materials that were used in the molecular identification in this case were not complete due to the short sequence of tef1 from the ex-type strain of T. atrobrunneum (Comment 3). The identification was precise, but inaccurate and ambiguous. Therefore, the strain ITEM 908 can be identified as T. cf. atrobrunneum.
(15) T. virens Gv29-8, type strain (Tables 1 and 4; Fig. 10)
Identification: not required for the type strain
The pairwise sequence similarity of ITS between strain Gv29-8 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain Gv29-8 belongs to the genus Trichoderma. Strain Gv29-8 is the ex-type strain of species T. virens. The similarity of rpb2 between strain Gv29-8 and the most closely related species T. virens (itself) that was found at this locus was 100.00% (Table 2; Fig. 10), and the similarity of tef1 between strain Gv29-8 and the most closely related species T. virens (itself) that was found at this locus was 100.00% (Table 2; Fig. 10) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete.
(16) FT-333 (Tables 1 and 4; Fig. 10)
Identification: The pairwise sequence similarity of ITS between strain FT-333 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain FT-333 belongs to the genus Trichoderma. The similarity of rpb2 between strain FT-333 and the most closely related species T. virens (strain Gv29-8) that is found at this locus was 100.00% (Table 2; Fig. 10), and the similarity of tef1 between strain FT-333 and the most closely related species T. virens (strain Gv29-8) that is found at this locus was 100.00% (Table 2; Fig. 10) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain FT-333 can be identified as T. virens.
(17) Tv-1511 (Tables 1 and 4; Fig. 10)
Identification: The pairwise sequence similarity of ITS between strain Tv-1511 and the references that were given in ITS56 dataset showed several values > 76%, which indicated that strain Tv-1511 belongs to the genus Trichoderma. The similarity of rpb2 between strain Tv-1511 and the most closely related species T. virens (strain Gv29-8) that is found at this locus was 100.00% (Table 2; Fig. 10), and the similarity of tef1 between strain Tv-1511 and the most closely related species T. virens (strain Gv29-8) that were found at this locus was 99.80% (Table 2; Fig. 10) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate and unambiguous. Therefore, the strain Tv-1511 can be identified as T. virens.
(18) IMI 304061 (Tables 1 and 4; Fig. 10)
Identification: The pairwise sequence similarity of ITS between strain IMI 304061 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain IMI 304061 belongs to the genus Trichoderma. The similarity of rpb2 between strain IMI 304061 and the most closely related species T. neocrassum (strain G.J.S. 01–227) that is found at this locus was 99.26% (Table 2; Fig. 10), while the similarity of tef1 between strain IMI 304061 and the most closely related species T. virens (strain Gv29-8) that is found at this locus was 97.26% (Table 2; Fig. 10) (i.e., the condition ∃!(rpb299 ≅ tef197) was not met). The similarity assessment and phylogeny of the tef1 locus indicated that strain IMI 304061 can be recognized as a putative new species other than T. virens and T. neocrassum (Comment 3).
Validation: The reference materials used in the molecular identification in this case were not complete due to the short sequence of tef1 from the ex-type strain of T. neocrassum (strain G.J.S. 01-227, Comment 3). The identification is precise and accurate but ambiguous. Therefore, the strain IMI 304061 can be identified as T. sp. aff. neocrassum IMI 304061.
(19) T. reesei QM 6a, type strain (Tables 1 and 4; Fig. 11)
Identification: not required for the type strain
The pairwise sequence similarity of ITS between strain QM 6a and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain QM 6a belongs to the genus Trichoderma. Strain QM 6a is the ex-type strain of species T. reesei. The similarity of rpb2 between strain QM 6a and the most closely related species T. reesei (itself) that is found at this locus was 100.00% (Table 2; Fig. 11), and the similarity of tef1 between strain QM 6a and the most closely related species T. reesei (itself) that is found at this locus was 100.00% (Table 2; Fig. 11) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete.
(20) CBS 999.97 (Tables 1 and 4; Fig. 11)
Identification: The pairwise sequence similarity of ITS between strain CBS 999.97 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain CBS 999.97 belongs to the genus Trichoderma. The similarity of rpb2 between strain CBS 999.97 and the most closely related species T. reesei (strain QM 6a) that is found at this locus was 99.75% (Table 2; Fig. 11), and the similarity of tef1 between strain CBS 999.97 and the most closely related species T. reesei (strain QM 6a) that is found at this locus was 99.60% (Table 2; Fig. 11) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain CBS 999.97 can be identified as T. reesei.
(21) T. parareesei CBS 125925, type strain (Tables 1 and 4; Fig. 11)
Identification: not required for the type strain
The pairwise sequence similarity of ITS between strain CBS 125925 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain QM 6a belongs to the genus Trichoderma. Strain CBS 125925 is the ex-type strain of species T. parareesei. The similarity of rpb2 between strain CBS 125925 and the most closely related species T. parareesei (itself) that is found at this locus was 100.00% (Table 2; Fig. 11), and the similarity of tef1 between strain CBS 125925 and the most closely related species T. parareesei (itself) that is found at this locus was 100.00% (Table 2; Fig. 11) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain CBS 125925 can be identified as T. parareesei.
(22) T. longibrachiatum CBS 816.68, type stain (Tables 1 and 4; Fig. 11)
Identification: not required for the type strain
The pairwise sequence similarity of ITS between strain CBS 816.68 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain CBS 816.68 belongs to the genus Trichoderma. Strain CBS 816.68 is the ex-type strain of species T. longibrachiatum. The similarity of rpb2 between strain CBS 816.68 and the most closely related species T. longibrachiatum (itself) that is found at this locus was 100.00% (Table 2; Fig. 11), and the similarity of tef1 between strain CBS 816.68 and the most closely related species T. longibrachiatum (itself) that is found at this locus was 100.00% (Table 2; Fig. 11) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain CBS 816.68 can be identified as T. longibrachiatum.
(23) SMF2 (Tables 1 and 4; Fig. 11)
Identification: The pairwise sequence similarity of ITS between strain SMF2 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain SMF2 belongs to the genus Trichoderma. The similarity of rpb2 between strain SMF2 and the most closely related species T. longibrachiatum (strain CBS 816.68) that is found at this locus was 99.88% (Table 2; Fig. 11), and the similarity of tef1 between strain SMF2 and the most closely related species T. longibrachiatum (strain CBS 816.68) that is found at this locus was 98.97% (Table 2; Fig. 11) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain SMF2 can be identified as T. longibrachiatum.
(24) MK1 (Tables 1 and 4; Fig. 11)
Identification: The pairwise sequence similarity of ITS between strain MK1 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain MK1 belongs to the genus Trichoderma. The similarity of rpb2 between strain MK1 and the most closely related species T. longibrachiatum (strain CBS 816.68) that is found at this locus was 99.75% (Table 2; Fig. 11), and the similarity of tef1 between strain MK1 and the most closely related species T. longibrachiatum (strain CBS 816.68) that is found at this locus was 100.00% (Table 2; Fig. 11) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain MK1 can be identified as T. longibrachiatum.
(25) JCM 1883 (Tables 1 and 4; Fig. 11)
Identification: The pairwise sequence similarity of ITS between strain JCM 1883 and the references that are given in the ITS56 dataset showed several values > 76%, which indicated that strain JCM 1883 belongs to the genus Trichoderma. The similarity of rpb2 between strain MK1 and the most closely related species T. longibrachiatum (strain CBS 816.68) that is found at this locus was 99.75% (Table 2; Fig. 11), and the similarity of tef1 between strain JCM 1883 and the most closely related species T. longibrachiatum (strain CBS 816.68) that is found at this locus was 100.00% (Table 2; Fig. 11) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain JCM 1883 can be identified as T. longibrachiatum.
(26) TUCIM 6016 (Tables 1 and 4; Fig. 11)
Identification: The pairwise sequence similarity of ITS between strain TUCIM 6016 and the references that are given in the ITS56 dataset showed several values > 76%, which indicated that strain TUCIM 6016 belongs to the genus Trichoderma. The similarity of rpb2 between strain TUCIM 6016 and the most closely related species including T. citrinoviride (strain CBS 258.85) that is found at this locus was 90.84% (Table 2; Fig. 11), while the similarity of tef1 between strain TUCIM 6016 and the most closely related species T. citrinoviride (strain CBS 258.85) that is found at this locus was 99.81% (Table 2; Fig. 11) (i.e., the condition ∃!(rpb299 ≅ tef197) was not met).
Validation: The reference materials used in the molecular identification in this case were not complete due to the short sequence of rpb2 from the ex-type strain (Comment 3). The identification was precise, but inaccurate and ambiguous. Therefore, the strain TUCIM 6016 can be identified as T. cf. citrinoviride.
(27) T. atroviride IMI 206040, (Tables 1 and 4; Fig. 12)

Molecular identification of genome-sequenced strains from the Section Trichoderma using maximum-likelihood (ML) phylogeny and pairwise sequence similarity calculation. The ML phylograms of rpb2 and tef1 were constructed in IQ-TREE 1.6.12 (bootstrap replicates = 1000) using the nucleotide substitution models of TNe + G4 and HKY + F + G4. Circles at the nodes indicate ultrafast bootstrap values > 80 given by IQ-TREE. Genome sequenced strains were shaded in green. The reference strains were provided with the GenBank accessions and the strain name, among which the strains with uncompleted reference information were shaded in orange. Results of the pairwise sequence similarity were illustrated on the dashed lines between the query strain and its closely related species (arrows point to the reference strains). The pairwise sequence similarity calculation was performed using the online tool ClustalOMEGA (https://www.ebi.ac.uk/Tools/msa/clustalo/)
The pairwise sequence similarity of ITS between strain IMI 206040 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain IMI 206040 belongs to the genus Trichoderma. Strain IMI 206040 is not the ex-type strain of species T. atroviride but is considered as a reference strain (Kubicek et al. 2011, 2019). The similarity of rpb2 between strain IMI 206040 and the most closely related species T. atroviride (itself) that is found at this locus was 100.00% (Table 2; Fig. 12), and the similarity of tef1 between strain IMI 206040 and the most closely related species T. atroviride (itself) that is found at this locus was 100.00% (Table 2; Fig. 12) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials were complete. The identification was precise, accurate and ambiguous. Therefore, the strain IMI 206040 can be identified as T. atroviride.
(28) P1, (Tables 1 and 4; Fig. 12)
Identification: The pairwise sequence similarity of ITS between strain P1 and the references that were given in ITS56 dataset showed several values > 76%, which indicated that strain P1 belongs to the genus Trichoderma. The similarity of rpb2 between strain P1 and the most closely related species T. atroviride (strain IMI 206040) that is found at this locus was 100.00% (Table 2; Fig. 12), and the similarity of tef1 between strain P1 and the most closely related species T. atroviride (strain IMI 206040) that is found at this locus was 99.43% (Table 2; Fig. 12) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete.
Note: this is a laboratory strain that was obtained in vitro from “T. harzianum 107” (DNA Barcoding was not available) by screening towards increased resistance to xenobiotics (Tronsmo, 1991). The strain is frequently treated as a wild-type isolate in research related to plant protection and growth promotion (biocontrol).
(29) XS2015 (Tables 1 and 4; Fig. 12)
Identification: The pairwise sequence similarity of ITS between strain XS2015 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain XS2015 belongs to the genus Trichoderma. The similarity of rpb2 between strain XS2015 and the most closely related species T. atroviride (strain IMI 206040) that is found at this locus was 100.00% (Table 2; Fig. 12), and the similarity of tef1 between strain XS2015 and the most closely related species T. atroviride (strain IMI 206040) that is found at this locus was 100.00% (Table 2; Fig. 12) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain XS2015 can be identified as T. atroviride.
(30) F7 (Tables 1 and 4; Fig. 12)
Identification: The pairwise sequence similarity of ITS between strain F7 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain F7 belongs to the genus Trichoderma. The similarity of rpb2 between strain F7 and the most closely related species T. atroviride (strain IMI 206040) that is found at this locus was 100.00% (Table 2; Fig. 12), and the similarity of tef1 between strain F7 and the most closely related species T. atroviride (strain IMI 206040) that is found at this locus was 99.43% (Table 2; Fig. 12) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification is precise, accurate, and unambiguous. Therefore, the strain F7 can be identified as T. atroviride.
(31) B10 (Tables 1 and 4; Fig. 12)
Identification: The pairwise sequence similarity of ITS between strain B10 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain B10 belongs to the genus Trichoderma. The similarity of rpb2 between strain B10 and the most closely related species T. atroviride (strain IMI 206040) that is found at this locus was 100.00% (Table 2; Fig. 12), and the similarity of tef1 between strain B10 and the most closely related species T. atroviride (strain IMI 206040) that is found at this locus was 99.43% (Table 2; Fig. 12) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate and unambiguous. Therefore, the strain B10 can be identified as T. atroviride.
(32) JCM 9410 (Tables 1 and 4; Fig. 12)
Identification: The pairwise sequence similarity of ITS between strain JCM 9410 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain JCM 9410 belongs to the genus Trichoderma. The similarity of rpb2 between strain JCM 9410 and the most closely related species T. atroviride (strain IMI 206040) that is found at this locus was 99.02% (Table 2; Fig. 12), and the similarity of tef1 between strain JCM 9410 and the most closely related species T. atroviride (strain IMI 206040) that is found at this locus was 100.00% (Table 2; Fig. 12) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain JCM 9410 can be identified as T. atroviride.
(33) LY357 (Tables 1 and 4; Fig. 12)
Identification: The pairwise sequence similarity of ITS between strain LY357 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain LY357 belongs to the genus Trichoderma. The similarity of rpb2 between strain LY357 and the most closely related species including T. paratroviride (strain CBS 136489) and T. atroviride (strain IMI 206040) that are found at this locus were 98.65% and 97.79%, respectively (Table 2; Fig. 12), and the similarity of tef1 between strain LY357 and the most closely related species including T. paratroviride (strain CBS 136489) and T. atroviride (strain IMI 206040) that are found at this locus were 83.37% and 91.29%, respectively (Table 2; Fig. 12) (i.e., the condition ∃!(rpb299 ≅ tef197) was not met). This indicates that strain LY357 can be recognized as a putative new species (Comment 3.4).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain LY357 can be identified as T. sp. LY357.
(34) T6085 (Tables 1 and 4; Fig. 12)
Identification: The pairwise sequence similarity of ITS between strain T6085 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain T6085 belongs to the genus Trichoderma. The similarity of rpb2 between strain T6085 and the most closely related species T. gamsii (strain G.J.S. 04-09) that is found at this locus was 99.38% (Table 2; Fig. 12), and the similarity of tef1 between strain T6085 and the most closely related species T. gamsii (strain G.J.S. 04-09) that is found at this locus was 97.31% (Table 2; Fig. 12) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain T6085 can be identified as T. gamsii.
(35) A5MH (Tables 1 and 4; Fig. 12)
Identification: The pairwise sequence similarity of ITS between strain A5MH and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain A5MH belongs to the genus Trichoderma. The similarity of rpb2 between strain A5MH and the most closely related species T. gamsii (strain G.J.S. 04-09) that is found at this locus was 99.63% (Table 2; Fig. 12), and the similarity of tef1 between strain A5MH and the most closely related species T. gamsii (strain G.J.S. 04-09) that is found at this locus was 95.98% (Table 2; Fig. 12) (i.e., the condition ∃!(rpb299 ≅ tef197) was not met).Thus, A5MH is a putative new species that is closely related to T. gamsii (Comment 3).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain A5MH can be identified as T. sp. aff. gamsii A5MH.
(36) POS7 (Tables 1 and 4; Fig. 12)
Identification: The pairwise sequence similarity of ITS between strain POS7 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain POS7 belongs to the genus Trichoderma. The similarity of rpb2 between strain POS7 and the most closely related species T. koningiopsis (strain CBS 119075) that is found at this locus was 98.89% (Table 2; Fig. 12), and the similarity of tef1 between strain POS7 and the most closely related species T. koningiopsis (strain CBS 119075) that is found at this locus was 96.71% (Table 2; Fig. 12) (i.e., the condition ∃!(rpb299 ≅ tef197) was not met). This indicates that strain POS7 can be recognized as a putative new species closely related to T. koningiopsis (Comment 3).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain POS7 can be identified as T. sp. aff. koningiopsis POS7.
(37) T. asperellum CBS 433.95, type strain (Tables 1 and 4; Fig. 12)
Identification: not required for the type strain
The pairwise sequence similarity of ITS between strain CBS 433.95 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain CBS 433.95 belongs to the genus Trichoderma. Strain CBS 433.95 is the ex-type strain of species T. atroviride. The similarity of rpb2 between strain CBS 433.95 and the most closely related species T. asperellum (itself) that is found at this locus was 100.00% (Table 2; Fig. 12), and the similarity of tef1 between strain CBS 433.95 and the most closely related species T. asperellum (itself) that is found at this locus was 100.00% (Table 2; Fig. 12) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain CBS 433.95 can be identified as T. asperellum.
(38) B05 (Tables 1 and 4; Fig. 12)
Identification: The pairwise sequence similarity of ITS between strain B05 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain B05 belongs to the genus Trichoderma. The similarity of rpb2 between strain B05 and the most closely related species including T. kunmingense (strain YMF1.02659) and T. asperellum (strain CBS 433.97) that are found at this locus were 99.88% and 99.51%, respectively (Table 2; Fig. 12), and the similarity of tef1 between strain B05 and the most closely related species including T. kunmingense (strain YMF1.02659) and T. asperellum (strain CBS 433.97) that are found at this locus were 91.92% and 99.81%, respectively (Table 2; Fig. 12) (i.e., the condition ∃!(rpb299 ≅ tef197) was not met).
Validation: The reference materials that were used in the molecular identification in this case were not complete due to the short sequences of tef1 from the ex-type strain of T. kunmingense (strain YMF1.02659) (Comment 3), indicating that strain B05 can be recognized as T. asperellum without excluding its possibility of being T. kunmingense (Comment 3). Thus, the group of T. asperellum and the species closely related to it may need a critical taxonomic revision. The identification was precise, but inaccurate and ambiguous. Therefore, the strain B05 can be identified as T. cf. asperellum.
(39) TR356 (Tables 1 and 4; Fig. 12)
Identification: The pairwise sequence similarity of ITS between strain TR356 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain TR356 belongs to the genus Trichoderma. The similarity of rpb2 between strain TR356 and the most closely related species T. asperelloides (strain G.J.S. 04-111) that is found at this locus was 99.88% (Table 2; Fig. 12), and the similarity of tef1 between strain TR356 and the most closely related species T. asperelloides (strain G.J.S. 04-111) that is found at this locus was 100.00% (Table 2; Fig. 12) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain TR356 can be identified as T. asperelloides.
(40) Ts93 (Tables 1 and 4; Fig. 12)
Identification: The pairwise sequence similarity of ITS between strain Ts93 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain Ts93 belongs to the genus Trichoderma. The similarity of rpb2 between strain Ts93 and the most closely related species T. asperelloides (strain G.J.S. 04-111) that is found at this locus was 99.75% (Table 2; Fig. 12), and the similarity of tef1 between strain Ts93 and the most closely related species T. asperelloides (strain G.J.S. 04-111) that is found at this locus was 99.62% (Table 2; Fig. 12) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain Ts93 can be identified as T. asperelloides.
(41) GD12 (Tables 1 and 4; Fig. 12)
Identification: The pairwise sequence similarity of ITS between strain GD12 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain GD12 belongs to the genus Trichoderma. The similarity of rpb2 between strain GD12 and the most closely related species T. hamatum (strain DAOM 167057) that is found at this locus was 100.00% (Table 2; Fig. 12), and the similarity of tef1 between strain GD12 and the most closely related species T. hamatum (strain DAOM 167057) that is found at this locus was 99.22% (Table 2; Fig. 12) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain GD12 can be identified as T. hamatum.
(42) IBT 40837 (Tables 1 and 4; Fig. 13)

Molecular identification of genome-sequenced strains from the Brevicompactum clades using maximum-likelihood (ML) phylogeny and pairwise sequence similarity calculation. The ML phylograms of rpb2 and tef1 were constructed in IQ-TREE 1.6.12 (bootstrap replicates = 1000) using the nucleotide substitution models of TIM2e and HKY + F + I. Circles at the nodes indicate ultrafast bootstrap values > 80 given by IQ-TREE. Genome sequenced strains were shaded in green. The reference strains were provided with the GenBank accessions and the strain name, among which, strains with uncompleted reference information were shaded in orange. Results of the pairwise sequence similarity were illustrated on the dashed lines between the query strain and its closely related species (arrows point to the reference strains). The pairwise sequence similarity calculation was performed using the online tool of ClustalOMEGA (https://www.ebi.ac.uk/Tools/msa/clustalo/)
Identification: The pairwise sequence similarity of ITS between strain IBT 40837 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain IBT 40837 belongs to the genus Trichoderma. The similarity of rpb2 between strain IBT 40837 and the most closely related species T. arundinaceum (strain CBS 119575) that is found at this locus was 100.00% (Table 2; Fig. 13), and the similarity of tef1 between strain IBT 40837 and the most closely related species T. arundinaceum (strain CBS 119575) that is found at this locus was 100.00% (Table 2; Fig. 13) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification was precise, accurate, and unambiguous. Therefore, the strain IBT 40837 can be identified as T. arundinaceum.
(43) IBT 40841 (Tables 1 and 4; Fig. 13)
Identification: The pairwise sequence similarity of ITS between strain IBT 40841 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain IBT 40841 belongs to the genus Trichoderma. The similarity of rpb2 between strain IBT 40841 and the most closely related species T. brevicompactum (strain CBS 109720) that is found at this locus was 100.00% (Table 2; Fig. 13), and the similarity of tef1 between strain IBT 40841 and the most closely related species including T. brevicompactum (strain CBS 109720) that is found at this locus was 93.67% (Table 2; Fig. 13) (i.e., the condition ∃!(rpb299 ≅ tef197) was not met).
Validation: The reference materials that were used in the molecular identification in this case were not complete due to the short sequences of tef1 from the ex-type strain of T. brevicompactum (strain CBS 109720) (Comment 3). The identification was precise and accurate but ambiguous. Therefore, the strain IBT 40841 can be identified as T. cf. brevicompactum.
(44) TW21990_1 (Tables 1 and 4)
Identification: The pairwise sequence similarity of ITS between strain TW21990_1 and the references that were given in the ITS56 dataset showed several values > 76%, which indicated that strain CBS 433.95 belongs to the genus Trichoderma. Strain TW21990_1 is the ex-type strain of species T. cyanodichotomus. The similarity of rpb2 between strain TW21990_1 and the most closely related species T. cyanodichotomus (itself) that is found at this locus was 100.00% (Table 2), and the similarity of tef1 between strain TW21990_1 and the most closely related species T. cyanodichotomus (itself) that is found at this locus was 100.00% (Table 2) (i.e., the condition ∃!(rpb299 ≅ tef197) was met).
Validation: The reference materials are complete. The identification is precise, accurate and unambiguous. Therefore, the strain TW21990_1 can be identified as T. cyanodichotomus.
Thus, the molecular identification protocol that was applied to 44 Trichoderma strains resulted in unambiguous identification of 38 (86%) strains and allowed assignment of 31 species names (including seven ex-type strains) (70%) and the proposal of eight new species (18%). Six (14%) identifications remained ambiguous because of either incomplete reference material or ambiguous taxonomy of the related species. Together, this result indicates the urgent need to achieve an agreement on the genus-wide criteria that are suitable to allow recognition of the species in Trichoderma and the requirement to complete the reference materials based on these criteria. Furthermore, the recognition of a considerable amount of putative new species indicates further rapid growth of Trichoderma diversity in the near future.
Discussion and suggestions
This study shows that identification of species is challenging for us and for most experts. As shown by the survey, Trichoderma researchers spent an average of one hour identifying the two strains based on three DNA barcodes for each, and achieved 50% accuracy. The rate of new species descriptions in the genus of Trichoderma was as high as approximately 50 per year, and this number is expected to increase faster in the future. Therefore, we selected a white paper format to present a detailed review on Trichoderma taxonomy, exploring the problem of molecular identification and proposing a possible solution in a form of an authoritative guideline.
We aimed to develop a protocol for the molecular identification of Trichoderma that should reflect the contemporary taxonomy of the genus. This means that where possible, we avoided an option of a taxonomic revision for a particular group or the entire genus (see for example, Houbraken et al. 2020). Instead, we considered Trichoderma to be a genus in its privileged taxonomic position because most of its species have been delimitated after the introduction of DNA-based methods. Trichoderma has received much attention from fungal taxonomists, which has resulted in the ample new species descriptions based on the newest (DNA-based) technologies and concepts (Seifert and Rossman 2010) that were mainly published over the last decade (Fig. 2). The “recently taxonomically resolved” state for Trichoderma taxonomy (that we believe is a correct assessment) was also considered to be an argument in support of the initiation of the whole-genus genomics project for Trichoderma (https://genome.jgi.doe.gov/portal/Genwidrichoderma/Genwidrichoderma.info.html) as taxonomy underlines all biological studies. Thus, our intention was to “measure” genetic similarities and dissimilarities that have already been used by the Trichoderma taxonomy providers and incorporate them into the DNA Barcoding protocol. In this manner, we hoped to balance the contradiction between the ultimate subjectivity in the species recognition and the need for the exact species identification that is crucial for applications, patenting, and research purposes. The availability of such a protocol should facilitate the accurate, precise, and unambiguous identification of Trichoderma species and beneficially contribute to the development of applications and research on these fungi.
We previously proposed an automated oligonucleotide DNA Barcoding tool for Trichoderma (Druzhinina et al. 2005; Kopchinskiy et al. 2005) that was based on ITS for approximately 100 species and was widely appreciated by the researchers for its unambiguous results and simplicity. Due to the insufficient variability of ITS between Trichoderma species (see above), this tool is no longer functional. The current study reveals the following features and their combination that impeded the simplicity of the molecular identification protocol that is presented here:
-
1.
Most Trichoderma species cannot be identified by a sequence similarity search or by the multiloci phylogenetic analysis if it is applied alone.
-
2.
The identification procedure requires three DNA barcoding loci, and sequences have to be prepared (trimmed) for the analysis.
-
3.
The retrieval of reference materials and the calculations of the pairwise similarities are tedious and they frequently need to be performed manually.
-
4.
In silico results require validation against the availability of reference materials (Figs. 10–13).
It is now evident for all DNA barcoded fungi that any molecular identification requires its biological verification as a necessary step (Lücking et al. 2020). The combination of several in silico methods was already appreciated by Trichoderma experts because 27 of the 47 respondents who completed our online survey did not rely on any of the methods alone, and instead, they used all the available tools. Therefore, the users of Trichoderma taxonomy are expected to have skills not only in mycology, fungal taxonomy, basic DNA techniques, but also sequence analysis.
To date, the sufficient training of taxonomy users is essential because there is no clear distinction between taxonomy users and taxonomy providers. The diversity of Trichoderma is such that the initial taxonomy users frequently detect potentially novel species and start their descriptions, i.e., become taxonomy providers. Conversely, taxonomy providers are usually the most dedicated users of existing taxonomy, but the work of taxonomy providers is essentially more laborious and is associated with more responsibility because the outcome (the taxonomic and nomenclatural acts, e.g., the formally described taxonomic entities) influences the development of taxonomic standards that are applied to a particular group of organisms. This study demonstrates how the results that were obtained by a few Trichoderma taxonomy providers in the last decade strongly impact the ambiguity of Trichoderma species identification and the application of species recognition criteria.
The transformations of the genealogical concordance species concept in Trichoderma taxonomy
Regardless of the species recognition criteria that are used, fungal taxonomy allows room for subjectivity in the assessment of species borders. In Trichoderma, this can be exemplified by many cases when taxonomists observed a considerable genetic, ecological, and phenotypic polymorphism within a particular group, but they did not find it sufficient for the species delimitations [see T. harzianum sensu Chaverri and Samuels (2003) or T. guizhouense sensu Chaverri et al. (2015)]. It is also possible that the same researchers change their assessment of species borders as more materials are studied [see the revision of the Harzianum Clade by Chaverri et al. (2015)]. However, numerous morphologically identical and genetically highly similar species have been named and formally described as cryptic taxa based on subtle genetic distance [e.g., T. bissettii was delimited from T. longibrachiatum (Sandoval-Denis et al. 2014) and T. kunmingense was separated from T. asperellum (Qiao et al. 2018)].
The ambiguity of taxonomy reflects the diversity of species recognition criteria that are applicable for fungi [recently reviewed by Lücking et al. (2020)]. However, only a few could be potentially suitable for the genus Trichoderma (Druzhinina and Kubicek 2005). Among them, the morphological species concept is no longer suitable for this genus because even the largest infrageneric groups, sections, are not always morphologically distinguishable [for example, see the transfer of the famous biocontrol strain P1 from T. harzianum (Tronsmo 1991) to T. atroviride (Mach et al. 1999)]. The high ambiguity of morphological identification of Trichoderma is no longer discussed. The biological species concept that is verifiable through in vitro mating is restricted to a single species T. reesei (Seidl et al. 2009) because none of the other species that have been found to date could repeatedly produce fruiting bodies in vitro. Therefore, the genealogical concordance phylogenetic species recognition (GCPSR) concept (Taylor et al. 2000) is the most widely claimed approach in this genus (see references below). After detecting many cryptic species, GCPSR became the only suitable option for species delimitation in Trichoderma. Although this concept was shown to be a powerful tool for species delimitation (Druzhinina and Kubicek 2005; Druzhinina et al. 2005; Jaklitsch 2009, 2011; Jaklitsch et al. 2013; Jaklitsch and Voglmayr 2015; Chen and Zhuang 2017a, b, d; Qin and Zhuang 2017), the two decades of its application, at least in Trichoderma, revealed several shortcomings. GCPSR requires the concordance of phylogram topologies from at least two unlinked loci that are not contradicted by the other loci (Taylor et al. 2000) (Fig. 14). In practice, the application of GCPSR assumes (i) the consideration of individual trees and (ii) sets of several strains per each species, which ultimately include reference materials for all species in questions. For example, Druzhinina et al. (2008) studied the evolutionary relationships between such species as T. longibrachiatum, T. orientale, and several related strains (Fig. 14a). They constructed single locus phylograms for tef1, chi18-5 (ech42), and cal1 (Box 1). The topologies and statistical supports for HTUs (hypothetical taxonomic units, internal nodes on phylograms) for tef1 and chi18-5 were highly concordant and revealed four monophyletic phylogenetic species (PS I–PS IV, Fig. 14a), which were supported by statistically significant posterior probabilities. The topology of cal1 did not contradict this conclusion. However, the resolution in cal1 phylogram was low. Nevertheless, this analysis allowed the application of GCPSR and the conclusion that individual PSs corresponded to four phylogenetic species (Druzhinina et al. 2008) that were then taxonomically described (Druzhinina et al. 2012; Samuels et al. 2012). Conversely, a similar analysis that was performed in the Harzianum Clade [a “demon” of Trichoderma taxonomy, Druzhinina et al. (2010b)], which revealed that GCPSR could not be applied to this group (Fig. 14b) because all strains “jumped” from clade to clade in single loci phylograms (Fig. 14b). Thus, no clades seen on a combined phylogram (based on the concatenated three loci) were apparent on single-gene phylograms. Based on the species delimitation proposal (Taylor et al. 2000), the whole clade represented a single species [that was provisionally named as “pseudoharzianum matrix,” (Druzhinina et al. 2010b)] because the phylograms of tested loci contradicted each other at this level. To explain the cases of concordant phylogenies for the analysis of Section Longibrachiatum and non-concordant for the Harzianum Clade, the authors of each study performed analyses of genetic recombination. This showed that T. longibrachiatum s. s. is likely a (clonal) agamospecies, while T. orientale is most likely holomorphic (Druzhinina et al. 2008). However, the evidence of intensive sexual recombination was obtained for most of the strains in the Harzianum Clade, except the T. harzianum s. s. subclade (Druzhinina et al. 2010b) explaining “jumping” positions of individual strains on single-loci phylograms. This result prevented the authors from delimiting the Harzianum Clade in several species because the GCPSR concept was not applicable. There were no other species recognition criteria available, therefore no taxonomic acts were performed. Thus, these examples illustrated one frequent shortcoming when applying for GCPSR in Trichoderma. The analysis of single loci phylograms is a critical and compulsory step in the application of GCPSR. Additionally, ambiguous cases can be verified by the in silico tests for sexual recombination (Rossman et al. 2016) or other analyses.

Examples of genealogical concordance in the genus Trichoderma. a Phylogenetic trees taken from Druzhinina et al. (2008) that describe phylogenetic concordance of the three loci (tef1, chi18-5, and cal1) in the Section Longibrachiatum. PS I–PS IV correspond to phylogenetic species. Colors indicate statistically supported clades of the concatenated phylogram of the three loci. See Druzhinina et al. (2008) for details. b Phylogenetic trees taken from Druzhinina et al. (2010b) describing the lack of phylogenetic concordance of the three loci (tef1, chi18-5 and cal1) in the Harzianum Clade. Colors indicate clades seen on the concatenated phylogram of the three loci. See Druzhinina et al. (2008) for details
Unfortunately, in a rapidly increasing number of studies, the new Trichoderma species are delineated and described based on the analysis of a combined phylogram that was obtained from a concatenated alignment of several loci (Chaverri et al. 2011, 2015; Chen and Zhuang 2017a, b, d; Qin and Zhuang 2016c; Jaklitsch 2009, 2011; Jaklitsch and Voglmayr, 2015) without consideration of the single locus trees. Such studies do not rely of genealogical concordance. Although GCPSR is usually cited and claimed, species are delimitated based on the topology of a single tree, i.e. based on the phylogenetic species concept (Box 1). The authors use such parameters as the branch length, and statistical support for individual HTU to assign a species rank to a group of strains, or even frequently to a single strain (see below). One example is the delimitation of the Harzianum Clade (mentioned above) in a dozen new species based on the combined phylogram of act, tef1, cal1, and ITS (Chaverri et al. 2015). Our evaluation of the sequences provided by the authors showed that the taxonomic act was largely completed based on the phylogenetic signal mainly obtained from polymorphism of an approximately 250 bp-long fragments of the tef1 gene. This is because the three other loci (ITS, act, and cal1) were sampled for roughly 60% of isolates, and act and ITS were highly conserved. Because individual phylograms were not assessed, the strict sense GCPSR was not applied in that study. Moreover, the monographs of Jaklitsch (2009, 2011) on European species of Hypocrea and the work on Trichoderma diversity in Southern Europe (Jaklitsch and Voglmayr 2015) also do not contain single loci trees, but species were delimitated mainly (not only) based on the strict consensus phylogram of tef1 and rpb2. Moreover, W. Jaklitsch used not the hypervariable fragment of tef1 considered above, but the sixth exon from the tef1 gene, although he noted that it “shows less variability among species than rpb2” (Jaklitsch 2009). [Refer to Fig. 4c and respective text above describes that rpb2 is already highly conserved and species are delimited based on minor (< 1%) dissimilarity. It means that the polymorphism of tef1 exon fragment is neglectable.] This algorithm based on the concatenated phylograms of the two conserved loci (tef1 and rpb2) was then adopted in more recent taxonomic studies on Trichoderma that assigned > 90 new species (Chen and Zhuang 2016, 2017a, b, c; d; Qin and Zhuang 2016a, b, c, 2017). The drawback of this approach is the lack of the third marker that is strictly required for GCPSR. Moreover, the use of combined phylogeny does not allow evaluation of the concordance between the two loci and does not reveal their polymorphism. If one of the combined markers is not sufficiently variable or conserved, it does not contribute to the structure of the combined tree. The recombination and incongruences between tree topologies have become neglected. Thus, despite claims in the publications, species resolved based on the combined phylograms of the two or sometimes even several loci were not recognized based on the strict sense of the GCPSR concept, although a phylogenetic species recognition (PSR) concept was applied (See Definitions in Box 1). If we consider that these are at least 200 species described by the groups of W.M. Jaklitsch and W.Y. Zhuang, we can conclude that GCPSR, the most powerful and widely accepted species concept for fungi, have not been applied for the delimitation of the majority of Trichoderma species. Because the choice of a species criteria and concepts are not determined in the Code, we refrained from any evaluation of the rationale for some of species delimitations. Instead, we used this example to show how the work of taxonomy providers influences the applicability of species recognition criteria.
For about the first 10 years since its introduction by Taylor et al. (2000), the GCPSR concept in Trichoderma was implemented in its strictest sense (Atanasova and Druzhinina 2010; Druzhinina et al. 2008, 2010b; Komoń-Zelazowska et al. 2007; Jaklitsch et al. 2008a, b; Chaverri and Samuels 2003; Lu et al. 2004; Samuels et al. 2000, 2010; Degenkolb et al. 2008), which resulted in the deposition of DNA barcoding sequences for additional loci such as chi18-5 (ech42), acl1, cal1, act, and some others (Table 2). However, the shift to the two loci that was initiated during the last decade and the massive introduction of new species without consideration for the supplementary barcodes and frequently also without ITS (Table 2), reduced the usability of these supplementary DNA barcodes almost to zero.
The second drawback that comes from the non-strict application of GCPSR appears when species are recognized based on a few or even a single isolate. In this case, it is not possible to distinguish between species and populations. Therefore, multiple Trichoderma species that were described based on a single available isolate are ambiguous unless the unique ecophysiological or morphological features were detected.
The factual retreat in Trichoderma taxonomy from the application of GCPSR to the less powerful PSR has practical and theoretical explanations and consequences for precision and accuracy of taxonomy. First, the genus-wide taxonomic revisions [such as that performed by (Jaklitsch 2009, 2011)] require the simultaneous analysis of several hundred isolates and sequences of several hundred reference strains. Ideally, GCPSR could be applied if tef1 and rpb2 phylograms could be confronted. However, because the intron-containing tef1 DNA barcode locus is highly polymorphic, respective sequences cannot be aligned across the genus. The analysis will require the construction of numerous smaller separate phylogenetic trees for individual sections (such as those shown in Figs. 10–13). The conserved exon-containing tef1 fragment that was selected by Jaklitsch (2009, 2011) allowed the avoidance of multiple phylograms because it was suitable for alignment across the genus. However, the poor resolution of resulting trees was shown before (Chaverri and Samuels 2003) and also mentioned by this author. We would like to warn the researchers who are aiming at identification of the large collections of Trichoderma strains that the correct application of GCPSR will require the construction and analysis of numerous phylograms.
Second reason why the GCPSR concept was replaced by the PSR, is theoretical. GCPSR alone does not allow a decision to be made on the rank of concordant clades. For cryptic species, even the strict application of GCPSR cannot distinguish between taxa of different ranks (such as populations, species, or genera). For this reason, T. aggressivum and T. caribbaeum consist of ambiguously defined varieties (Samuels et al. 2002, 2006). Thus, we can conclude that although GCPSR is considered to be the most powerful concept (Nguyen et al. 2015a), it did not yet find its broad application in Trichoderma taxonomy.
As it has been already explained above, the revision of the distribution of DNA barcoding loci revealed that the currently available material for species identification within the genus Trichoderma (Table 2) makes DNA Barcoding limited to the three loci analysis among which, the concordance rpb2 ≅ tef1 should not be contradicted by ITS. Unexpectedly, it further raised the taxonomic value of ITS. In fungi, ITS fragments have numerous features that limit its taxonomic applicability [reviewed by Lücking et al. (2020)], but most of them are not known for Trichoderma. To the best of our knowledge, there were no reports on intragenomic polymorphism of this locus. However, a high number of homoplasious sites was demonstrated (Druzhinina et al. 2005) and there was insufficient polymorphism between many related species (Druzhinina and Kubicek 2005). Therefore, we do not recommend using ITS for phylogenetic analysis, but we suggest the similarity analysis for this locus that can be applied for assigning the genus delimitation.
The search for the best phylogenetic markers by Trichoderma taxonomy providers resulted in the mosaic and incomplete distribution of DNA barcoding loci and the genus phylogram (Table 2). These gaps can be filled if taxonomists worldwide have easy access to the reference strains’ cultures for additional sequencing. However, the practice shows that in some countries where fungal taxonomy develops very fast (such as China), the acquisition of reference strains from culture collections abroad is overly burdensome and costly such that it cannot be accomplished by most researchers. Conversely, shipment of reference strains, even from the authorized collections in China to other countries, is also complicated, expensive, and time-consuming. These non-scientific obstacles result in a bottleneck for the development of Trichoderma taxonomy and lead to the emergence of ambiguous species descriptions and increase the incomplete distribution of phylogenetic markers.
The only solution that we can propose is cooperation within the community of Trichoderma taxonomists. For example, a colleague “A” who is working on the taxonomic description of a species “X” that is related to species “Y”, which was described by colleague “B,” can request the latter person to provide sequences of additional DNA barcoding loci (Table 2) for species “Y”. For example, for T. changbaiense in a group of species that are related to T. fertile, providing either four missing chi18-5 (ech42) or three missing acl1 sequences could allow the application of the GCPSR concept and unambiguous species recognition. The current state of T. changbaiense species is ambiguous because its description does not correspond to the recommendations for the new fungal species description (Seifert and Rossman 2010). It has been described based on a single strain and the concatenated analysis of the two loci (Chen and Zhuang 2017a). Moreover, the morphology of T. changbaiense did not correspond to the related morphospecies, which also suggests the need for further sampling. Thus, the cooperation between taxonomists can aid in the in silico analysis. However, the exchange of sequence data will not replace the need to perform the comparative analysis of phenotypes and ecophysiological features will require consideration of the reference cultures in vitro, not only in silico.
In summary, sequencing of ITS, tef1, and rpb2 is currently the minimum sufficient set of phylogenetic markers that is required for the application of the GCPSR concept. In those cases, when these markers are not concordant (see examples above), consideration of other loci is required. The WGSs can provide enough material to resolve evolutionary positions of species with non-concordant phylogenies of rpb2 and tef1 and shared (identical) ITS sequences. However, because only 10% for Trichoderma species have been whole genome sequenced to date, the phylogenomic analysis for Trichoderma will not be available in the near future. We anticipate many new species that will be described based only on a few phylogenetic markers.
Testing the identifiability of every new species using the currently available materials for related strains is essential for species recognition. Comparative analysis of ecophysiological traits along with multiparametric phenotypes of a putative new species and the closely related taxa along with the application of the GCPSR concept will result in the most reliable species delimitation practice, a polyphasic approach (Lücking et al. 2020).
Comparative ecology aids identification of Trichoderma species
The reliability of species recognition in Trichoderma can be further aided by the analysis of DNA barcodes that are deposited for environmental samples and corresponding metadata that are recorded in public databases. In almost all cases, it will include the analysis of ITS. In fortunate cases where there are unique ITS sequences [e.g., T. asperelloides delimitated from T. asperellum, Samuels et al. (2010)], the sequences of new species can be searched in public databases for their occurrence in various habitats and ecosystems worldwide. The sequence similarity search in public databases that is performed with tef1 and rpb2 can also reveal other strains of a given new taxon among the pool of nearly 2000 taxonomically undefined records that were deposited as “Trichoderma sp.” in public databases (July 2020). The metadata for such records of the respective sequences can also serve as a useful supporting material for species description. Because most Trichoderma DNA barcodes were deposited in public databases within the last two decades, the authors of most sequences can be contacted, and a collaboration can frequently be established. For example, in our earlier study of Trichoderma diversity in Mediterranean sponges that was performed in collaboration with Oded Yarden’s group (Israel), we identified several potentially new species of Trichoderma (Gal-Hemed et al. 2011). The sequence similarity search in the NCBI GenBank revealed that strains with identical or highly similar DNA barcodes were already deposited by Karin Jacobs’ group (South Africa). These findings essentially supported our new species hypotheses because highly similar strains were found on the other continent. We contacted Professor Jacobs’ group and the cooperation between the three groups and the active exchange of materials between Austria, Israel, and South Africa resulted in the joint description of five new species (du Plessis et al. 2018). This cooperation arose from the analysis of sequences and respective metadata for strains deposited in a public database.
Suggestions for Trichoderma diversity studies
The popularity of the large-scale biodiversity surveys among mycologists worldwide and the relative ease of Trichoderma sampling and isolation attract many new researchers in this area. Based on our personal communications, at least several groups throughout the world, in particular, but not only, in China, are possessing collections consisting of several hundred or even thousands of Trichoderma isolates pending their taxonomic evaluations. As described above, the GCPSR (Taylor et al. 2000) and concept of cryptic fungal species (Struck et al. 2018) together with the broad availability of basic DNA techniques (PCR amplification and Sanger sequencing) result in the relative simplicity of the new species delimitation in this genus. Our assessments allow foreseeing the description of a considerable number of new species in the near future and urge us to propose genus-wide standards to discuss at the upcoming nomenclatural and taxonomic meetings. The most active providers of Trichoderma systematics are a few groups of highly experienced fungal taxonomists (Fig. 2, Table 2) who are invited to share their skills and knowledge with the beginners [see also fungi-wide recommendations in Lücking et al. (2020)]. The International Commission on Trichoderma Taxonomy (ICTT, www.trichoderma.info) or regular meetings such as the International Workshop on Trichoderma and Gliocladium or the Trichoderma Workshop that satellites the European Conference on Fungal Genetics (ECFG) offer opportunities for such exchanges. In Box 2 and below, we summarize practical recommendations that arose from this study and that can be useful for Trichoderma scientists that shift their research interest towards a taxonomy and hold collections of unidentified isolates.
We also propose that genus-wide standardization of species criteria that can be achieved if every new species hypothesis is to be first submitted to the ICTT board for the review and approval before committing to a taxonomic and nomenclatural act. In this way, the researchers can effectively communicate, exchange their Trichoderma experience and methods, and also compose the UpToDate global list of Trichoderma species names that is started in this study. The regulations and principles of such approvals can be discussed at the upcoming international meeting in consultation with the members of the parental International Commission on Taxonomy of Fungi (ICTF) (www.fungaltaxonomy.org), and the conclusions can be recorded in ICTT statues.
The responsible curation of deposited material upon the taxonomic and nomenclatural acts is another essential recommendation that should be given to the providers of Trichoderma taxonomy. This practice will result in reduced ambiguity in Trichoderma taxonomy. It is strongly suggested to revise species identifications for all DNA barcoding materials upon the release of species names. As shown above, the names of several dozen Trichoderma species have not been updated in the NCBI Taxonomy Browser (Table 2). Therefore, they are not visible in a sequence similarity search and may be easily overlooked by the beginner users of Trichoderma taxonomy.
Another (repeated) recommendation is the ultimate provision of ITS sequences for all Trichoderma species, including those that have already been described. Although species can be recognized based on the use of other phylogenetic markers in some cases, ITS should be provided to record this taxon in metagenomic studies. Even if the ITS phylotype of a given species is not unique, it is essential to associate all possible taxonomic names with each phylotype of ITS. Because the resolution of metabarcoding is expected to improve with the integration of new technologies and longer reads (Feng et al. 2015; Rhoads and Au 2015), ITS sequences will gain further value in the diversity research of all fungi, including Trichoderma. Furthermore, ITS can serve as the third locus, complementing the strict GCPSR that is applied for tef1 and rpb2 (see above).
Description of a new species that is based on a single strain is not recommended (Seifert and Rossman 2010). Exceptional cases require justification and a clear statement that genealogical concordance was not accessed (see above). The need for the nomenclatural act for a single isolate (assigning of a new name) can be considered to be convincing if the specimen was collected in a habitat that cannot be further sampled [as from clinical material (Druzhinina et al. 2008)], if the strain has some unique and clearly distinguishing ecophysiological properties [T. cyanodichotomous, (Li et al. 2018)], if it is particularly relevant for applications [T. taxi, (Zhang et al. 2007)], or if it has pathological significance. Single strains can be assigned as putative new species and communicated using their strain ID. Thus, in this study, we refrain from describing the strain that was used as an example, T. sp. TUCIM 5640, as a formal new species because it meets all but this criterion (see above). The formal taxonomic description should be completed when more samples become available. Unfortunately, a formal taxonomic description based on a single isolate is still common in Trichoderma taxonomy (Chen and Zhuang 2017a; Jaklitsch 2009, 2011; Jaklitsch and Voglmayr 2015), which frequently results in ambiguous species that can also not be unambiguously identified. It is recommended that measures should be taken to perform additional sampling and search public databases, strain collections, fungaria, and herbaria for the specimens and cultures with matching properties and/or DNA barcodes.
Besides the increasing number of the WGS strains in the Trichoderma spp., the applicability of WGS in taxonomy and DNA Barcoding did not reach its potential importance. Researchers repeatedly select strains that belong to the same species for WGS (Table 4). Thus, for now, three whole genomes of T. harzianum s. s., four genomes of T. longibrachiatum, and seven genomes of T. atroviride are available in public databases (see references in Tables 1 and 4).
The diversity surveys of Trichoderma are now frequently based on large samples of several hundred or even thousands of isolates (Migheli et al. 2009; Ma et al. 2020). The development of the protocol for handling such datasets requires a bioinformatic approach that will be presented elsewhere. However, we would like to specify the need to perform biological verification of the identification results that were obtained in silico. For example, if the soil is not sampled, the most common species in the genus in Europe are T. europaeum and its sister species T. mediterraneum (Jaklitsch and Voglmayr 2015). However, isolation-based surveys and metagenomic diversity studies did not identify these species or the closely related T. minutisporum in bulk soil or rhizosphere (Friedl and Druzhinina 2012; Hagn et al. 2007; Meincke et al. 2010). This does not mean that isolation of these species from the soil is not expected, but that identification of one of these species that is isolated from bulk soil requires critical evaluation. Generally, most of the infrageneric diversity of the genus Trichoderma is found in habitats other than soil (Jaklitsch 2009, 2011; Jaklitsch and Voglmayr 2015; Qin and Zhuang 2016c) and only a limited number of highly environmentally opportunistic Trichoderma species can establish in this environment (Friedl and Druzhinina 2012; Hagn et al. 2007; Meincke et al. 2010).
Similarly, T. reesei is a common and cosmopolitan species with a distribution that is limited to 20° south and north of the equator (Druzhinina et al. 2010; Druzhinina and Kubicek. 2016). The abundant detection of this species in temperate soils in Austria reported by HinterdoblerFootnote 1 requires verification by repeated sampling and consideration of artifacts.
The aspects of the Trichoderma lifecycle can also be considered to verify the in silico identification. Thus, T. longibrachiatum s. s. is a common species with a cosmopolitan distribution. Its isolates are known from all continents, including Antarctica, and subjected to several molecular evolutionary investigations that revealed that this was most likely a clonal species (agamospecies) (Druzhinina et al. 2008). Consequently, molecular identification of a teleomorph-derived isolate as T. longibrachiatum should be questioned and verified.
Concluding remarks and outlooks: Trichoderma genomics and polyphasic approach
For two centuries, the identification of Trichoderma (and other common cultivable fungi) required microscopic preparations, scientific drawings, and growth observation on multiple nutritional media. It was a laborious practice that frequently resulted in ambiguous species assignments (Fig. 15). The introduction of DNA-based techniques first slightly complicated the process by the need to equip mycological labs with molecular biological devices, but then it resulted in a drastic decrease in the labor that was required for the identification (DNA Barcoding). In a few years, the commercial kits for DNA extraction, ready PCR mixes, well-optimized PCR components, and the broad availability of Sanger sequencing service made DNA Barcoding a widely accepted technique. Additionally, the public databases of DNA sequences became powered by automated sequence analysis tools such as BLAST (Ye et al. 2006). Some online identification tools also become available for individual genera and fungal groups [TrichoKey, (Druzhinina et al. 2005); MIST, (Dou et al. 2020); UNITE, (Nilsson et al. 2019)]. Together with the GCPSR and PSR concepts, this prepared a simple methodological framework for the relative ease of species delimitation and triggered the ongoing boost of Trichoderma taxonomy (Fig. 3). Within a short time, the labor that was subsequently required for species identification sharply increased (Fig. 15), and the rapid growth of newly described species also contributed to the increased ambiguity of species diagnosis. Based on our estimation, 40% of Trichoderma species can not be unambiguously identified because either the respective reference materials are incomplete or species criteria that were used for the species delimitation has become ambiguous. The standardization of species recognition criteria and an agreement between Trichoderma taxonomy providers will allow us to avoid reaching the level when unambiguous species diagnosis will become rare or impossible (Fig. 15).

A schematic diagram showing the changes of labor related to species identification in Trichoderma over > 230 years
The current diversity of Trichoderma species is mostly recognized based on tef1 and rpb2 polymorphisms and supported by ITS allowing the development of the molecular identification protocol that will result in the frequent proposal of putative new species. Thus, we anticipate the future rapid growth of Trichoderma species to 1000 in the next decade. We agree that the particular species delimitation allows the precise identification and prediction of useful properties. However, we also hope that advances in taxonomy will improve rather than hinder our understanding of fungal biology and evolution.
Favorable opportunity and venture of the whole-genus genomics
Compared to some other ubiquitous fungi, the genus Trichoderma is relatively young. Its origin likely coincided with the Cretaceous–Paleogene extinction event, which was roughly 66 million years ago (mya) (Kubicek et al. 2019). It was approximately 15 million years after the putative origin of Aspergillus (81.7 mya) and about 10 million years after the formation of the ancestor of Penicillium. (73.6 mya) (Steenwyk et al. 2019). However, compared to the evolution of other groups, 66 million years are long. It includes the time passed from the end of the Cretaceous period and the entire Mesozoic Era, which was sufficient for the evolution of Hominidae (humans and other higher apes) from the placental mammalians similar to a rat-sized Purgatorius (O’Leary et al. 2013) that hardly had any features of modern primates. In contrast to mammals, fungal taxonomy is complicated by the lack of distinctive features (either phenotypic or DNA-barcodes) and fossils. However, the immense evolutionary time that has passed since the genus’ origin is reflected in the diversity of Trichoderma genomes (Kubicek et al. 2011, 2019). In the first comparative genomic study, syntenic orthologs of Trichoderma spp. were evaluated to be only 70% (T. reesei versus T. atroviride) to 78% (T. reesei versus T. virens) similar, which is comparable to the similarity between species of other fungal genera [69% for Aspergillus fumigatus versus A. niger (Galagan et al. 2005)] and to those between fish and man (Nadeau and Taylor 1984; Fedorova et al. 2008). Our more recent genomic investigations of a dozen Trichoderma spp. showed that the formation of the three major infrageneric groups, Section Longibrachiatum, Section Trichoderma (sensu Viride Clade), and the Harzianum–Virens Clades started 20–30 mya. Thus, these lineages were already separated by millions of years of independent evolution. The divergence between sister species, such as T. reesei and T. parareesei (Section Longibrachiatum), cryptic species T. harzianum, T. afroharzianum, and T. guizhouense happened several mya (4 to 8 mya) (Kubicek et al. 2019). In that study, Kubicek et al. (2019) found this evolutionary distance to be a supportive argument for delimitation of respective lineages in separate species (Druzhinina et al. 2010a; Atanasova et al. 2010; Chaverri et al. 2015). However, this judgment remained subjective because no standards on genomic or genetic similarities or the length of evolutionary distance were proposed that were sufficient to recognize a species. The number of intraspecific genomic studies for Trichoderma spp. remains limited. In the same work, the divergence between the two strains of the putative agamospecies T. harzianum sensu stricto (Druzhinina et al. 2010b) (the ex-type strain from the UK and a strain isolated from Brazil) was calculated to have occurred approximately 460,000 years ago. By all taxonomic means described in this study, these strains are not distinguishable. However, probably the most taxonomically-relevant and remarkable finding of the comparative genomics is the detection of 1699 genes in the genome of the ex-type T. harzianum strain CBS 226.95 (12% of the entire genome) that were absent from TR274 strain, and 1419 genes that were present in the latter (10.1%) were absent from the type strain. Most of these genes encoded orphan proteins for the species, and a function could only be predicted for less than 200 of them (Kubicek et al. 2019). Notably, the lack or presence of > 1000 entire genes in an individual genome a more significant distinction that 1–3% dissimilarity between rpb2 or tef1 DNA barcoding markers, which was used to identify species above. Thus, the level of taxonomic precision can be strongly influenced by the resolution of the method. Because the separation of species due to the long evolutionary history can be further powered by the high resolution of advanced -omics techniques, such as genomics, transcriptomics, epigenomics, metabolomics, or phenomics, the distinctions between any individual strains will appear deeper as more such tools become available for taxonomic studies, but the decision of the boundaries for particular fungal species may remain subjective.
The availability of the genomes opened an avenue for ecological genetics, which is the study of the role of individual genes and proteins in fungal fitness that was largely impeded in pre-genomic time. Cai et al. (2020) revealed that a single gene encoding the amphiphilic surface-active protein hydrophobin (HFB4) that covers Trichoderma conidia could drastically influence species-specific traits of T. guizhouense and T. harzianum that are related to spore dispersal and stress resistance. The results of that research pointed to another dimension that can be applied to distinguish between the two species that were previously considered to be cryptic and sympatric (Druzhinina et al. 2010b; Li et al. 2013; Chaverri et al. 2015). The ecophysiological profiling of HFB-deletion mutants suggested that T. guizhouense has features of anemophilous aero-aquatic fungi, while the T. harzianum has evolved towards pluviophilous dispersal (by rain droplets) and is adapted to habitats that are not flooded by water (soil or plant tissues) (Cai et al. 2020).
Thus, the application of the modern techniques will ultimately reveal more differences between individual fungal taxa (of all ranks) than similarities and, thus, improve cladistics (search of clades within clades) and phylogenetic resolution. Besides the differences, taxonomy also aims to reveal similarities between the organisms and, thus, improve our understanding of relationships and evolutionary history. Therefore, we anticipate that Trichoderma taxonomy and DNA Barcoding will be further challenged by choices between the biological accuracy and high precision of genetic delimitation of species and possibly subsequent identification. The results of the on-going whole-genus genomic project for Trichoderma (https://genome.jgi.doe.gov/portal/Genwidrichoderma/Genwidrichoderma.info.html), which aims for whole-genome sequencing of all Trichoderma spp., will drastically increase the precision of strain recognition. However, it may result in the distinction on the level of populations and even individual isolates rather than species and, thus, severely jeopardize the identifiability of Trichoderma species and ecological studies that are crucial for understanding the genomes. The urgent task for the Trichoderma community is to achieve an agreement on the genus-wide criteria that are used to recognize species and, thus, prepare for the release of massive genomic data.
Polyphasic approach and the work of John Bissett
Lücking et al. (2020) wrote that “the lack of accuracy of fungal identifications cannot be excused by the lack of adequate tools, and so the availability of tools determines which fungi can be studied. However, lack of molecular tools can be partially balanced by expertise: talented and knowledgeable mycologists may provide more accurate species identifications through non-molecular approaches than unexperienced users do through DNA-based identifications.”
We dedicate this work to the distinguished Trichoderma taxonomist John Bissett (1948–2020). Almost immediately after the introduction of DNA-based techniques in Trichoderma diversity studies, he proposed the integration of these tools with the advanced semiquantitative phenotypic characterization of individual strains and species. Today, the urgent need for the comprehensive implementation of such an approach—a polyphasic approach in species recognition, i.e. the combination of molecular phylogeny, phenotyping and ecology—is highly supported by fungal taxonomists including members of the ICTF [see Lücking et al. (2020)].
J. Bissett developed a fungal version of the microplate-based simultaneous characterization of fungi growth on 95 carbon sources and water (Phenotype MicroArrays). For Trichoderma, the system was first applied to the collection of South-East Asian isolates (Kubicek et al. 2003), and then this concept was used for the taxonomic description and characterization of numerous species (Atanasova et al. 2010; Ding et al. 2020; Druzhinina et al. 2006, 2008, 2010a, b; López-Quintero et al. 2013), strain collections (Komoń-Zelazowska et al. 2007; Gal-Hemed et al. 2011; Hatvani et al. 2019; Friedl and Druzhinina 2012; Cai et al. 2020), or individual mutants (Friedl et al. 2008; Seidl et al. 2006, 2008; Schuster and Schmoll 2010; Derntl et al. 2017, Wang and Zhuang 2020). The principle of semiquantitative phenotype profiling based on spectrophotometric or nephelometric measurements (Joubert et al. 2010) is becoming accepted in research on Trichoderma and other fungi [see Atanasova and Druzhinina (2010) for the review]. Cai et al. (2020) introduced REPAINT, which is the advanced version of Phenotype Microarrays that is powered by the artificial intelligence algorithm for the semiquantitative assessment of the reproductive potential such as production of aerial hyphae and conidiation. We propose that these or similar quantitative or semiquantitative tools for multiparametric automated phenotyping can rapidly find its applicability in the formal taxonomy of Trichoderma and of other fungi. It will allow the development of standardized phenotypic databases that are available for taxonomy and identification, and thus, prepare for the use of upcoming wave of Trichoderma “Big Data”.
Materials and methods
Strains, cultivation conditions, PCR, and sequencing
In this study, the two Trichoderma isolates (TUCIM 5640 and TUCIM 10063) from our collection were used as test material for a DNA barcoding exercise. For DNA extraction, Trichoderma cultures were maintained on potato dextrose agar (PDA; Becton, Dickinson and Company, Franklin Lakes, NJ, USA) plates at 25 °C in darkness. Fungal strains used for DNA Barcoding were cultivated for 48 h on PDA plates in darkness. Genomic DNA was extracted using a Phire Plant Direct PCR kit (Thermo Scientific, Waltham, Massachusetts, USA), according the manufacturer’s instructions. PCR amplification of the phylogenetic markers corresponding to ITS 1 and 2 of the rRNA gene cluster (ITS, including the 5.8S rRNA), the fragments of RNA polymerase II subunit B gene (rpb2), and the translation elongation factor 1-α (tef1) were set as described in Table 3. Amplicons were sent for Sanger sequencing.
Online survey
To estimate the molecular identifiability of Trichoderma spp. by the experts, we performed an on-line survey (the detailed questions can be seen in https://www.surveymonkey.com/r/?sm=hgTrOEkKaUnBxAsJkS5pSw_3D_3D) that was titled “Trichoderma 20x20”. The respondents were shown two sets of DNA barcoding markers (ITS, rpb2, and tef1) for two unknown isolates that had not been deposited into public databases. The questions concerned species identification or each strain, time spent, methods and loci used, and self-estimation of the respondent’s experience in the area of Trichoderma research and fungal taxonomy. The survey could have been completed anonymously or the respondents could leave their name and comments. The link to the survey was sent to > 200 respondents using the mailing list from the regular International Workshop on Trichoderma and Gliocladium.
Retrieval of taxonomic data
The information regarding taxonomy of the genus Trichoderma, including species names, publication year, and author names were exported from Index Fungorum (http://www.indexfungorum.org/), Mycobank (http://www.mycobank.org/), and the National Center for Biotechnology Information (NCBI) Taxonomy Browser (https://www.ncbi.nlm.nih.gov/taxonomy/). The latter was manually screened for all loci that were deposited per each taxonomic name of Trichoderma. Sequences that were assigned to undefined species of Trichoderma were not sampled. In our survey, we omitted Hypocrea names that were not transferred to Trichoderma according to Rossman et al. (2013) because they do not currently contribute to the molecular identification of Trichoderma.
The reference sequences of each marker locus for each type strain was retrieved from the NCBI database, which is based on the information that was provided by the NCBI RefSeq Targeted Loci Project (Robbertse et al. 2017) or from related publications (Bissett et al. 2015). Overall, 42 Trichoderma genomes (listed in Table 1) that were publicly available from the NCBI and the Joint Genome Institute (JGI) databases were used as the sequence resources for strain identification with author’s permissions for yet unpublished records. The respective sequences of each marker from T. reesei QM 6a, T. harzianum CBS 226.95, and T. asperellum CBS 433.97 were used in BLASTn when querying the genomes.
Online tools supporting Trichoderma taxonomy
The retrieved taxonomic data from the above three resources were manually confirmed and summarized in Table 2, which is also shown on the official website of the International Commission on Trichoderma Taxonomy (ICTT, https://www.Trichoderma.info (Fig. 16) as well as on https://www.trichokey.com (Fig. 17). The list of Trichoderma species contains species names that were valid as of July 2020, including those that are currently invalid species that lack DNA Barcoding information.

www.Trichoderma.info. A snapshot showing the design and content of the website of the International Subcommission of Taxonomy of Trichoderma (ICTT)

www.Trichokey.com. A snapshot of the Trichoderma taxonomy 2020 page containing the digitally searchable and sortable copy of Table 2 described in this study
Due to the lack of consistency within the Trichoderma community as to which primers to use for amplifying and sequencing of marker loci, there is considerable variation in the length and fragment area of sequences that are deposited into public databases under the same locus name. Additionally, a partial, rather than the whole fragment, of the marker locus is informative for molecular identification (Druzhinina and Kubicek 2005; Druzhinina et al. 2005; Kopchinskiy et al. 2005). Thus, we released the updated on-line tool TrichoMARK 2020 (https://trichokey.com/index.php/trichomark), by which the diagnostic area of each phylogenetic marker (ITS, rpb2, and tef1) with no flanking fragments can be retrieved. As described in Kopchinskiy et al. (2005), TrichoMARK is a specifically script-written tool for detecting and retrieving phylogenetic markers in query sequences, and it is based on genus specific oligonucleotides both on 5ʹ and 3ʹ ends of the marker.
We also developed and updated another online tool TrichoBLAST 2020 (https://trichokey.com/index.php/trichoblast), which covers all 361 currently genetically characterized species of Trichoderma and contains almost complete sets of the diagnostic fragments of the rpb2 locus from these 361 species and ITS sequences from the 56 type strains of each species that were representatively distributed in the whole genus. TrichoBLAST is a publicly available database that supports the similarity search tool to find the “best hit” of the query strain (sequence) within the genus that is based on a single locus of rpb2 or ITS. With respect to ITS as the marker locus harboring the largest dataset for fungal identification, TrichoBLAST, with 56 representative ITS sequences, allows estimation of whether a query strain belongs to the genus of Trichoderma (based on the current scope) if the subsequent calculation of the similarity between the query sequence (after trimmed by TrichoMARK) and the “best hit” is performed afterwards (see below).
Phylogenetic analysis
Sequences of each marker from the query strains and from the reference strains were consistently trimmed using TrichoMARK. The processed sequences were then aligned using Muscle 3.8.31 (Edgar 2004) available Aliview 1.23 (Larsson 2014). Maximum-likelihood (ML) phylogeny was performed using IQ-TREE 1.6.12 (Nguyen et al. 2015b). Statistical bootstrapping support was computed with 1000 replicates. The nucleotide substitution model was selected by ModelFinder (Kalyaanamoorthy et al. 2017) integrated in IQ-TREE, based on the Bayesian Information Criterion (BIC). Phylogenetic trees were visualized in FigTree v1.4.2 and annotated using CorelDraw 2017 (Corel, Ottawa, Ontario, Canada).
Pairwise similarity calculation
The multiple sequence alignment matrix of each locus was submitted to the online tool, Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/), for pairwise similarity calculation between two sequences.
Statistical analysis
The pairwise similarity data for each locus was illustrated using heatmaps that were generated by R (v3.6.1). The distribution of the data matrix was analyzed using STATISTICA 6 (StatSoft, Hamburg, Germany). One-way analysis of variance (ANOVA) and Tukey honest significance difference multiple comparison were set at the significance threshold P ≤ 0.05.
Data availability
All data are available as supplementary materials and on accessory websites www.trichoderma.info and www.trichokey.com.
Notes
The talk of Wolfgang Hinterdobler who presented W. Hinterdobler, J. Scholda, G. Li, S. Böhmdorfer, M. Schmoll “Trichoderma spp. impact mycotoxin production of the plant pathogen Fusarium graminearum” on the ECFG15 Satellite Workshop "Trichoderma, Clonostachys and other biocontrol fungi” (February, 2020, Rome, Italy). The abundant detection of T. reesei in a soil sample from Austria was also reported earlier by the same group, e.g. on the 15th International Trichoderma and Gliocladium Workshop (June, 2018, Salamanca, Spain). A respective publication is anticipated (W. Hinterdobler, personal communication).
References
Abdelfattah A, Li Destri Nicosia MG, Cacciola SO, Droby S, Schena L (2015) Metabarcoding analysis of fungal diversity in the phyllosphere and carposphere of Olive (Olea europaea). PLoS ONE 10(7):e0131069. https://doi.org/10.1371/journal.pone.0131069
Alves A, Crous PW, Correia A, Phillips AJL (2008) Morphological and molecular data reveal cryptic speciation in Lasiodiplodia theobromae. v.28
Atanasova L, Druzhinina IS (2010) Review: global nutrient profiling by Phenotype MicroArrays: a tool complementing genomic and proteomic studies in conidial fungi. J Zhejiang Univ Sci B 11(3):151–168. https://doi.org/10.1631/jzus.B1000007
Atanasova L, Jaklitsch WM, Komon-Zelazowska M, Kubicek CP, Druzhinina IS (2010) Clonal species Trichoderma parareesei sp. nov. likely resembles the ancestor of the cellulase producer Hypocrea jecorina/T. reesei. Appl Environ Microb 76(21):7259–7267. https://doi.org/10.1128/Aem.01184-10
Atanasova L, Druzhinina IS, Jaklitsch WM (2013) Two hundred Trichoderma species recognized on the basis of molecular phylogeny. In: Mukherjee PK, Horwitz BA, Singh US, Mukherjee M, Schmoll M (eds) Trichoderma: biology and applications. CABI, Croydon, pp 10–42. https://doi.org/10.1079/9781780642475.0010
Bajpai A, Rawat S, Johri BN (2019) Fungal diversity: global perspective and ecosystem dynamics. In: Satyanarayana T, Johri BN, Das SK (eds) Microbial diversity in ecosystem sustainability and biotechnological applications: microbial diversity in normal & extreme environments, vol 1. Springer, Singapore, pp 83–113. https://doi.org/10.1007/978-981-13-8315-1_4
Baroncelli R, Piaggeschi G, Fiorini L, Bertolini E, Zapparata A, Pe ME, Sarrocco S, Vannacci G (2015) Draft whole-genome sequence of the biocontrol agent Trichoderma harzianum T6776. Genome Announc 3(3):e00647-15. https://doi.org/10.1128/genomeA.00647-15
Baroncelli R, Zapparata A, Piaggeschi G, Sarrocco S, Vannacci G (2016) Draft whole-genome sequence of Trichoderma gamsii T6085, a promising biocontrol agent of Fusarium head blight on wheat. Genome Announc 4(1):e01747-15. https://doi.org/10.1128/genomeA.01747-15
Bissett J, Gams W, Jaklitsch W, Samuels GJ (2015) Accepted Trichoderma names in the year 2015. IMA Fungus 6(2):263–295. https://doi.org/10.5598/imafungus.2015.06.02.02
Cai F, Gao R, Zhao Z, Ding M, Jiang S, Yagtu C, Zhu H, Zhang J, Ebner T, Mayrhofer-Reinhartshuber M, Kainz P, Chenthamara K, Akcapinar GB, Shen Q, Druzhinina IS (2020) Evolutionary compromises in fungal fitness: hydrophobins can hinder the adverse dispersal of conidiospores and challenge their survival. ISME J 14:2610–2624. https://doi.org/10.1038/s41396-020-0709-0
Carbone I, Kohn LM (1999) A method for designing primer sets for speciation studies in filamentous ascomycetes. Mycologia 91(3):553–556. https://doi.org/10.1080/00275514.1999.12061051
Castrillo ML, Bich GA, Modenutti C, Turjanski A, Zapata PD, Villalba LL (2017) First whole-genome shotgun sequence of a promising cellulase secretor, Trichoderma koningiopsis strain POS7. Genome Announc 5(37):e00823-17. https://doi.org/10.1128/genomeA.00823-17
Chaverri P, Samuels GJ (2003) Hypocrea/Trichoderma (Ascomycota, Hypocreales, Hypocreaceae): species with green ascospores. Stud Mycol 48:1–116
Chaverri P, Gazis RO, Samuels GJ (2011) Trichoderma amazonicum, a new endophytic species on Hevea brasiliensis and H. guianensis from the Amazon basin. Mycologia 103(1):139–151. https://doi.org/10.3852/10-078
Chaverri P, Branco-Rocha F, Jaklitsch W, Gazis R, Degenkolb T, Samuels GJ (2015) Systematics of the Trichoderma harzianum species complex and the re-identification of commercial biocontrol strains. Mycologia 107(3):558–590. https://doi.org/10.3852/14-147
Chen K, Zhuang W-Y (2016) Trichoderma shennongjianum and Trichoderma tibetense, two new soil-inhabiting species in the Strictipile clade. Mycoscience 57(5):311–319. https://doi.org/10.1016/j.myc.2016.04.005
Chen K, Zhuang W-Y (2017a) Seven new species of Trichoderma from soil in China. Mycosystema 36(11):1441–1462. https://doi.org/10.13346/j.mycosystema.170134
Chen K, Zhuang WY (2017b) Discovery from a large-scaled survey of Trichoderma in soil of China. Sci Rep 7(1):9090. https://doi.org/10.1038/s41598-017-07807-3
Chen K, Zhuang WY (2017c) Three new soil-inhabiting species of Trichoderma in the Stromaticum clade with test of their antagonism to pathogens. Curr Microbiol 74(9):1049–1060. https://doi.org/10.1007/s00284-017-1282-2
Chen K, Zhuang W-Y (2017d) Seven soil-inhabiting new species of the genus Trichoderma in the Viride clade. Phytotaxa 312(1):28–46. https://doi.org/10.11646/phytotaxa.312.1.2
Chenthamara K, Druzhinina IS, Rahimi MJ, Grujic M, Cai F (2020) Ecological genomics and evolution of Trichoderma reesei. In: Mach-Aigner A, Martzy R (eds) Trichoderma reesei—methods and protocols. Springer, Puducherry
Choi J, Kim SH (2017) A genome tree of life for the Fungi kingdom. Proc Natl Acad Sci USA 114(35):9391–9396. https://doi.org/10.1073/pnas.1711939114
Compant S, Gerbore J, Antonielli L, Brutel A, Schmoll M (2017) Draft genome sequence of the root-colonizing fungus Trichoderma harzianum B97. Genome Announc 5(13):e00137-17. https://doi.org/10.1128/genomeA.00137-17
De Queiroz K (2007) Species concepts and species delimitation. Syst Biol 56(6):879–886. https://doi.org/10.1080/10635150701701083
Degenkolb T, Dieckmann R, Nielsen KF, Gräfenhan T, Theis C, Zafari D, Chaverri P, Ismaiel A, Brückner H, von Döhren H, Thrane U, Petrini O, Samuels GJ (2008) The Trichoderma brevicompactum clade: a separate lineage with new species, new peptaibiotics, and mycotoxins. Mycol Prog 7(3):177–219. https://doi.org/10.1007/s11557-008-0563-3
Derntl C, Kluger B, Bueschl C, Schuhmacher R, Mach RL, Mach-Aigner AR (2017) Transcription factor Xpp1 is a switch between primary and secondary fungal metabolism. Proc Natl Acad Sci USA 114(4):E560. https://doi.org/10.1073/pnas.1609348114
Ding MY, Chen W, Ma XC, Lv BW, Jiang SQ, Yu YN, Rahimi MJ, Gao RW, Zhao Z, Cai F, Druzhinina IS (2020) Emerging salt marshes as a source of Trichoderma arenarium sp. nov. and other fungal bioeffectors for biosaline agriculture. J Appl Microbiol. https://doi.org/10.1111/jam.14751
Dou K, Lu Z, Wu Q, Ni M, Yu C, Wang M, Li Y, Wang X, Xie H, Chen J, Zhang C (2020) MIST: a multiloci identification system for Trichoderma. Appl Environ Microbiol. https://doi.org/10.1128/AEM.01532-20
Druzhinina I, Kubicek CP (2005) Species concepts and biodiversity in Trichoderma and Hypocrea: from aggregate species to species clusters? J Zhejiang Univ Sci B 6(2):100–112. https://doi.org/10.1631/jzus.2005.B0100
Druzhinina IS, Kubicek CP (2016) Chapter two—familiar stranger: ecological genomics of the model saprotroph and industrial enzyme producer Trichoderma reesei breaks the stereotypes. In: Sariaslani S, Michael Gadd G (eds) Advances in applied microbiology, vol 95. Academic Press, New York, pp 69–147. https://doi.org/10.1016/bs.aambs.2016.02.001
Druzhinina IS, Kopchinskiy AG, Komon M, Bissett J, Szakacs G, Kubicek CP (2005) An oligonucleotide barcode for species identification in Trichoderma and Hypocrea. Fungal Genet Biol 42(10):813–828. https://doi.org/10.1016/j.fgb.2005.06.007
Druzhinina IS, Kopchinskiy AG, Kubicek CP (2006) The first 100 Trichoderma species characterized by molecular data. Mycoscience 47(2):55–64. https://doi.org/10.1007/S10267-006-0279-7
Druzhinina IS, Komoń-Zelazowska M, Kredics L, Hatvani L, Antal Z, Belayneh T, Kubicek CP (2008) Alternative reproductive strategies of Hypocrea orientalis and genetically close but clonal Trichoderma longibrachiatum, both capable of causing invasive mycoses of humans. Microbiology 154(11):3447–3459. https://doi.org/10.1099/mic.0.2008/021196-0
Druzhinina IS, Komon-Zelazowska M, Atanasova L, Seidl V, Kubicek CP (2010a) Evolution and ecophysiology of the industrial producer Hypocrea jecorina (Anamorph Trichoderma reesei) and a new sympatric agamospecies related to it. PLoS ONE 5(2):e0009191. https://doi.org/10.1371/journal.pone.0009191
Druzhinina IS, Kubicek CP, Komon-Zelazowska M, Mulaw TB, Bissett J (2010b) The Trichoderma harzianum demon: complex speciation history resulting in coexistence of hypothetical biological species, recent agamospecies and numerous relict lineages. BMC Evolut Biol. https://doi.org/10.1186/1471-2148-10-94
Druzhinina IS, Seidl-Seiboth V, Herrera-Estrella A, Horwitz BA, Kenerley CM, Monte E, Mukherjee PK, Zeilinger S, Grigoriev IV, Kubicek CP (2011) Trichoderma: the genomics of opportunistic success. Nat Rev Microbiol 9(10):749–759. https://doi.org/10.1038/nrmicro2637
Druzhinina IS, Komoń-Zelazowska M, Ismaiel A, Jaklitsch W, Mullaw T, Samuels GJ, Kubicek CP (2012) Molecular phylogeny and species delimitation in the section Longibrachiatum of Trichoderma. Fungal Genet Biol 49(5):358–368. https://doi.org/10.1016/j.fgb.2012.02.004
Druzhinina IS, Kopchinskiy AG, Kubicek EM, Kubicek CP (2016) A complete annotation of the chromosomes of the cellulase producer Trichoderma reesei provides insights in gene clusters, their expression and reveals genes required for fitness. Biotechnol Biofuels 9:75. https://doi.org/10.1186/s13068-016-0488-z
Druzhinina IS, Chenthamara K, Zhang J, Atanasova L, Yang DQ, Miao YZ, Rahimi MJ, Grujic M, Cai F, Pourmehdi S, Abu Salim K, Pretzer C, Kopchinskiy AG, Henrissat B, Kuo A, Hundley H, Wang M, Aerts A, Salamov A, Lipzen A, LaButti K, Barry K, Grigoriev IV, Shen QR, Kubicek CP (2018) Massive lateral transfer of genes encoding plant cell wall-degrading enzymes to the mycoparasitic fungus Trichoderma from its plant-associated hosts. PLoS Genet 14(4):e1007322. https://doi.org/10.1371/journal.pgen.1007322
du Plessis IL, Druzhinina IS, Atanasova L, Yarden O, Jacobs K (2018) The diversity of Trichoderma species from soil in South Africa, with five new additions. Mycologia 110(3):559–583. https://doi.org/10.1080/00275514.2018.1463059
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform 5:113. https://doi.org/10.1186/1471-2105-5-113
Fanelli F, Liuzzi VC, Logrieco AF, Altomare C (2018) Genomic characterization of Trichoderma atrobrunneum (T. harzianum species complex) ITEM 908: insight into the genetic endowment of a multi-target biocontrol strain. BMC Genomics 19(1):662. https://doi.org/10.1186/s12864-018-5049-3
Fedorova ND, Khaldi N, Joardar VS, Maiti R, Amedeo P, Anderson MJ, Crabtree J, Silva JC, Badger JH, Albarraq A, Angiuoli S, Bussey H, Bowyer P, Cotty PJ, Dyer PS, Egan A, Galens K, Fraser-Liggett CM, Haas BJ, Inman JM, Kent R, Lemieux S, Malavazi I, Orvis J, Roemer T, Ronning CM, Sundaram JP, Sutton G, Turner G, Venter JC, White OR, Whitty BR, Youngman P, Wolfe KH, Goldman GH, Wortman JR, Jiang B, Denning DW, Nierman WC (2008) Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet 4(4):e1000046. https://doi.org/10.1371/journal.pgen.1000046
Feng Y, Zhang Y, Ying C, Wang D, Du C (2015) Nanopore-based fourth-generation DNA sequencing technology. Genomics Proteomics Bioinform 13(1):4–16. https://doi.org/10.1016/j.gpb.2015.01.009
Fontaine B, van Achterberg K, Alonso-Zarazaga MA, Araujo R, Asche M, Aspock H, Aspock U, Audisio P, Aukema B, Bailly N, Balsamo M, Bank RA, Belfiore C, Bogdanowicz W, Boxshall G, Burckhardt D, Chylarecki P, Deharveng L, Dubois A, Enghoff H, Fochetti R, Fontaine C, Gargominy O, Gomez Lopez MS, Goujet D, Harvey MS, Heller KG, van Helsdingen P, Hoch H, De Jong Y, Karsholt O, Los W, Magowski W, Massard JA, McInnes SJ, Mendes LF, Mey E, Michelsen V, Minelli A, Nieto Nafria JM, van Nieukerken EJ, Pape T, De Prins W, Ramos M, Ricci C, Roselaar C, Rota E, Segers H, Timm T, van Tol J, Bouchet P (2012) New species in the Old World: Europe as a frontier in biodiversity exploration, a test bed for 21st century taxonomy. PLoS ONE 7(5):e36881. https://doi.org/10.1371/journal.pone.0036881
Friedl MA, Druzhinina IS (2012) Taxon-specific metagenomics of Trichoderma reveals a narrow community of opportunistic species that regulate each other’s development. Microbiology 158(Pt 1):69–83. https://doi.org/10.1099/mic.0.052555-0
Friedl MA, Schmoll M, Kubicek CP, Druzhinina IS (2008) Photostimulation of Hypocrea atroviridis growth occurs due to a cross-talk of carbon metabolism, blue light receptors and response to oxidative stress. Microbiology 154(Pt 4):1229–1241. https://doi.org/10.1099/mic.0.2007/014175-0
Galagan JE, Henn MR, Ma L-J, Cuomo CA, Birren B (2005) Genomics of the fungal kingdom: insights into eukaryotic biology. Genome Res 15(12):1620–1631. https://doi.org/10.1101/gr.3767105
Gal-Hemed I, Atanasova L, Komon-Zelazowska M, Druzhinina IS, Viterbo A, Yarden O (2011) Marine isolates of Trichoderma spp. as potential halotolerant agents of biological control for arid-zone agriculture. Appl Environ Microbiol 77(15):5100–5109. https://doi.org/10.1128/AEM.00541-11
Galtier N, Daubin V (2008) Dealing with incongruence in phylogenomic analyses. Philos Trans R Soc B 363(1512):4023–4029. https://doi.org/10.1098/rstb.2008.0144
Garnett ST, Christidis L, Conix S, Costello MJ, Zachos FE, Bánki OS, Bao Y, Barik SK, Buckeridge JS, Hobern D, Lien A, Montgomery N, Nikolaeva S, Pyle RL, Thomson SA, van Dijk PP, Whalen A, Zhang Z-Q, Thiele KR (2020) Principles for creating a single authoritative list of the world’s species. PLoS Biol 18(7):e3000736. https://doi.org/10.1371/journal.pbio.3000736
Grafenhan T, Schroers HJ, Nirenberg HI, Seifert KA (2011) An overview of the taxonomy, phylogeny, and typification of nectriaceous fungi in Cosmospora, Acremonium, Fusarium, Stilbella, and Volutella. Stud Mycol 68:79–113. https://doi.org/10.3114/sim.2011.68.04
Grujic M, Dojnov B, Potocnik I, Atanasova L, Duduk B, Srebotnik E, Druzhinina IS, Kubicek CP, Vujcic Z (2019) Superior cellulolytic activity of Trichoderma guizhouense on raw wheat straw. World J Microbiol Biotechnol 35(12):194. https://doi.org/10.1007/s11274-019-2774-y
Hagn A, Wallisch S, Radl V, Charles Munch J, Schloter M (2007) A new cultivation independent approach to detect and monitor common Trichoderma species in soils. J Microbiol Methods 69(1):86–92. https://doi.org/10.1016/j.mimet.2006.12.004
Harman GE, Howell CR, Viterbo A, Chet I, Lorito M (2004) Trichoderma species—opportunistic, avirulent plant symbionts. Nat Rev Microbiol 2(1):43–56. https://doi.org/10.1038/nrmicro797
Hatvani L, Homa M, Chenthamara K, Cai F, Kocsubé S, Atanasova L, Mlinaric-Missoni E, Manikandan P, Revathi R, Dóczi I, Bogáts G, Narendran V, Büchner R, Vágvölgyi C, Druzhinina IS, Kredics L (2019) Agricultural systems as potential sources of emerging human mycoses caused by Trichoderma: a successful, common phylotype of Trichoderma longibrachiatum in the frontline. FEMS Microbiol Lett 366(21):fnz246. https://doi.org/10.1093/femsle/fnz246
Hawksworth DL, Lücking R (2017) Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol Spectr 5(4):79–95. https://doi.org/10.1128/microbiolspec.FUNK-0052-2016
Houbraken J, Samson RA (2011) Phylogeny of Penicillium and the segregation of Trichocomaceae into three families. Stud Mycol 70(1):1–51. https://doi.org/10.3114/sim.2011.70.01
Houbraken J, Kocsube S, Visagie CM, Yilmaz N, Wang XC, Meijer M, Kraak B, Hubka V, Bensch K, Samson RA, Frisvad JC (2020) Classification of Aspergillus, Penicillium, Talaromyces and related genera (Eurotiales): an overview of families, genera, subgenera, sections, series and species. Stud Mycol 95:5–169. https://doi.org/10.1016/j.simyco.2020.05.002
Hoyos-Carvajal L, Orduz S, Bissett J (2009) Genetic and metabolic biodiversity of Trichoderma from Colombia and adjacent neotropic regions. Fungal Genet Biol 46(9):615–631. https://doi.org/10.1016/j.fgb.2009.04.006
Hyde KD, Xu J, Rapior S, Jeewon R, Lumyong S, Niego AGT, Abeywickrama PD, Aluthmuhandiram JVS, Brahamanage RS, Brooks S, Chaiyasen A, Chethana KWT, Chomnunti P, Chepkirui C, Chuankid B, de Silva NI, Doilom M, Faulds C, Gentekaki E, Gopalan V, Kakumyan P, Harishchandra D, Hemachandran H, Hongsanan S, Karunarathna A, Karunarathna SC, Khan S, Kumla J, Jayawardena RS, Liu J-K, Liu N, Luangharn T, Macabeo APG, Marasinghe DS, Meeks D, Mortimer PE, Mueller P, Nadir S, Nataraja KN, Nontachaiyapoom S, O’Brien M, Penkhrue W, Phukhamsakda C, Ramanan US, Rathnayaka AR, Sadaba RB, Sandargo B, Samarakoon BC, Tennakoon DS, Siva R, Sriprom W, Suryanarayanan TS, Sujarit K, Suwannarach N, Suwunwong T, Thongbai B, Thongklang N, Wei D, Wijesinghe SN, Winiski J, Yan J, Yasanthika E, Stadler M (2019) The amazing potential of fungi: 50 ways we can exploit fungi industrially. Fungal Divers 97(1):1–136. https://doi.org/10.1007/s13225-019-00430-9
Jaklitsch WM (2009) European species of Hypocrea Part I. The green-spored species. Stud Mycol 63:1–91. https://doi.org/10.3114/sim.2009.63.01
Jaklitsch WM (2011) European species of Hypocrea part II: species with hyaline ascospores. Fungal Divers 48(1):1–250. https://doi.org/10.1007/s13225-011-0088-y
Jaklitsch WM, Voglmayr H (2012) Hypocrea britdaniae and H. foliicola: two remarkable new European species. Mycologia 104(5):1213–1221. https://doi.org/10.3852/11-429
Jaklitsch WM, Voglmayr H (2013) New combinations in Trichoderma (Hypocreaceae, Hypocreales). Mycotaxon 126:143–156. https://doi.org/10.5248/126.143
Jaklitsch WM, Voglmayr H (2015) Biodiversity of Trichoderma (Hypocreaceae) in Southern Europe and Macaronesia. Stud Mycol 80:1–87. https://doi.org/10.1016/j.simyco.2014.11.001
Jaklitsch WM, Komon M, Kubicek CP, Druzhinina IS (2005) Hypocrea voglmayrii sp. nov. from the Austrian Alps represents a new phylogenetic clade in Hypocrea/Trichoderma. Mycologia 97(6):1365–1378. https://doi.org/10.3852/mycologia.97.6.1365
Jaklitsch WM, Samuels GJ, Dodd SL, Lu B-S, Druzhinina IS (2006) Hypocrea rufa/Trichoderma viride: a reassessment, and description of five closely related species with and without warted conidia. Stud Mycol 56:135–177. https://doi.org/10.3114/sim.2006.56.04
Jaklitsch WM, Gruber S, Voglmayr H (2008a) Hypocrea seppoi, a new stipitate species from Finland. Karstenia 48(1):1–11. https://doi.org/10.29203/ka.2008.423
Jaklitsch WM, Kubicek CP, Druzhinina IS (2008b) Three European species of Hypocrea with reddish brown stromata and green ascospores. Mycologia 100(5):796–815. https://doi.org/10.3852/08-039
Jaklitsch WM, Stadler M, Voglmayr H (2012) Blue pigment in Hypocrea caerulescens sp. nov. and two additional new species in sect. Trichoderma. Mycologia 104(4):925–941. https://doi.org/10.3852/11-327
Jaklitsch WM, Samuels GJ, Ismaiel A, Voglmayr H (2013) Disentangling the Trichoderma viridescens complex. Persoonia 31:112–146. https://doi.org/10.3767/003158513X672234
Jaklitsch WM, Lechat C, Voglmayr H (2014) The rise and fall of Sarawakus (Hypocreaceae, Ascomycota). Mycologia 106(1):133–144. https://doi.org/10.3852/13-117
Joubert A, Calmes B, Berruyer R, Pihet M, Bouchara J-P, Simoneau P, Guillemette T (2010) Laser nephelometry applied in an automated microplate system to study filamentous fungus growth. Biotechniques 48(5):399–404. https://doi.org/10.2144/000113399
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14(6):587–589. https://doi.org/10.1038/nmeth.4285
Kohli J (1987) Genetic nomenclature and gene list of the fission yeast Schizosaccharomyces pombe. Curr Genet 11(8):575–589. https://doi.org/10.1007/BF00393919
Komoń-Zelazowska M, Bissett J, Zafari D, Hatvani L, Manczinger L, Woo S, Lorito M, Kredics L, Kubicek CP, Druzhinina IS (2007) Genetically closely related but phenotypically divergent Trichoderma species cause green mold disease in oyster mushroom farms worldwide. Appl Environ Microbiol 73(22):7415–7426. https://doi.org/10.1128/AEM.01059-07
Kopchinskiy A, Komon M, Kubicek CP, Druzhinina IS (2005) TrichoBLAST: a multilocus database for Trichoderma and Hypocrea identifications. Mycol Res 109(Pt 6):658–660. https://doi.org/10.1017/s0953756205233397
Kubicek CP, Bissett J, Druzhinina I, Kullnig-Gradinger C, Szakacs G (2003) Genetic and metabolic diversity of Trichoderma: a case study on South-East Asian isolates. Fungal Genet Biol 38(3):310–319. https://doi.org/10.1016/s1087-1845(02)00583-2
Kubicek CP, Komon-Zelazowska M, Druzhinina IS (2008) Fungal genus Hypocrea/Trichoderma: from barcodes to biodiversity. J Zhejiang Univ Sci B 9(10):753–763. https://doi.org/10.1631/jzus.B0860015
Kubicek CP, Herrera-Estrella A, Seidl-Seiboth V, Martinez DA, Druzhinina IS, Thon M, Zeilinger S, Casas-Flores S, Horwitz BA, Mukherjee PK, Mukherjee M, Kredics L, Alcaraz LD, Aerts A, Antal Z, Atanasova L, Cervantes-Badillo MG, Challacombe J, Chertkov O, McCluskey K, Coulpier F, Deshpande N, von Dohren H, Ebbole DJ, Esquivel-Naranjo EU, Fekete E, Flipphi M, Glaser F, Gomez-Rodriguez EY, Gruber S, Han C, Henrissat B, Hermosa R, Hernandez-Onate M, Karaffa L, Kosti I, Le Crom S, Lindquist E, Lucas S, Lubeck M, Lubeck PS, Margeot A, Metz B, Misra M, Nevalainen H, Omann M, Packer N, Perrone G, Uresti-Rivera EE, Salamov A, Schmoll M, Seiboth B, Shapiro H, Sukno S, Tamayo-Ramos JA, Tisch D, Wiest A, Wilkinson HH, Zhang M, Coutinho PM, Kenerley CM, Monte E, Baker SE, Grigoriev IV (2011) Comparative genome sequence analysis underscores mycoparasitism as the ancestral life style of Trichoderma. Genome Biol 12(4):R40. https://doi.org/10.1186/gb-2011-12-4-r40
Kubicek CP, Steindorff AS, Chenthamara K, Manganiello G, Henrissat B, Zhang J, Cai F, Kopchinskiy AG, Kubicek EM, Kuo A, Baroncelli R, Sarrocco S, Noronha EF, Vannacci G, Shen Q, Grigoriev IV, Druzhinina IS (2019) Evolution and comparative genomics of the most common Trichoderma species. BMC Genomics 20(1):485. https://doi.org/10.1186/s12864-019-5680-7
Kuhls K, Lieckfeldt E, Samuels GJ, Kovacs W, Meyer W, Petrini O, Gams W, Börner T, Kubicek CP (1996) Molecular evidence that the asexual industrial fungus Trichoderma reesei is a clonal derivative of the ascomycete Hypocrea jecorina. Proc Natl Acad Sci USA 93(15):7755–7760. https://doi.org/10.1073/pnas.93.15.7755
Larsson A (2014) AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30(22):3276–3278. https://doi.org/10.1093/bioinformatics/btu531
Li QR, Tan P, Jiang YL, Hyde KD, Mckenzie EHC, Bahkali AH, Kang JC, Wang Y (2013) A novel Trichoderma species isolated from soil in Guizhou, T. guizhouense. Mycol Prog 12(2):167–172. https://doi.org/10.1007/s11557-012-0821-2
Li J, Wu Y, Chen K, Wang Y, Hu J, Wei Y, Yang H (2018) Trichoderma cyanodichotomus sp. nov., a new soil-inhabiting species with a potential for biological control. Can J Microbiol 64(12):1020–1029. https://doi.org/10.1139/cjm-2018-0224
Lieckfeldt E, Cavignac Y, Fekete C, Borner T (2000) Endochitinase gene-based phylogenetic analysis of Trichoderma. Microbiol Res 155(1):7–15. https://doi.org/10.1016/S0944-5013(00)80016-6
Liu YJ, Whelen S, Hall BD (1999) Phylogenetic relationships among ascomycetes: evidence from an RNA polymerse II subunit. Mol Biol Evol 16(12):1799–1808. https://doi.org/10.1093/oxfordjournals.molbev.a026092
López-Quintero CA, Atanasova L, Franco-Molano AE, Gams W, Komon-Zelazowska M, Theelen B, Muller WH, Boekhout T, Druzhinina I (2013) DNA barcoding survey of Trichoderma diversity in soil and litter of the Colombian lowland Amazonian rainforest reveals Trichoderma strigosellum sp. nov. and other species. Antonie Van Leeuwenhoek 104(5):657–674. https://doi.org/10.1007/s10482-013-9975-4
Lu B, Druzhinina IS, Fallah P, Chaverri P, Gradinger C, Kubicek CP, Samuels GJ (2004) Hypocrea/Trichoderma species with pachybasium-like conidiophores: teleomorphs for T. minutisporum and T. polysporum and their newly discovered relatives. Mycologia 96(2):310–342
Lücking R, Aime MC, Robbertse B, Miller AN, Ariyawansa HA, Aoki T, Cardinali G, Crous PW, Druzhinina IS, Geiser DM, Hawksworth DL, Hyde KD, Irinyi L, Jeewon R, Johnston PR, Kirk PM, Malosso E, May TW, Meyer W, Opik M, Robert V, Stadler M, Thines M, Vu D, Yurkov AM, Zhang N, Schoch CL (2020) Unambiguous identification of fungi: where do we stand and how accurate and precise is fungal DNA barcoding? Ima Fungus 11:14. https://doi.org/10.1186/s43008-020-00033-z
Lutzoni F, Kauff F, Cox CJ, McLaughlin D, Celio G, Dentinger B, Padamsee M, Hibbett D, James TY, Baloch E, Grube M (2004) Where are we in assembling the fungal tree of life, classifying the fungi, and understanding the evolution of their subcellular traits? Am J Bot 91:1446–1480
Ma J, Tsegaye E, Li M, Wu B, Jiang X (2020) Biodiversity of Trichoderma from grassland and forest ecosystems in Northern Xinjiang, China. 3 Biotech 10(8):362. https://doi.org/10.1007/s13205-020-02301-6
Mach RL, Peterbauer CK, Payer K, Jaksits S, Woo SL, Zeilinger S, Kullnig CM, Lorito M, Kubicek CP (1999) Expression of two major chitinase genes of Trichoderma atroviride (T. harzianum P1) is triggered by different regulatory signals. Appl Environ Microb 65(5):1858–1863. https://doi.org/10.1128/AEM.65.5.1858-1863.1999
Martinez D, Berka RM, Henrissat B, Saloheimo M, Arvas M, Baker SE, Chapman J, Chertkov O, Coutinho PM, Cullen D, Danchin EG, Grigoriev IV, Harris P, Jackson M, Kubicek CP, Han CS, Ho I, Larrondo LF, de Leon AL, Magnuson JK, Merino S, Misra M, Nelson B, Putnam N, Robbertse B, Salamov AA, Schmoll M, Terry A, Thayer N, Westerholm-Parvinen A, Schoch CL, Yao J, Barabote R, Nelson MA, Detter C, Bruce D, Kuske CR, Xie G, Richardson P, Rokhsar DS, Lucas SM, Rubin EM, Dunn-Coleman N, Ward M, Brettin TS (2008) Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat Biotechnol 26(5):553–560. https://doi.org/10.1038/nbt1403
May TW, Redhead SA, Bensch K, Hawksworth DL, Lendemer J, Lombard L, Turland NJ (2019) Chapter F of the International Code of Nomenclature for algae, fungi, and plants as approved by the 11th International Mycological Congress, San Juan, Puerto Rico, July 2018. Ima Fungus 10:21. https://doi.org/10.1186/s43008-019-0019-1
Meincke R, Weinert N, Radl V, Schloter M, Smalla K, Berg G (2010) Development of a molecular approach to describe the composition of Trichoderma communities. J Microbiol Methods 80(1):63–69. https://doi.org/10.1016/j.mimet.2009.11.001
Migheli Q, Balmas V, Komon-Zelazowska M, Scherm B, Fiori S, Kopchinskiy AG, Kubicek CP, Druzhinina IS (2009) Soils of a Mediterranean hot spot of biodiversity and endemism (Sardinia, Tyrrhenian Islands) are inhabited by pan-European, invasive species of Hypocrea/Trichoderma. Environ Microbiol 11(1):35–46. https://doi.org/10.1111/j.1462-2920.2008.01736.x
Nadeau JH, Taylor BA (1984) Lengths of chromosomal segments conserved since divergence of man and mouse. Proc Natl Acad Sci USA 81(3):814–818. https://doi.org/10.1073/pnas.81.3.814
Nguyen HDT, Jančič S, Meijer M, Tanney JB, Zalar P, Gunde-Cimerman N, Seifert KA (2015a) Application of the phylogenetic species concept to Wallemia sebi from house dust and indoor air revealed by multi-locus genealogical concordance. PLoS ONE 10(3):e0120894. https://doi.org/10.1371/journal.pone.0120894
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ (2015b) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32(1):268–274. https://doi.org/10.1093/molbev/msu300
Nilsson RH, Larsson KH, Taylor AFS, Bengtsson-Palme J, Jeppesen TS, Schigel D, Kennedy P, Picard K, Glockner FO, Tedersoo L, Saar I, Koljalg U, Abarenkov K (2019) The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res 47(D1):D259–D264. https://doi.org/10.1093/nar/gky1022
O’Donnell K, Ward TJ, Robert VARG, Crous PW, Geiser DM, Kang S (2015) DNA sequence-based identification of Fusarium: current status and future directions. Phytoparasitica 43(5):583–595. https://doi.org/10.1007/s12600-015-0484-z
O’Leary MA, Bloch JI, Flynn JJ, Gaudin TJ, Giallombardo A, Giannini NP, Goldberg SL, Kraatz BP, Luo Z-X, Meng J, Ni X, Novacek MJ, Perini FA, Randall ZS, Rougier GW, Sargis EJ, Silcox MT, Simmons NB, Spaulding M, Velazco PM, Weksler M, Wible JR, Cirranello AL (2013) The placental mammal ancestor and the post–K-Pg radiation of placentals. Science 339(6120):662. https://doi.org/10.1126/science.1229237
Overton BE, Stewart EL, Geiser DM (2006) Taxonomy and phylogenetic relationships of nine species of Hypocrea with anamorphs assignable to Trichoderma section Hypocreanum. Stud Mycol 56:39–65. https://doi.org/10.3114/sim.2006.56.02
Perkins DD (1999) Neurospora genetic nomenclature. Fungal Genet Newslett 46:34–41
Persoon CH (1794) Neurospora genetic nomenclature. Romers Neues Mag Bot 1:81–128
Proctor RH, McCormick SP, Kim H-S, Cardoza RE, Stanley AM, Lindo L, Kelly A, Brown DW, Lee T, Vaughan MM, Alexander NJ, Busman M, Gutiérrez S (2018) Evolution of structural diversity of trichothecenes, a family of toxins produced by plant pathogenic and entomopathogenic fungi. PLoS Pathog 14(4):e1006946. https://doi.org/10.1371/journal.ppat.1006946
Qiao M, Du X, Zhang Z, Xu J, Yu Z (2018) Three new species of soil-inhabiting Trichoderma from southwest China. MycoKeys 44:63–80
Qin W-T, Zhuang W-Y (2016a) Four new species of Trichoderma with hyaline ascospores from central China. Mycol Prog 15(8):811–825. https://doi.org/10.1007/s11557-016-1211-y
Qin W-T, Zhuang W-Y (2016b) Four new species of Trichoderma with hyaline ascospores in the Brevicompactum and Longibrachiatum clades. Mycosystema 35(11):e160158. https://doi.org/10.13346/j.mycosystema.160158
Qin WT, Zhuang WY (2016c) Seven wood-inhabiting new species of the genus Trichoderma (Fungi, Ascomycota) in Viride clade. Sci Rep 6:27074. https://doi.org/10.1038/srep27074
Qin W-T, Zhuang W-Y (2017) Seven new species of Trichoderma (Hypocreales) in the Harzianum and Strictipile clades. Phytotaxa 305(3):121–139. https://doi.org/10.11646/phytotaxa.305.3.1
Rahimi MJ, Cai F, Grujic M, Chenthamara K, Druzhinina IS (2020) Molecular identification of Trichoderma reesei. In: Mach-Aigner A, Martzy R (eds) Trichoderma reesei—methods and protocols. Springer, Puducherry
Redhead SA, Norvell LL (2012) News. IMA Fungus 3(2):44–47. https://doi.org/10.1007/BF03449512
Rhoads A, Au KF (2015) PacBio sequencing and its applications. Genomics Proteomics Bioinform 13(5):278–289. https://doi.org/10.1016/j.gpb.2015.08.002
Rifai MA (1969) A revision of the genus Trichoderma. Mycol Pap 116:1–54
Rivera-Méndez W, Obregón M, Morán-Diez ME, Hermosa R, Monte E (2020) Trichoderma asperellum biocontrol activity and induction of systemic defenses against Sclerotium cepivorum in onion plants under tropical climate conditions. Biol Control 141:104145. https://doi.org/10.1016/j.biocontrol.2019.104145
Robbertse B, Strope PK, Chaverri P, Gazis R, Ciufo S, Domrachev M, Schoch CL (2017) Improving taxonomic accuracy for fungi in public sequence databases: applying ‘one name one species’ in well-defined genera with Trichoderma/Hypocrea as a test case. Database. https://doi.org/10.1093/database/bax072
Röhrich CR, Jaklitsch WM, Voglmayr H, Iversen A, Vilcinskas A, Nielsen KF, Thrane U, von Dohren H, Bruckner H, Degenkolb T (2014) Front line defenders of the ecological niche! Screening the structural diversity of peptaibiotics from saprotrophic and fungicolous Trichoderma/Hypocrea species. Fungal Divers 69(1):117–146. https://doi.org/10.1007/s13225-013-0276-z
Rossman AY, Seifert KA, Samuels GJ, Minnis AM, Schroers HJ, Lombard L, Crous PW, Poldmaa K, Cannon PF, Summerbell RC, Geiser DM, Zhuang WY, Hirooka Y, Herrera C, Salgado-Salazar C, Chaverri P (2013) Genera in Bionectriaceae, Hypocreaceae, and Nectriaceae (Hypocreales) proposed for acceptance or rejection. IMA Fungus 4(1):41–51. https://doi.org/10.5598/imafungus.2013.04.01.05
Rossman AY, Allen WC, Braun U, Castlebury LA, Chaverri P, Crous PW, Hawksworth DL, Hyde KD, Johnston P, Lombard L, Romberg M, Samson RA, Seifert KA, Stone JK, Udayanga D, White JF (2016) Overlooked competing asexual and sexually typified generic names of Ascomycota with recommendations for their use or protection. IMA Fungus 7(2):289–308. https://doi.org/10.5598/imafungus.2016.07.02.09
Samuels GJ, Pardo-Schultheiss R, Hebbar KP, Lumsden RD, Bastos CN, Costa JC, Bezerra JL (2000) Trichoderma stromaticum sp. nov., a parasite of the cacao witches broom pathogen. Mycol Res 104(6):760–764. https://doi.org/10.1017/S0953756299001938
Samuels GJ, Dodd SL, Gams W, Castlebury LA, Petrini O (2002) Trichoderma species associated with the green mold epidemic of commercially grown Agaricus bisporus. Mycologia 94(1):146–170. https://doi.org/10.1080/15572536.2003.11833257
Samuels GJ, Dodd SL, Lu BS, Petrini O, Schroers HJ, Druzhinina IS (2006) The Trichoderma koningii aggregate species. Stud Mycol 56:67–133. https://doi.org/10.3114/sim.2006.56.03
Samuels GJ, Ismaiel A, Bon M-C, De Respinis S, Petrini O (2010) Trichoderma asperellum sensu lato consists of two cryptic species. Mycologia 102(4):944–966. https://doi.org/10.3852/09-243
Samuels GJ, Ismaiel A, Mulaw TB, Szakacs G, Druzhinina IS, Kubicek CP, Jaklitsch WM (2012) The Longibrachiatum Clade of Trichoderma: a revision with new species. Fungal Divers 55(1):77–108. https://doi.org/10.1007/s13225-012-0152-2
Sandoval-Denis M, Sutton DA, Cano-Lira JF, Gene J, Fothergill AW, Wiederhold NP, Guarro J (2014) Phylogeny of the clinically relevant species of the emerging fungus Trichoderma and their antifungal susceptibilities. J Clin Microbiol 52(6):2112–2125. https://doi.org/10.1128/JCM.00429-14
Schoch CL, Sung GH, Lopez-Giraldez F, Townsend JP, Miadlikowska J, Hofstetter V, Robbertse B, Matheny PB, Kauff F, Wang Z, Gueidan C, Andrie RM, Trippe K, Ciufetti LM, Wynns A, Fraker E, Hodkinson BP, Bonito G, Groenewald JZ, Arzanlou M, de Hoog GS, Crous PW, Hewitt D, Pfister DH, Peterson K, Gryzenhout M, Wingfield MJ, Aptroot A, Suh SO, Blackwell M, Hillis DM, Griffith GW, Castlebury LA, Rossman AY, Lumbsch HT, Lucking R, Budel B, Rauhut A, Diederich P, Ertz D, Geiser DM, Hosaka K, Inderbitzin P, Kohlmeyer J, Volkmann-Kohlmeyer B, Mostert L, O’Donnell K, Sipman H, Rogers JD, Shoemaker RA, Sugiyama J, Summerbell RC, Untereiner W, Johnston PR, Stenroos S, Zuccaro A, Dyer PS, Crittenden PD, Cole MS, Hansen K, Trappe JM, Yahr R, Lutzoni F, Spatafora JW (2009) The Ascomycota tree of life: a phylum-wide phylogeny clarifies the origin and evolution of fundamental reproductive and ecological traits. Syst Biol 58(2):224–239. https://doi.org/10.1093/sysbio/syp020
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W, Fungal Barcoding C, Fungal Barcoding Consortium Author L (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci USA 109(16):6241–6246. https://doi.org/10.1073/pnas.1117018109
Schoch CL, Ciufo S, Domrachev M, Hotton CL, Kannan S, Khovanskaya R, Leipe D, McVeigh R, O’Neill K, Robbertse B, Sharma S, Soussov V, Sullivan JP, Sun L, Turner S, Karsch-Mizrachi I (2020) NCBI taxonomy: a comprehensive update on curation, resources and tools. Database. https://doi.org/10.1093/database/baaa062
Schuster A, Schmoll M (2010) Biology and biotechnology of Trichoderma. Appl Microbiol Biotechnol 87(3):787–799. https://doi.org/10.1007/s00253-010-2632-1
Seidl V, Druzhinina IS, Kubicek CP (2006) A screening system for carbon sources enhancing β-N-acetylglucosaminidase formation in Hypocrea atroviridis (Trichoderma atroviride). Microbiology 152(7):2003–2012. https://doi.org/10.1099/mic.0.28897-0
Seidl V, Gamauf C, Druzhinina IS, Seiboth B, Hartl L, Kubicek CP (2008) The Hypocrea jecorina (Trichoderma reesei) hypercellulolytic mutant RUT C30 lacks a 85 kb (29 gene-encoding) region of the wild-type genome. BMC Genomics 9(1):327. https://doi.org/10.1186/1471-2164-9-327
Seidl V, Seibel C, Kubicek CP, Schmoll M (2009) Sexual development in the industrial workhorse Trichoderma reesei. Proc Natl Acad Sci USA 106(33):13909. https://doi.org/10.1073/pnas.0904936106
Seifert KA, Rossman AY (2010) How to describe a new fungal species. IMA Fungus 1(2):109–116. https://doi.org/10.5598/imafungus.2010.01.02.02
Sherkhane PD, Bansal R, Banerjee K, Chatterjee S, Oulkar D, Jain P, Rosenfelder L, Elgavish S, Horwitz BA, Mukherjee PK (2017) Genomics-driven discovery of the gliovirin biosynthesis gene cluster in the plant beneficial fungus Trichoderma virens. ChemistrySelect 2(11):3347–3352. https://doi.org/10.1002/slct.201700262
Shi-Kunne X, Seidl MF, Faino L, Thomma BP (2015) Draft genome sequence of a strain of cosmopolitan fungus Trichoderma atroviride. Genome Announc 3(3):e00287-15. https://doi.org/10.1128/genomeA.00287-15
Sklenar F, Jurjevic Z, Zalar P, Frisvad JC, Visagie CM, Kolarik M, Houbraken J, Chen AJ, Yilmaz N, Seifert KA, Coton M, Deniel F, Gunde-Cimerman N, Samson RA, Peterson SW, Hubka V (2017) Phylogeny of xerophilic aspergilli (subgenus Aspergillus) and taxonomic revision of section Restricti. Stud Mycol 88:161–236. https://doi.org/10.1016/j.simyco.2017.09.002
Stasz TE, Harman GE, Weeden NF (1988) Protoplast preparation and fusion in two biocontrol strains of Trichoderma Harzianum. Mycologia 80(2):141–150. https://doi.org/10.1080/00275514.1988.12025515
Steenwyk JL, Shen XX, Lind AL, Goldman GH, Rokas A (2019) A robust phylogenomic time tree for biotechnologically and medically important fungi in the genera Aspergillus and Penicillium. mBio 10(4):e00925. https://doi.org/10.1128/mBio.00925-19
Stielow JB, Levesque CA, Seifert KA, Meyer W, Iriny L, Smits D, Renfurm R, Verkley GJ, Groenewald M, Chaduli D, Lomascolo A, Welti S, Lesage-Meessen L, Favel A, Al-Hatmi AM, Damm U, Yilmaz N, Houbraken J, Lombard L, Quaedvlieg W, Binder M, Vaas LA, Vu D, Yurkov A, Begerow D, Roehl O, Guerreiro M, Fonseca A, Samerpitak K, van Diepeningen AD, Dolatabadi S, Moreno LF, Casaregola S, Mallet S, Jacques N, Roscini L, Egidi E, Bizet C, Garcia-Hermoso D, Martin MP, Deng S, Groenewald JZ, Boekhout T, de Beer ZW, Barnes I, Duong TA, Wingfield MJ, de Hoog GS, Crous PW, Lewis CT, Hambleton S, Moussa TA, Al-Zahrani HS, Almaghrabi OA, Louis-Seize G, Assabgui R, McCormick W, Omer G, Dukik K, Cardinali G, Eberhardt U, de Vries M, Robert V (2015) One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Persoonia 35:242–263. https://doi.org/10.3767/003158515X689135
Struck TH, Feder JL, Bendiksby M, Birkeland S, Cerca J, Gusarov VI, Kistenich S, Larsson KH, Liow LH, Nowak MD, Stedje B, Bachmann L, Dimitrov D (2018) Finding evolutionary processes hidden in cryptic species. Trends Ecol Evol 33(3):153–163. https://doi.org/10.1016/j.tree.2017.11.007
Studholme DJ, Harris B, Le Cocq K, Winsbury R, Perera V, Ryder L, Ward JL, Beale MH, Thornton CR, Grant M (2013) Investigating the beneficial traits of Trichoderma hamatum GD12 for sustainable agriculture-insights from genomics. Front Plant Sci 4:258. https://doi.org/10.3389/fpls.2013.00258
Taylor JW (2011) One Fungus = One Name: DNA and fungal nomenclature twenty years after PCR. Ima Fungus 2(2):113–120. https://doi.org/10.5598/imafungus.2011.02.02.01
Taylor JW, Jacobson DJ, Kroken S, Kasuga T, Geiser DM, Hibbett DS, Fisher MC (2000) Phylogenetic species recognition and species concepts in fungi. Fungal Genet Biol 31(1):21–32. https://doi.org/10.1006/fgbi.2000.1228
Tedersoo L, Bahram M, Polme S, Koljalg U, Yorou NS, Wijesundera R, Villarreal Ruiz L, Vasco-Palacios AM, Thu PQ, Suija A, Smith ME, Sharp C, Saluveer E, Saitta A, Rosas M, Riit T, Ratkowsky D, Pritsch K, Poldmaa K, Piepenbring M, Phosri C, Peterson M, Parts K, Partel K, Otsing E, Nouhra E, Njouonkou AL, Nilsson RH, Morgado LN, Mayor J, May TW, Majuakim L, Lodge DJ, Lee SS, Larsson KH, Kohout P, Hosaka K, Hiiesalu I, Henkel TW, Harend H, Guo LD, Greslebin A, Grelet G, Geml J, Gates G, Dunstan W, Dunk C, Drenkhan R, Dearnaley J, De Kesel A, Dang T, Chen X, Buegger F, Brearley FQ, Bonito G, Anslan S, Abell S, Abarenkov K (2014) Fungal biogeography. Global diversity and geography of soil fungi. Science 346(6213):1256688. https://doi.org/10.1126/science.1256688
Tisch D, Pomraning KR, Collett JR, Freitag M, Baker SE, Chen CL, Hsu PW, Chuang YC, Schuster A, Dattenbock C, Stappler E, Sulyok M, Bohmdorfer S, Oberlerchner J, Wang TF, Schmoll M (2017) Omics analyses of Trichoderma reesei CBS999.97 and QM6a indicate the relevance of female fertility to carbohydrate-active enzyme and transporter levels. Appl Environ Microbiol 83(22):e01578. https://doi.org/10.1128/AEM.01578-17
Tomah AA, Abd Alamer IS, Li B, Zhang J-Z (2020) A new species of Trichoderma and gliotoxin role: a new observation in enhancing biocontrol potential of T. virens against Phytophthora capsici on chili pepper. Biol Control 145:104261. https://doi.org/10.1016/j.biocontrol.2020.104261
Tronsmo AM (1991) Biological and integrated controls of Botrytis cinerea on apple with Trichoderma harzianum. Biol Control 1(1):59–62. https://doi.org/10.1016/1049-9644(91)90102-6
Valentini A, Pompanon F, Taberlet P (2009) DNA barcoding for ecologists. Trends Ecol Evol 24(2):110–117. https://doi.org/10.1016/j.tree.2008.09.011
Vu D, Groenewald M, de Vries M, Gehrmann T, Stielow B, Eberhardt U, Al-Hatmi A, Groenewald JZ, Cardinali G, Houbraken J, Boekhout T, Crous PW, Robert V, Verkley GJM (2019) Large-scale generation and analysis of filamentous fungal DNA barcodes boosts coverage for kingdom fungi and reveals thresholds for fungal species and higher taxon delimitation. Stud Mycol 92:135–154. https://doi.org/10.1016/j.simyco.2018.05.001
Wain HM, Bruford EA, Lovering RC, Lush MJ, Wright MW, Povey S (2002) Guidelines for human gene nomenclature. Genomics 79(4):464–470. https://doi.org/10.1006/geno.2002.6748
Wang C, Zhuang WY (2020) Carbon metabolic profiling of Trichoderma strains provides insight into potential ecological niches. Mycologia 112(2):213–223. https://doi.org/10.1080/00275514.2019.1698246
White TJ, Bruns T, Lee S, Taylor J (1990) 38—amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols. Academic Press, San Diego, pp 315–322. https://doi.org/10.1016/B978-0-12-372180-8.50042-1
Xie BB, Qin QL, Shi M, Chen LL, Shu YL, Luo Y, Wang XW, Rong JC, Gong ZT, Li D, Sun CY, Liu GM, Dong XW, Pang XH, Huang F, Liu W, Chen XL, Zhou BC, Zhang YZ, Song XY (2014) Comparative genomics provide insights into evolution of Trichoderma nutrition style. Genome Biol Evol 6(2):379–390. https://doi.org/10.1093/gbe/evu018
Yang D, Pomraning K, Kopchinskiy A, Karimi Aghcheh R, Atanasova L, Chenthamara K, Baker SE, Zhang R, Shen Q, Freitag M, Kubicek CP, Druzhinina IS (2015) Genome sequence and annotation of Trichoderma parareesei, the ancestor of the cellulase producer Trichoderma reesei. Genome Announc 3(4):e00885-15. https://doi.org/10.1128/genomeA.00885-15
Ye J, McGinnis S, Madden TL (2006) BLAST: improvements for better sequence analysis. Nucleic Acids Res 34(2):W6–W9. https://doi.org/10.1093/nar/gkl164
Yoder OC, Valent B, Chumley F (1986) Genetic nomenclature and practice for plant pathogenic fungi. Phypotaphology 76:383–385
Yu Z-F, Qiao M, Zhang Y, Zhang K-Q (2007) Two new species of Trichoderma from Yunnan, China. Antonie Van Leeuwenhoek 92(1):101–108. https://doi.org/10.1007/s10482-006-9140-4
Yu Z, Armant O, Yu Z, Armant O, Fischer R (2016) Fungi use the SakA (HogA) pathway for phytochrome-dependent light signalling. Nat Microbiol 1:16019. https://doi.org/10.1038/nmicrobiol.2016.19
Zhang C-L, Liu S-P, Lin F-C, Kubicek CP, Druzhinina IS (2007) Trichoderma taxi sp nov, an endophytic fungus from Chinese yew Taxus mairei. FEMS Microbiol Lett 270(1):90–96. https://doi.org/10.1111/j.1574-6968.2007.00659.x
Zhou Y, Wang Y, Chen K, Wu Y, Hu J, Wei Y, Li J, Yang H, Ryder M, Denton MD (2020) Near-complete genomes of two Trichoderma species: a resource for biological control of plant pathogens. Mol Plant-Microbe Interact 33(8):1036–1039. https://doi.org/10.1094/MPMI-03-20-0076-A
Zhu Z-X, Zhuang W-Y (2014) Two new species of Trichoderma (Hypocreaceae) from China. Mycosystema 33(6):1168–1174. https://doi.org/10.13346/j.mycosystema.140049
Acknowledgements
We highly value the time and effort of Trichoderma researchers from Argentina, Austria, China, Estonia, Ethiopia, Italy, Mexico, New Zealand, Spain, Turkey, USA and other countries that completed the online survey on the identification of the two Trichoderma strains. We thank Alexey Kopchinskiy (Vienna, Austria) for his efforts to design the websites that are dedicated to Trichoderma taxonomy and DNA Barcoding. Mohammad J. Rahimi is acknowledged for isolation of the two test Trichoderma cultures. We thank Matteo Lorito, Sheridan Woo, Gelsomina, Gelsomina Manganiello, and Francesco Vinale (University of Naples Federico II, Italy) for permission to use rpb2 and tef1 sequences from unpublished genomes of Trichoderma strains deposited in DOE JGI MycoCosm. Equally, we appreciate the permission of Peter Urban (University of Pecs, Hungary) and Laszlo Kredics (University of Szeged, Hungary) for their agreement to use DNA Barcoding sequences present in genomes of green mold Trichoderma spp. deposited in GenBank. We thank Olga Druzhinina (Moscow, Russia) for her contribution to the proofreading of the tables, and we thank all FungiG students (www.fungig.org, Nanjing Agricultural University, Nanjing, China) for their support.
Funding
Open access funding provided by TU Wien (TUW). The work of FC was supported by the grants from the National Science Foundation of China (31801939) and the Fundamental Research Funds for the Central Universities (KYXK202012).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cai, F., Druzhinina, I.S. In honor of John Bissett: authoritative guidelines on molecular identification of Trichoderma. Fungal Diversity 107, 1–69 (2021). https://doi.org/10.1007/s13225-020-00464-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13225-020-00464-4