
Abstract
The great diversity of nanomaterials provides ample opportunities for constructing effective agents for biomedical applications ranging from biosensing to drug delivery. Multifunctional nanoagents that combine several features in a single particle are of special interest due to capabilities that substantially exceed those of molecular drugs. An ideal theranostic agent should simultaneously be an advanced biosensor to identify a disease and report the diagnosis and a biomedical actuator to treat the disease. While many approaches were developed to load a nanoparticle with various drugs for actuation of the diseased cells (e.g., to kill them), the nanoparticle-based approaches for the localized biosensing with real-time reporting of the marker concentration severely lag behind. Here, we show a smart in situ nanoparticle-based biosensor/actuator system that dynamically and reversibly changes its structural and optical properties in response to a small molecule marker to allow real-time monitoring of the marker concentration and adjustment of the system ability to bind its biomedical target. Using the synergistic combination of signal readout based on the localized surface plasmon resonance and an original method of fabrication of smart ON/OFF-switchable nanoagents, we demonstrate reversible responsiveness of the system to a model small molecule marker (antibiotic chloramphenicol) in a wide concentration range. The proposed approach can be used for the development of advanced multifunctional nanoagents for theranostic applications.
Similar content being viewed by others

Introduction
Over the past decade, nanoparticle-based techniques have experienced the second birth due to emerging applications in biosensing (Holzinger et al. 2014), personalized nanomedicine (Jo et al. 2016) and theranostics (Kneževic and Kaluderovic 2017). Evidently, nanoparticles can become a major instrument in addressing the grand challenges in these fields due to their unique ability to implement multiple functions in a single agent. For example, nanoparticles can be sensors (Howes et al. 2014), actuators (Govorov et al. 2006), vectors (Cheng et al. 2008) and markers (Lin et al. 2005). Besides, their properties and performance can be easily adjusted by changing composition, coating or functional molecules immobilized on their surface (Wu et al. 2016). While their therapeutic applications only start to develop, nanoparticles are already widely used for biosensing in various areas ranging from food safety to in vitro diagnostics (Peng et al. 2015; Rocha-Santos 2014).
The vast majority of the commercial tests represent ex situ sensors, e.g., lateral flow assay or enzyme-linked immunosorbent assay that detect analyte in a specially processed sample. Small size and reliable detectability make nanoparticles extremely attractive for the development of in situ biosensors that perform non-invasive analysis directly in cell culture or tissue slices (Shipunova et al. 2016; Lee et al. 2007; Gandra et al. 2016). In situ monitoring of changes of biomolecule concentration in the natural or physiological environment is highly demanded in many fields of biology and medicine for efficient analysis of various biological processes. Even more needed, though, would be a sensor/actuator system that could not only report the analyte concentration but also react to changes in this concentration by a certain biological action, for instance, binding to a specific cell or any other biomolecular targets (Vitol et al. 2012; Polyak et al. 2016; Kneževic et al. 2011).
Development of reliable in situ biosensors for long-term measurements has been one of the most sophisticated tasks. The current analytical toolbox is quite limited due to degradation of the majority of molecular labels during measurements or of the sample during preparation. For example, the use of fluorescently labeled probes (DNA, RNA, antibodies, etc.) permits immediate sensitive and specific detection of an analyte or cell surface marker. However, they are rarely employed for monitoring of long-term processes because of progressive fading of a dye. Other optical methods such as digital holographic microscopy can resolve this issue but lack the necessary specificity (Kim 2011). On the contrary, the combination of microscopy and mass spectrometry has excellent specificity but requires elaborate sample preparation and sophisticated equipment (Setou and Kurabe 2011). The robust biosensing label-free methods based on magnetometry (Orlov et al. 2016b, 2017), interferometry (Nikitin et al. 2003), Raman spectroscopy (Millo et al. 2011) and localized surface plasmon resonance (Anker et al. 2008) represent a good alternative to microscopy. Other systems are constructed via self-assembly of nanoparticles and biomolecules in a layered fashion (Aghayeva et al. 2013). Operation of such agents is based on programming the routes of disassembly of these layers. Such assembly/disassembly feature uniquely suits for gold nanoparticles and their enhanced optical properties due ability to detect co-localization or aggregation of the particles via the localized surface plasmon resonance (SPR) phenomenon (Shevchenko et al. 2017; Pollet et al. 2009; Yuk et al. 2003; Kim et al. 2007).
In this study, we have developed a smart assembly/disassembly nanoparticle-based system for the reversible in situ SPR-based biosensing of small molecules within a relatively wide range of concentrations. The system will contribute to the research toolbox in cell biology.
Materials and methods
Materials
The following reagents and materials were used: chloramphenicol (CAP), bovine serum albumin (BSA), phosphate-buffered saline (PBS) buffer, acetate buffer, dimethyl sulfoxide (DMSO), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysulfosuccinimide sodium salt (sulfo-NHS), biotin N-hydroxysuccinimide ester (BIO-NHS), carboxymethyl dextran (CMD; Cat. No. 86524); 3,3′,5,5′-Tetramethylbenzidine (TMB) (Sigma-Aldrich, Germany); streptavidin (STR), horseradish peroxidase (HRP), Zeba™ Spin Desalting Columns, 7 K MWCO, 0.5 mL (ThermoFisher Scientific); Fetal Bovine Serum (FBS) (GE HealthCare and Life Sciences, USA); Mouse anti-chloramphenicol antibodies (anti-CAP, Russian Research Center for Molecular Diagnostics and Therapy, Russia); Estapor MH1-020/50 230 nm carboxylated polystyrene coated magnetic beads (Merck Millipore, France); chlorauric acid (Dragtsvetmet, Russia). Conjugates of biotin (BIO-BSA) and chloramphenicol (CAP-BSA) with BSA were prepared as previously (Nikitin et al. 2014). Other auxiliary reagents were of “analytical grade” quality.
Instruments
Absorption spectra as well as optical density (OD) were measured using a CLARIOstar® multimode microplate reader (BMG Labtech, Germany).
Nanoparticle synthesis
The 40 ± 5 nm gold nanoparticles (AuNPs) were prepared by citrate reduction of HAuCl4 solution according to the modified standard protocol (Turkevich et al. 1951). Briefly, 100 μL of 1% HAuCl4 solution in 40 mL water was boiled under reflux with vigorous stirring. Then, 400 μL of 1% trisodium citrate was rapidly added. The solution was further boiled for 10 min under reflux and then for another 20 min without condenser to concentrate the nanoparticles. The obtained suspension was cooled down to room temperature, and its optical density at 530 nm was adjusted to 2.00 ± 0.05 (ca. 135 pM) by MilliQ water. Iron oxyhydroxide particles (IOH) were synthesized according to the previously reported method (Nikitin et al. 2014) with modification. Briefly, 300 mg of FeCl3·6H2O was dissolved in 5 mL of MilliQ water. Then, 1.25 mL of 30% NH4OH was quickly added and the resulting solution was heated to 90 °C with following 2 h incubation. The obtained suspension was precipitated by centrifugation for 5 min at 3500×g. For peptization of the particles, 0.6 M of HNO3 was added and the pellet was resuspended. The particles were thrice washed with MilliQ water by centrifugation at 18,000×g for 30 min. The remaining aggregates were removed by 30 min centrifugation at 4000×g. The coating of nanoparticles was performed with carboxymethyl dextran (CMD) solution at 300 g/L in MilliQ water, which was added to the particle suspension to get the final concentration of 50 g/L. Then, the solution was incubated for 4 h at 80 °C. The non-bound CMD was washed thrice with MilliQ water by centrifugation at 18,000×g for 2 h.
Nanoparticle-protein conjugates
Magnetic particles (MP)
The conjugates of magnetic core nanoparticles with CAP-BSA and BIO-BSA (MP-CAP-BSA:BIO-BSA) were prepared as described earlier (Orlov et al. 2016a). Briefly, the solution of 30 mg of EDC and 12 mg of sulfo-NHS in 125 μL of MES buffer (50 mM, pH 5.0) was added to 2 mg of Estapor MH1 magnetic nanoparticles. The nanoparticles were incubated for 20 min with sonication and then magnetically washed once in MilliQ water. Next, the following protein mixture in 20 μL of borate buffer (0.1 M, pH 8.6) was added: 10 μg of CAP-BSA, 8 μg of BIO-BSA, and 22 μg of BSA. After overnight incubation in a shaker, the MP-CAP-BSA:BIO-BSA conjugates were magnetically washed thrice with 1% BSA in PBS buffer (pH 7.4).
Iron oxyhydroxide nanoparticles (IOH)
The conjugates of iron oxyhydroxide nanoparticles with STR and HRP (IOH-STR:HRP) were prepared as follows. The solution of 20 mg of EDC and 12 mg of sulfo-NHS in 60 μL of MES buffer (50 mM, pH 5.0) was added to 1 mg of IOH and incubated for 20 min with sonication. The nanoparticles were washed in MilliQ water and separated by centrifugation (15 min at 15,000×g). Next, the protein mixture of 60 μg of STR and 90 μg of HRP in 60 μL of HEPES buffer (10 mM, pH 4.6) was added to the particles and incubated overnight in a shaker. The IOH conjugates were washed thrice by centrifugation (15 min at 15,000×g) with 1% BSA in PBS buffer (pH 7.4).
Gold nanoparticles
The conjugates of gold nanoparticles with anti-CAP antibodies (AuNP-anti-CAP) were prepared as follows. The solution of 48.6 μg of antibody against chloramphenicol was added to 1.6 mL of gold nanoparticle suspension (OD = 2.0 at 530 nm) with pH = 9.0 (adjusted with NaOH), and the resulting mixture was incubated for 3 min. After addition of 0.4 mL of 5% BSA in PBS, the AuNP conjugates were washed four times with 1% BSA in PBS by centrifugation (10 min at 10,000×g).
Assembly and performance of smart system
The described nanoparticle-based smart system was constructed as follows. 6 µg of MP-CAP-BSA:BIO-BSA conjugate was co-incubated with 13.5 nM of AuNP-anti-CAP in 50 μL of 1% BSA in PBS or in 50 μL of 10% FBS in PBS for 1 h. For system disassembly, various concentrations of CAP solution in DMSO were added and incubated for 1 h. To inhibit system assembly, various concentrations of CAP solution in DMSO were added to AuNP-anti-CAP, incubated for 5 min, then added to MP conjugates and additionally incubated for 1 h. Free CAP was removed from the environment by passing the nanoparticle complexes through a desalting column in accordance with the manufacturer’s recommendations. The SPR assay was performed in 384-well plates.
To perform horseradish peroxidase assay (HRP assay), 10 μg of IOH-STR:HRP was added to the structure suspension and incubated for 15 min in the shaker. The unreacted IOH conjugates were removed by magnetic washing (five times) with 1% BSA in PBS buffer. Then, TMB substrate solution was added. After color development, MP were magnetically separated, and substrate oxidation in supernatant was stopped with 2 M sulfuric acid. The optical density of the solution was measured at 450 nm with a spectrophotometer.
Scanning electron microscopy (SEM)
SEM images were obtained with a Tescan MAIA3 scanning electron microscope (Tescan, Czech Republic). The specimens were prepared as follows. The assembled structures were magnetically washed with PBS (five times) and with MilliQ water (also five times) sequentially with 20 min incubation between the washings. Then, they were transferred onto a silicon wafer pretreated with a “piranha” etching solution (a 1:3 (v/v) mixture of 25% hydrogen peroxide and 98% sulfuric acid) to remove organic impurities. The microscope images were obtained at 5.0 kV in secondary electron (SE) and back scattered electron (BSE) modes.
Data processing
All experiments were performed at least thrice. For convenience of UV–Vis spectral data representation, all spectra were subjected to baseline correction (as the baseline, we regarded the average absorbance spectra of MP in 1% BSA or 10% FBS solution in PBS buffer). In all graphs, the values represent the average, and error bars show standard deviations.
Results
System design
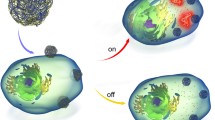
The developed nanoparticle-based smart system is multifunctional and reacts to the presence of a molecular input as a sensor and an actuator simultaneously. The system is self-assembled via a non-covalent molecular interface from two types of particles: larger magnetic one (a “core”) and smaller AuNPs “shielding” nanoparticles. The detectable analyte disrupts the non-covalent bond between the core and shielding nanoparticles, which results in system disassembly. That leads to an SPR shift due to spatial separation of AuNPs from the initial condensed state on the core nanoparticle and to the exposure of the “output” receptor of the core particle conjugate previously sterically inaccessible to the agent’s target due to concealment under the layer of the shielding particles (Fig. 1). Therefore, the system may serve as a biosensor sensitive to any type of biological molecules (DNA, RNA, protein, small molecules, etc.). Here we used antigen–antibody bond between the input receptor on the core particle and its ligand on the shielding particle. Particularly, the magnetic nanoparticle with CAP-BSA and BIO-BSA immobilized on its surface was used as a core, and AuNPs functionalized with anti-CAP antibodies were employed as the shielding particles. Additionally, to demonstrate the actuation modality of the agents, namely ability to bind with a target, we used model reporter IOH nanoparticles. The reporter particles had streptavidin molecules (which readily associated with BIO-BSA on the core particles) and HRP on their surface. After interaction between the core and reporter particles, formation of the nanocomplex was detected by adding the TMB substrate, which produced coloration due to HRP oxidation (Fig. 1). Evidently, instead of these reporter particles the agent could bind cells or other biomedically relevant targets through the agent’s output receptor (as we showed in Nikitin et al. 2014). Hence, with employment of a proper antibody or other cell-actuating receptor as the output receptor the binding of such agent with the cell could trigger specific cell signaling pathways, for example, actuate MAPK-JNK signaling pathways with anti-CD33 or anti-CDw328 antibodies (see Douglas et al. 2012).

Design of the multifunctional system and methods for evaluation of its performance. The system assembles from two types of nanoparticles (left). In this state, when AuNPs are densely settled on the surface of MPs, the SPR spectrum of the system shifts to the longer wavelengths with respect to that of the unbound gold nanoparticles (right). At the same time, the BIO-BSA molecule is sterically shielded from binding to its target on the surface of IOH particles. The system disassembles in the presence of a free molecular CAP as it replaces the immobilized CAP-BSA in the antigen–antibody complex that links the core and shielding particles. This produces both a shift in the SPR spectrum, which is recorded by UV–Vis spectrometry, and an increase of binding of IOH particles to the core particles, which is registered by the HRP assay
First, to evaluate functionality of the selected interface, we measured the SPR absorbance maximum and HRP signals for the assembled and disassembled states of the system. As a negative control, we used such configuration of the system when assembly of AuNPs-anti-CAP on the surface of MP-CAP-BSA:BIO-BSA was inhibited by free CAP in solution (“assembly block”). Briefly, AuNPs-anti-CAP were pre-incubated for 5 min with free CAP at the concentration sufficient for saturating all antibody molecules immobilized on the surface of the particles. The SPR spectral maximum for such configuration equaled 528 ± 1.0 nm. The maximal SPR shift for fully assembled system was evaluated after 1 h of MP-CAP-BSA:BIO-BSA co-incubation with AuNP-anti-CAP to be 564 ± 1.0 nm. Therefore, the dynamic range of SPR shift was 36.0 ± 1.0 nm. The difference in HRP signals for the assembled and disassembled (upon assembly block) states and, therefore, the difference in the efficacy of IOH binding by the system was 3.7-fold.
Addition of free CAP to the pre-assembled system resulted in its disassembly accompanied by the SPR shift back to the initial value and increased HRP signals. These effects were driven by disruption of the antigen–antibody complex that links AuNPs and the core particles by free CAP molecules. When present, free CAP substituted BSA-CAP molecules immobilized on the surface of core nanoparticle in the complex due to the low avidity of the used clone of anti-CAP antibody. To select antibody with a necessary affinity and avidity parameters for building the system, we analyzed avidity of different anti-CAP antibody clones, as well as stability of the built antigen–antibody complexes in the presence of free CAP by label-free spectral-correlation interferometry described in detail in Burenin et al. 2015. Eventually, we have chosen the CAP-B10 clone, which is known to be highly selective for chloramphenicol (Zvereva et al. 2015), as well as have demonstrated optimal affinity characteristics for construction of a reversible and simultaneously sensitive system. The concentration of CAP-BSA molecules (3.5 g/L) was picked up experimentally to allow the maximal SPR shift with preserving sufficient sensitivity of the system to free CAP. AuNPs concentration (13.5 nM) was also selected empirically so that they provided the necessary colloidal and sedimental stability of the nanocomplex but minimized the number of unbound gold nanoparticles in the sample.
Spectral and biochemical characterization of the system
The key factor that determines optical properties of the system is plasmonic nature of the AuNPs. Upon the system assembly, the density of immobilized particles is enough to shift the SPR absorbance spectrum to red area, whereas upon their dissociation the SPR peak shifts back. The shift magnitude is reversely proportional to the surface concentration of gold nanoparticles and directly proportional to CAP concentration. Thus, changes of biosensor spectral properties are gradual and depend on its molecular environment.
For testing the system sensitivity to free CAP, we performed a comparative analysis of the SPR shift in the one-step and two-step (“assembly block”) formats (Fig. 2b, c). In the one-step format, free hapten at different concentrations was added to the pre-assembled system, and a spectral analysis was performed after 1 h incubation. For the two-step format, AuNPs were first pre-incubated with free CAP in different concentrations for 5 min and then the core particles were added. Spectral measurements were performed 1 h after co-incubation of the two types of particles. As we expected, in both cases the SPR shift was reversely proportional to free CAP concentration (Fig. 2b). As can be observed from the dependence of absorption maximum on free CAP concentration (Fig. 2c), the studied formats exhibited different sensitivity ranges (the area of the maximal slope of the curve). For both formats, the ranges were located within higher (from 7.8·10−5 to 8.0·10−2 g/L) and lower (from 2.0·10−5 to 1.2·10−3 g/L) concentrations of free CAP, respectively. Moreover, the limit of detection (LOD) was objectively lower for the two-step format. Further alteration of CAP concentration to higher (> 0.1 g/L) or lower (< 10−5 g/L) values did not change the spectral properties of the system. The assembly and disassembly of the system were verified by SEM (Fig. 2a). To assess the potential of the system to function in complex media, we performed its assembly and disassembly in the 10% Fetal Bovine Serum (FBS) instead of the previously used 1% BSA in PBS buffer (Fig. 2d). The system behavior in FBS was similar to that observed previously: the SPR shift was specific and reversible, though, with the lesser amplitude (15 ± 1 nm).

Spectral characterization of the system: a Electron microphotographs of the system in the assembled and disassembled states; b–c Comparison of the system’s sensitivity in one-step (left) and two-step (right) formats of the analysis of CAP presence in the medium (1% BSA in PBS). For the one-step format, different concentrations of free CAP were added to the assembled system resulting in its disassembly. In the two-step format, the shielding nanoparticles were pre-incubated with free CAP in different concentrations and then added to the core particles to block the assembly; d The SPR shift and system performance in 10% Fetal Bovine Serum; e System reversibility upon addition and removal of free CAP from the medium (1% BSA in PBS)
The “actuator” function of the system was evaluated with the HRP assay to analyze binding of the system to its nanosized target (IOH particles) and its dependence on analyte concentration (CAP) in the medium. The peroxidase signal depended upon the degree of isolation of the core particles’ surface and was directly proportional to concentration of free CAP (Fig. 3a).

Biochemical characterization of the system: a HRP analysis of the system binding to its molecular target and its dependence on concentration of free CAP in the medium; b Reversibility of binding properties of the system depending on its state
As we see from the data, in such configuration of the system, the actuator function is of ON/OFF type, taking the limit values in the presence and absence of interactions between the particles. However, the signal of the system enhances intermittently upon cutoff CAP concentration (approximately 0.05 g/L) and is insensitive to its further increase.
Reversibility of the system’s functions
A characteristic feature of the developed system is dependence of its structure on the presence of non-covalent bond between the core and shielding particles. In combination with low avidity of the used antibody clone, which forms this bond, this feature might provide reversibility of changes in structure and optical properties of the system under a decrease of CAP concentration in the medium. To check this hypothesis, we removed free CAP from the disassembled system by express gel-chromatography on desalting columns. As a result of the CAP removal, the equilibrium shifted toward formation of a complex between antibody on the AuNPs surface and CAP-BSA on the core particle surface followed by the system assembly. Such re-assembly was accompanied by a reverse 10 nm shift of the SPR spectrum (Fig. 2e) and a significant drop of the HRP signal. The signal reduced to almost that of fully assembled system due to the rapid decrease in efficacy of IOH binding to the core due to its steric blocking by the shielding particles (Fig. 3b). Remarkably, after the second addition of free CAP, the system disassembled again and changed its optic and biochemical properties (Figs. 2e, 3b). Therefore, the developed biosensor changed its optical properties in reversible manner. Although, due to harsh conditions of CAP removal, the second SPR shift was smaller than during the first assembly.
Discussion
Here, we show a combination of several advanced approaches applied to development of a multifunctional nanoparticle-based smart system, which is capable to perform real-time monitoring of its microenvironment and influence it in a controllable way. The proposed system is an in situ biosensor, which can detect changes in concentration of target analyte in its natural environment without specific sample processing.
We have shown the responsiveness of the developed system to molecular ligands in the environment, investigated its sensitivity, and demonstrated the reversible character of biosensing well suitable for in situ monitoring. Indeed, most of the existing biosensors employ irreversible binding to analyte, and the sensing nanoparticles or molecules cannot be reused and become insensitive to subsequent changes in concentration of detectible molecules (Ivanov and Zubov 2016; Lin et al. 2016; Sokolov et al. 2017). Therefore, it is impossible to use such sensors for real-time measurements or monitoring.
In our system, reusability of its sensing elements and potential suitability for real-time measurements is due to disruption and recovery of low avidity antigen–antibody bond upon availability and removal of the antigen. We show regeneration of the sensing properties of the system by removing the free hapten via express-chromatography on desalting column. So far, we have shown only one regeneration cycle, which may possibly be due to the chosen method of free CAP removal from the environment. Throughout this process, the sample dilutes, and this is accompanied by loss of nanoparticle colloidal stability and their further removal from the reaction zone. We expect higher viability of the system and its ability to undergo many regenerative cycles if it has a more sophisticated design and is tested in milder conditions, where it is intended to be used in situ (e.g., to study physiological regulation of concentration of biologically active molecules in secretion and catabolism processes).
Another direction of possible improvements of the system is enhancing its sensitivity and dynamic range. Although the demonstrated characteristics of the reversible in situ biosensing are rather attractive (linear range of 2.5 orders and LOD about 10 mg/L), it is important to increase the sensitivity as high as possible and to make the smart system suitable for diagnostic applications. Its analytic characteristics could be further enhanced by optimization of the biomolecule immobilization on the core particle or improvement of the molecular receptors by adjusting the balance between the efficacy of nanoparticle assembly and capability of the system to disassemble upon presence of the input molecules. For antibody-based systems, this could be realized by adjusting the balance between affinity and avidity of the antibody. Tuning the plasmonic properties of the shielding nanoparticles by changing their size (Njoki et al. 2007), shape (Hu et al. 2006) or material (e.g., to silver or copper, see Khlebtsov and Dykman 2010) could also positively affect the LOD and dynamic range in the SPR sensing regime. When using the reporter IOH-HRP particles, the detectable signal can be substantially amplified via higher loading of the particles with HRP or via use of fluorescent or chemiluminescent substrates. Another promising route for the overall enhancement of the agents performance would be employment of other receptor types. In fact, selection of the most suitable sensing receptors is a critical factor for any biosensor. There is always a tradeoff between the receptor’s sensitivity and reversibility. Receptors that are more sensitive (hence, affine) would be less active in terms of rapid disassembly for reversal of the agent’s state. In this regard, DNA aptamers stand out due to easy control over their affinity to a target. Furthermore, aptamers have two different ways of binding a target: via either their oligonucleotide sequence or their secondary structure (Feng et al. 2014). Hence, there are two separate tools for adjustment of the receptor for assembly and disassembly. This unique feature may offer a notably better control over the system parameters than that demonstrated here with the antibodies.
Conclusion
Our system combines sensory and actuating functions; therefore, the range of potential applications of the system is significantly wider than for classic biosensors. These properties make our system attractive for use in different areas of biology and medicine with focus on cell biology, physiology and theranostics. Further development of this approach can yield nanoagents capable of non-invasive real-time profiling of biochemical markers in cell culture, tissue slices or in vivo with further response with a specific biomedical action.
References
Aghayeva UF, Nikitin MP, Lukash SV, Deyev SM (2013) Denaturation-resistant bifunctional colloidal superstructures assembled via the proteinaceous barnase–barstar interface. ACS Nano 7(2):950–961. https://doi.org/10.1021/nn302546v
Anker JN, Hall WP, Lyandres O, Shah NC, Zhao J, van Duyne RP (2008) Biosensing with plasmonic nanosensors. Nat Mater 7(6):442–453. https://doi.org/10.1038/nmat2162
Burenin AG, Urusov AE, Betin AV, Orlov AV, Nikitin MP, Ksenevich TI, Gorshkov BG, Zherdev AV, Dzantiev BB, Nikitin PI (2015) Direct immunosensing by spectral correlation interferometry: assay characteristics versus antibody immobilization chemistry. Anal Bioanal Chem 407(14):3955–3964. https://doi.org/10.1007/s00216-015-8600-y
Cheng Y, Samia AC, Meyers JD, Panagopoulos I, Fei B, Burda C (2008) Highly efficient drug delivery with gold nanoparticle vectors for in vivo photodynamic therapy of cancer. J Am Chem Soc 130:10643–10647. https://doi.org/10.1021/ja801631c
Douglas SM, Bachelet I, Church GM (2012) A logic-gated nanorobot for targeted transport of molecular payloads. Science 335:831–834. https://doi.org/10.1126/science.1214081
Feng C, Dai S, Wang L (2014) Optical aptasensors for quantitative detection of small biomolecules: a review. Biosens Bioelectron 59:64–74. https://doi.org/10.1016/j.bios.2014.03.014
Gandra N, Hendargo HC, Norton SJ, Fales AM, Palmer GM, Vo-Dinh T (2016) Tunable and amplified Raman gold nanoprobes for effective tracking (TARGET): in vivo sensing and imaging. Nanoscale 8(16):8486–8494. https://doi.org/10.1039/c5nr08980h
Govorov AO, Zhang W, Skeini T, Richardson H, Lee J, Kotov NA (2006) Gold nanoparticle ensembles as heaters and actuators: melting and collective plasmon resonances. Nanoscale Res Lett 1:84–90. https://doi.org/10.1007/s11671-006-9015-7
Holzinger M, Le Goff A, Cosnier S (2014) Nanomaterials for biosensing applications: a review//Nanomaterials for biosensing applications: a review. Front Chem 2:63. https://doi.org/10.3389/fchem.2014.00063
Howes PD, Chandrawati R, Stevens MM (2014) Bionanotechnology. Colloidal nanoparticles as advanced biological sensors. Science 346:1247390. https://doi.org/10.1126/science.1247390
Hu M, Chen J, Li Z-Y, Au L, Hartland GV, Li X, Marquez M, Xia Y (2006) Gold nanostructures: engineering their plasmonic properties for biomedical applications. Chem Soc Rev 35:1084–1094. https://doi.org/10.1039/B517615H
Ivanov AE, Zubov VP (2016) Smart polymers as surface modifiers for bioanalytical devices and biomaterials: theory and practice. Russ Chem Rev 85:565–584. https://doi.org/10.1070/RCR4567
Jo SD, Ku SH, Won Y-Y, Kim SH, Kwon IC (2016) Targeted nanotheranostics for future personalized medicine: recent progress in cancer therapy. Theranostics 6:1362–1377. https://doi.org/10.7150/thno.15335
Khlebtsov NG, Dykman LA (2010) Optical properties and biomedical applications of plasmonic nanoparticles. J Quant Spectrosc Radiat Transf 111:1–35. https://doi.org/10.1016/j.jqsrt.2009.07.012
Kim MK (2011) Digital Holographic Microscopy: Principles, Techniques, and Applications. Digital Holographic Microscopy. Springer, New York, p 240. https://doi.org/10.1007/978-1-4419-7793-9
Kim SJ, Gobi KV, Iwasaka H, Tanaka H, Miura N (2007) Novel miniature SPR immunosensor equipped with all-in-one multi-microchannel sensor chip for detecting low-molecular-weight analytes. Biosens Bioelectron 23(5):701–707. https://doi.org/10.1016/j.bios.2007.08.010
Knežević NŽ, Kaluderovic GN (2017) Silicon-based nanotheranostics. Nanoscale 9:12821–12829. https://doi.org/10.1039/C7NR04445C
Knežević NŽ, Trewyn BG, Lin VS (2011) Light- and pH-responsive release of doxorubicin from a mesoporous silica-based nanocarrier. Chemistry 17(12):3338–3342. https://doi.org/10.1002/chem.201002960
Lee D, Khaja S, Velasquez-Castano JC, Dasari M, Sun C, Petros J, Taylor WR, Murthy N (2007) In vivo imaging of hydrogen peroxide with chemiluminescent nanoparticles. Nat Mater 6(10):765–769. https://doi.org/10.1038/nmat1983
Lin Y-S, Tsai C-P, Huang H-Y, Kuo C-T, Hung Y, Huang D-M, Chen Y-C, Mou C-Y (2005) Well-ordered mesoporous silica nanoparticles as cell markers. Chem Mater 17:4570–4573. https://doi.org/10.1021/cm051014c
Lin L, Peng X, Wang M, Scarabelli L, Mao Z, Liz-Marzán LM, Becker MF, Zheng Y (2016) Light-directed reversible assembly of plasmonic nanoparticles using plasmon-enhanced thermophoresis. ACS Nano 10:9659–9668. https://doi.org/10.1021/acsnano.6b05486
Millo D, Harnisch F, Patil SA, Ly HK, Schröder U, Hildebrandt P (2011) In situ spectroelectrochemical investigation of electrocatalytic microbial biofilms by surface-enhanced resonance Raman spectroscopy. Angewandte Chemie Int Ed Engl 50(11):2625–2627. https://doi.org/10.1002/anie.201006046
Nikitin PI, Valeiko MV, Gorshkov BG (2003) New direct optical biosensors for multi-analyte detection. Sens Actuators, B 90:46–51. https://doi.org/10.1016/S0925-4005(03)00020-0
Nikitin MP, Shipunova VO, Deyev SM, Nikitin PI (2014) Biocomputing based on particle disassembly. Nat Nanotechnol 9(9):716–722. https://doi.org/10.1038/nnano.2014.156
Njoki PN, Lim I-IS, Mott D, Park H-Y, Khan B, Mishra S, Sujakumar R, Luo J, Zhong C-J (2007) Size correlation of optical and spectroscopic properties for gold nanoparticles. J Phys Chem C 111:14664–14669. https://doi.org/10.1021/jp074902z
Orlov AV, Znoyko SL, Cherkasov VR, Nikitin MP, Nikitin PI (2016a) Multiplex biosensing based on highly sensitive magnetic nanolabel quantification: rapid detection of botulinum neurotoxins A, B, and E in liquids. Anal Chem 88(21):10419–10426. https://doi.org/10.1021/acs.analchem.6b02066
Orlov AV, Bragina VA, Nikitin MP, Nikitin PI (2016b) Rapid dry-reagent immunomagnetic biosensing platform based on volumetric detection of nanoparticles on 3D structures. Biosens Bioelectron 79:423–429. https://doi.org/10.1016/j.bios.2015.12.049
Orlov AV, Burenin AG, Massarskaya NG, Betin AV, Nikitin MP, Nikitin PI (2017) Highly reproducible and sensitive detection of mycotoxins by label-free biosensors. Sens Actuators, B 246:1080–1084. https://doi.org/10.1016/j.snb.2016.12.071
Peng M, Li H, Luo Z, Kong J, Wan Y, Zheng L, Zhang Q, Niu H, Vermorken A, van de Ven W, Chen C, Zhang X, Li F, Guo L, Cui Y (2015) Dextran-coated superparamagnetic nanoparticles as potential cancer drug carriers in vivo. Nanoscale 7(25):11155–11162. https://doi.org/10.1039/C5NR01382H
Pollet J, Delport F, Janssen KP, Jans K, Maes G, Pfeiffer H, Wevers M, Lammertyn J (2009) Fiber optic SPR biosensing of DNA hybridization and DNA–protein interactions. Biosens Bioelectron 25(4):864–869. https://doi.org/10.1016/j.bios.2009.08.045
Polyak B, Medved M, Lazareva N, Steele L, Patel T, Rai A, Rotenberg MY, Wasko K, Kohut AR, Sensenig R, Friedman G (2016) Magnetic nanoparticle-mediated targeting of cell therapy reduces in-stent stenosis in injured arteries. ACS Nano. https://doi.org/10.1021/acsnano.6b04912
Rocha-Santos TA (2014) Sensors and biosensors based on magnetic nanoparticles. TrAC, Trends Anal Chem 62:28–36. https://doi.org/10.1016/j.trac.2014.06.016
Setou M, Kurabe N (2011) Mass microscopy: high-resolution imaging mass spectrometry. J Electron Microsc 60(1):47–56. https://doi.org/10.1093/jmicro/dfq079
Shevchenko KG, Cherkasov VR, Tregubov AA, Nikitin PI, Nikitin MP, Shevchenko KG, Cherkasov VR, Tregubov AA, Nikitin PI, Nikitin MP (2017) Surface plasmon resonance as a tool for investigation of non-covalent nanoparticle interactions in heterogeneous self-assembly & disassembly systems. Biosens Bioelectron 88:3–8. https://doi.org/10.1016/j.bios.2016.09.042
Shipunova VO, Nikitin MP, Nikitin PI, Deyev SM (2016) MPQ-cytometry: a magnetism-based method for quantification of nanoparticle-cell interactions. Nanoscale 8(25):12764–12772. https://doi.org/10.1039/c6nr03507h
Sokolov IL, Cherkasov VR, Tregubov AA, Buiucli SR, Nikitin MP (2017) Smart materials on the way to theranostic nanorobots: molecular machines and nanomotors, advanced biosensors, and intelligent vehicles for drug delivery. Biochimica et Biophysica Acta (BBA) - General Subjects 1861:1530–1544. https://doi.org/10.1016/j.bbagen.2017.01.027
Turkevich J, Stevenson PC, Hillier J (1951) A study of the nucleation and growth processes in the synthesis of colloidal gold Discuss. Faraday Soc. 11:55–75. https://doi.org/10.1039/DF9511100055
Vitol EA, Novosad V, Rozhkova EA (2012) Microfabricated magnetic structures for future medicine: from sensors to cell actuators. Nanomed (Lond) 7(10):1611–1624. https://doi.org/10.2217/nnm.12.133
Wu W, Jiang CZ, Roy VAL (2016) Designed synthesis and surface engineering strategies of magnetic iron oxide nanoparticles for biomedical applications. Nanoscale 8(47):19421–19474. https://doi.org/10.1039/c6nr07542h
Yuk JS, Yi S-J, Lee HG, Lee HJ, Kim Y-M, Ha K-S (2003) Characterization of surface plasmon resonance wavelength by changes of protein concentration on protein chips. Sens Actuators, B 94:161–164. https://doi.org/10.1016/S0925-4005(03)00345-9
Zvereva EA, Byzova NA, Sveshnikov PG, Zherdev AV, Dzantiev BB (2015) Cut-off on demand: adjustment of the threshold level of an immunochromatographic assay for chloramphenicol. Anal Methods 7:6378–6384. https://doi.org/10.1039/C5AY00835B
Acknowledgements
Different aspects and parts of this multidisciplinary research were partially supported by FASIE and the Russian Foundation for Basic Research (grants No 16-32-00791, 16-34-60230, 17-33-80176, 17-54-560024, 15-04-99499 and 15-02-07791).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shevchenko, K.G., Cherkasov, V.R., Nikitina, I.L. et al. Smart multifunctional nanoagents for in situ monitoring of small molecules with a switchable affinity towards biomedical targets. Appl Nanosci 8, 195–203 (2018). https://doi.org/10.1007/s13204-018-0659-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13204-018-0659-2